Working with water insoluble organic molecules in aqueous media: fluorene derivative-containing polymers as sensory materials for the colorimetric sensing of cyanide in water†
Saúl
Vallejos
,
Hamid
El Kaoutit
,
Pedro
Estévez
,
Félix Clemente
García
,
José Luis
de la Peña
,
Felipe
Serna
and
José Miguel
García
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Plaza de Misael Bañuelos s/n, 09001, Burgos, Spain. E-mail: jmiguel@ubu.es; Web: http://www2.ubu.es/quim/quimorg/polimeros/polimeros.htm Fax: +34 947 25 88 31; Tel: +34 947 25 80 85
First published on 4th March 2011
Abstract
This paper describes a strategy followed to achieve a sensing phenomenon in aqueous media using water-insoluble organic molecules. We have prepared a methacrylamide and a methacrylate with pendant cyanide chemosensors based on a fluorene-derivative motif, and we have fabricated highly hydrophilic membranes by means of copolymerising these hydrophobic monomers with others. Therefore, upon absorption of water in the membranes, solvated ions enter the membrane by a simple diffusion mechanism, reaching the hydrophobic chemosensor motifs and giving rise to a macroscopic sensing phenomenon. In this way, we have prepared solid materials (dense membranes or films) capable of selectively detecting cyanide, with an extremely low detection threshold, in aqueous solution by means of colour changes (naked-eye sensing) (13 ppb). Nevertheless, the key point of this research is the description of the possibilities of anchoring organic insoluble molecules (i.e., drugs, fungicides, bactericides, sensing probes, etc.) to solubilise them in water, or to prepare hydrogels, permitting the use of these molecules in aqueous media or in biological media for medical, biological or biochemical purposes.
Introduction
Molecular recognition is a widespread, naturally occurring key event in nature. The recognition between two or more molecules occurs when they are chemically and geometrically, or structurally, complementary. Thus, the “fit together” takes place spatially, and the binding to each other arises from intermolecular feeble interactions, including hydrogen bonds, electrostatic interactions, hydrophobic interactions and weak metal coordination.1,2 In nature, the processes take place in aqueous environments and are usually implicated in natural macromolecules or even in complex and organised systems, such as cells. Examples of the recognition process include the binding of an enzyme to a substrate,3 a drug to a biological target,4,5antigen/antibody recognition in the immune system,6,7 the formation of messenger RNA from DNA templates,8etc. In this context, the development of chemosensors for the detection of target chemicals is a field of current interest.9Naturally, recognition in biological systems takes place in aqueous environments. The active sites, for example in an enzyme, have the shape needed to fit the guest molecule, or a part of the guest molecule, and the interaction relies on feeble bonds that take place due to the hydrophobic microdomain in a hydrophilic general domain provided by the chemical and by the quaternary structure of a protein. In a very simplified manner, the competitiveness of the water molecules to establish these weak integrations is locally diminished or altered by the active site microsurroundings.
Mimicking nature, water insoluble receptors may be chemically anchored to a linear or crosslinked hydrophilic polymer, giving rise to water-soluble sensing polymers or to water-swelled sensing networks. The latter approach permits easy control of swelling by means of increasing the crosslinking ratio. That is, the hydrophilic character of a polymer, derived from the chemical composition of the monomers, may be controlled not only by the monomer nature, but also by increasing the crosslinking monomer content of the network; thus, mechanically impairing the water uptake. Thus, an induced partially hydrophobic character may be addressed with an overall hydrophilic polymer associated with the mechanical stretching of the network upon swelling with water, as we have previously described.10,11 Moreover, the polymers usually have good thermal and mechanical properties, can be easily transformed into end materials, e.g., into films or coatings obtained by casting, to produce cheap sensing devices (e.g., user-friendly “naked-eye” film sensors). These polymers may also be incorporated as coating at the end of an optic fibre connected to a portable UV/Vis diode-array or fluorescence detector.12–14
For example, cyanide is a highly toxic, chemical target currently used in many industrial processes.15–17 Due to its toxicity, the United States Environmental Protection Agency (EPA) has set the maximum contaminant level (MCL) for cyanide in drinkingwater at 0.2 ppm, rendering its detection important in terms of both environmental and industrial control.
The detection ofcyanide through various supramolecular approaches16 (the binding site-signalling subunit,18 the chemodosimeter19,20 or the displacement21–24) has been extensively reported. Nevertheless, few examples of sensing of the anion in aqueous solutions,20,22,24,25 some of them showing detection limits lower than the MCL,16,22 have been described. Most cyanide sensing molecules are insoluble in water because of their organic nature, preventing their use in aqueous media. We felt that detection ofcyanide by means of changes in optical signals upon interaction of a target guest with a sensor molecule could give rise to the development of inexpensive, portable devices for environmental testing inside or outside the laboratory.9,15 Prior to our investigation, few examples existed describing materials exhibiting colorimetric, or fluorescence, recognition with potential use as solid systems or practical kits for “dip-in” naked-eye cyanide detection (e.g., films or membranes,10,25,26quantum dots27 or supported materials28). We hypothesised that the chemical anchoring of a sensing motif to a hydrophilic polymer chain could provide a water-rich environment to the sensing moiety upon polymer swelling, thus permitting the solvated cyanide ions to reach the guest or the transducer cores, giving rise to the sensing phenomenon, and resulting in cyanide detection.
Experimental
All materials and solvents were commercially available and used as received, unless otherwise indicated. They included the following: sodium cyanide (Panreac, 98%), 2-isocyanato-9H-fluorene (Aldrich, 98%), ethanolamine (Merck, 98%), cesium carbonate (Aldrich, 99%), malononitrile (Aldrich, 99%), 2-nitro-9H-fluorene (Aldrich, 98%), Pd/C (Aldrich, 10%), methacryloyl chloride (Fluka, 97%), 2-(2-aminoethoxy ethanol) (Aldrich, 98%), fluorenone (Aldrich, 98%), ethylene glycol dimethacrylate (Aldrich, 98%), DMSO (Merck, 99%), acetone (Aldrich, 99%), ethanol (Aldrich, 99%), dichloromethane (Aldrich, 99%), DMF (Aldrich, 99%), hexanes (Aldrich, 99%), and ethyl acetate (Aldrich, 99%). Pyridine was dried under reflux over sodium hydroxide for 24 h and was distilled over 4 Å molecular sieves. Azo-bis-isobutyronitrile (AIBN, Aldrich, 99%) was recrystallised twice from methanol.Intermediates and monomers
The two methacrylamide and methacrylate monomers were synthesised following the reaction conditions described in Scheme 1.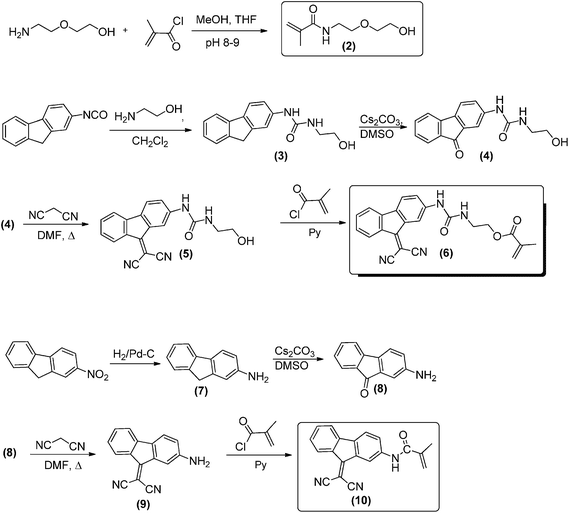 | ||
| Scheme 1 Monomer synthesis and labels. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 1704; δN–H: 1606.
o: 1656.
O: 1701.
O: 1671.
O: 1704; δN–H: 1606.
o: 1656.
O: 1701.
O: 1671.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (3:1). Mp: 212 ± 2 °C. Yield: 83%. 1H NMR, δ (400 MHz, DMSO-d6): 8.99 (s, 1H, NH); 8.30 (m, 1H, ArH); 8.12 (d, 1H, J = 7.8 Hz, ArH); 7.64 (m, 3H, ArH); 7.54 (t, 1H, J = 7.5 Hz, ArH); 7.32 (t, 1H, J = 7.7 Hz, ArH); 6.41 (t, 1H, J = 5.9 Hz, NH); 6.12 (s, 1H, CH2); 5.74 (s, 1H, CH2); 4.18 (t, 1H, J = 5.4 Hz, CH2); 1.93 (s, 3H, CH3). 13C NMR, δ (100.6 MHz, DMSO-d6): 167.17, 161.26, 155.71, 143.23, 142.26, 136.46, 135.80, 135.25, 134.86, 134.06, 128.69, 126.56, 123.90, 122.43, 121.15, 116.24, 114.27, 113.87, 81.67, 76.15, 64.61, 18.57. EI-LRMS m/z: 399.1 (M+, 2), 398.1 (8), 313.1 (1), 270.1 (7), 243.1 (100), 226.1 (1), 163.0 (2). FTIR [wavenumbers (cm−1)]: νNH: 3355; νCN: 2220; νCO: 1708.
O: 1701.
:ethyl acetate (1:1). Mp: 262 ± 1 °C.Yield before purification: 80%. Yield after purification: 25%. 1H NMR, δ (400 MHz, DMSO-d6): 8.07 (d, 1H, J = 7.8 Hz, ArH); 7.51–7.40 (m, 4H, ArH); 7.19 (dt, 1H, J = 1.9, 7.3 Hz, ArH); 6.71 (dd, 1H, J = 2.0, 8.1 Hz, ArH); 5.88 (s, 2H, NH2). 13C NMR, δ (100.6 MHz, DMSO-d6): 162.31, 151.04, 144.62, 135.84, 133.70, 129.86, 127.27, 126.49, 123.00, 119.95, 119.39, 114.55, 114.21, 112.18. EI-LRMS m/z: 242.1 (M+, 3), 243.1 (100), 217.1 (1), 215.1 (15), 189.1 (7), 78.0 (28). FTIR [wavenumbers (cm−1)]: νNH: 3480, 3370; νCN: 2220; νCO: 1613.
:ethyl acetate (2:1) and was washed in a Soxhlet assembly with ethanol. Mp: 224 ± 1 °C. Yield before purification: 87%. Yield after purification: 20%. 1H NMR, δ (400 MHz, DMSO-d6): 10.21 (s, 1H, NH); 8.78 (m, 1H, ArH); 8.19 (d, 1H, J = 7.8 Hz, ArH); 7.84–7.73 (m, 3H, ArH); 7.60 (t, 1H, J = 7.5 Hz, ArH); 7.40 (t, 1H, J = 7.7 Hz, ArH); 5.87 (s, 1H, CH2); 5.61 (s, 1H, CH2); 2.00 (s, 3H, CH3). 13C NMR, δ (100.6 MHz, DMSO-d6): 167.81, 160.92, 142.72, 140.75, 137.39, 135.84, 134.61, 134.27, 129.20, 126.80, 126.66, 122.22, 121.61, 121.15, 118.66, 114.36, 113.74, 76.56, 19.40. EI-LRMS m/z: 310.1 (M+, 1), 311.1 (73), 69.0 (100), 188.0 (9), 78.0 (5). FTIR [wavenumbers (cm−1)]: νNH: 3377; νCN: 2220; νCO: 1679.
:ethyl acetate (3:1). Mp: 212 ± 2 °C. Yield: 83%. 1H NMR, δ (400 MHz, DMSO-d6): 8.99 (s, 1H, NH); 8.30 (m, 1H, ArH); 8.12 (d, 1H, J = 7.8 Hz, ArH); 7.64 (m, 3H, ArH); 7.54 (t, 1H, J = 7.5 Hz, ArH); 7.32 (t, 1H, J = 7.7 Hz, ArH); 6.41 (t, 1H, J = 5.9 Hz, NH); 6.12 (s, 1H, CH2); 5.74 (s, 1H, CH2); 4.18 (t, 1H, J = 5.4 Hz, CH2); 1.93 (s, 3H, CH3). 13C NMR, δ (100.6 MHz, DMSO-d6): 167.17, 161.26, 155.71, 143.23, 142.26, 136.46, 135.80, 135.25, 134.86, 134.06, 128.69, 126.56, 123.90, 122.43, 121.15, 116.24, 114.27, 113.87, 81.67, 76.15, 64.61, 18.57. EI-LRMS m/z: 399.1 (M+, 2), 398.1 (8), 313.1 (1), 270.1 (7), 243.1 (100), 226.1 (1), 163.0 (2). FTIR [wavenumbers (cm−1)]: νNH: 3355; νCN: 2220; νCO: 1708.
O: 1701.
:ethyl acetate (1:1). Mp: 262 ± 1 °C.Yield before purification: 80%. Yield after purification: 25%. 1H NMR, δ (400 MHz, DMSO-d6): 8.07 (d, 1H, J = 7.8 Hz, ArH); 7.51–7.40 (m, 4H, ArH); 7.19 (dt, 1H, J = 1.9, 7.3 Hz, ArH); 6.71 (dd, 1H, J = 2.0, 8.1 Hz, ArH); 5.88 (s, 2H, NH2). 13C NMR, δ (100.6 MHz, DMSO-d6): 162.31, 151.04, 144.62, 135.84, 133.70, 129.86, 127.27, 126.49, 123.00, 119.95, 119.39, 114.55, 114.21, 112.18. EI-LRMS m/z: 242.1 (M+, 3), 243.1 (100), 217.1 (1), 215.1 (15), 189.1 (7), 78.0 (28). FTIR [wavenumbers (cm−1)]: νNH: 3480, 3370; νCN: 2220; νCO: 1613.
:ethyl acetate (2:1) and was washed in a Soxhlet assembly with ethanol. Mp: 224 ± 1 °C. Yield before purification: 87%. Yield after purification: 20%. 1H NMR, δ (400 MHz, DMSO-d6): 10.21 (s, 1H, NH); 8.78 (m, 1H, ArH); 8.19 (d, 1H, J = 7.8 Hz, ArH); 7.84–7.73 (m, 3H, ArH); 7.60 (t, 1H, J = 7.5 Hz, ArH); 7.40 (t, 1H, J = 7.7 Hz, ArH); 5.87 (s, 1H, CH2); 5.61 (s, 1H, CH2); 2.00 (s, 3H, CH3). 13C NMR, δ (100.6 MHz, DMSO-d6): 167.81, 160.92, 142.72, 140.75, 137.39, 135.84, 134.61, 134.27, 129.20, 126.80, 126.66, 122.22, 121.61, 121.15, 118.66, 114.36, 113.74, 76.56, 19.40. EI-LRMS m/z: 310.1 (M+, 1), 311.1 (73), 69.0 (100), 188.0 (9), 78.0 (5). FTIR [wavenumbers (cm−1)]: νNH: 3377; νCN: 2220; νCO: 1679.
The model compounds used to clarify the sensing mechanism were synthesised following the reaction conditions described in Scheme 4.
Membrane preparation
The membranes were prepared by radical polymerisation of mixtures of the 4 different co-monomers (1, 2, 6, 10) using ethylene glycol dimethacrylate as a cross-linking agent (1%) and AIBN (1 wt%) as a thermal radical initiator. The monomers (6) and (10) were added in DMF. Eight different polymer films were prepared in this fashion by varying the molar concentration of monomers. The reactions were performed in silanised glass moulds of 100 μm thickness in an oxygen-free atmosphere at 65 °C for 5 h. Upon demoulding, the film was air dried for two days and overnight under vacuum at rt.Measurements
1H and 13C NMR spectra were recorded with a Varian Inova 400 spectrometer operating at 399.92 and 100.57 MHz, respectively, with deuterated dimethyl sulfoxide (DMSO-d6) as solvent. Infrared spectra (FT-IR) were recorded with a Nicolet Impact spectrometer or with a JASCO FT/IT-4100 fitted with a PIKE TECH “Miracle” ATR. Thermogravimetric analysis (TGA) data were recorded on a 5 mg sample under a nitrogen or oxygen atmosphere on a TA Instrumenta Q50 TGA analyser at a scan rate of 10 °C min−1. UV-Vis spectra were recorded using a Varian Cary3-Bio UV-Visspectrophotometer. Sensing measurements performed in water were performed at physiological pH (7.5, buffered with Tris). Apparent response time was recorded using a digital stopwatch. The different polymeric films were dipped in a water solution of sodium cyanide (3 M), and the time was taken when the colour of the film turned colourless to the naked eye. Each experiment was repeated three times, and the results were averaged. The water-swelling percentage (WSP), the weight percentage of water uptake by the films upon soaking until equilibrium, in pure water, was obtained from the weights of a dry sample film (wd) and a water-swelled (the membranes were immersed in pure water at 20 °C until the swelled equilibrium was achieved) sample film (ws) as follows: 100 × [(ws − wd)/wd].Results and discussion
The main aim of this research was to extend some of the properties or characteristics of organic molecules, insoluble in water, to aqueous media by means of chemically incorporating them into a hydrophilic polymer network. To investigate our hypothesis, we prepared two methacrylic and methacrylamide monomers containing a fluorene derivative as a colorimetric cyanide sensing motif [(6) and (10)] and copolymerised them with either a hydrophobic or a hydrophilic monomer [(1) and (2), respectively]. The cross-linking agent 1,2-ethanedioldimethacrylate was used to obtain different, dense polymer films or membranes (Scheme 2 and Table 1), which were optically transparent and had good mechanical properties, even after water swelling, from which cyanide solid sensing strips could be easily obtained with a hand blade from the films.
| Membrane or film | 1 (x)a | 2 (y)a | 6 (z)a | 10 (z)a | WSP b | Raw filmc | Film + CN−c | Response time/sd |
|---|---|---|---|---|---|---|---|---|
| a Molar composition ratio of the monomers used for the preparation of the films. b Water-swelling percentage. c Digital picture of the films taken over a written paper upon soaking in pure water and in an aqueous solution of cyanide (10 equiv. per equiv. of the sensing fluorene derivative motif), respectively; the pictures have been taken on written paper to show the transparency and the different shades of the films. d Naked-eye response time (—: without response, n.a.: not applicable). | ||||||||
| (a) | 100 | — | 2 | — | 3 |

|

|
— |
| (b) | 80 | 20 | 2 | — | 20 |

|

|
— |
| (c) | 60 | 40 | 2 | — | 80 |

|

|
72 |
| (d) | 50 | 50 | 2 | — | 110 |

|
|
33 |
| (e) | — | 100 | 2 | — | 275 |

|

|
20 |
| (f) | — | 100 | — | 2 | 250 |

|

|
35 |
| (g) | 50 | 50 | 0.2 | — | 185 |
|

|
n.a. |
| (h) | 50 | 50 | — | 0.2 | 160 |

|

|
n.a. |
Monomer synthesis and characterisation
The reaction steps performed to obtain the new methacrylate (6) and methacrylamide (10) monomers incorporating the cyanide sensing motifs were straightforward and well documented from an organic chemistry viewpoint and produced the monomers from widely available chemicals in good yield and purity. These monomers were incorporated as comonomers in the dense crosslinked membranes at a molar percentage lower than 2%, meaning that their presence in the membrane network had a negligible impact in the mechanical properties, or handling, of the materials due to their low molar and weight contents. Purification by column chromatography yielded monomers of high purity, as demonstrated by NMR spectra. The intermediates and monomers were characterised by IR, 1H and 13C NMR spectroscopy, thereby fully confirming the chemical structure of all the products. As an illustrative example, Fig. 1 depicts the 1H NMR spectra of the monomers.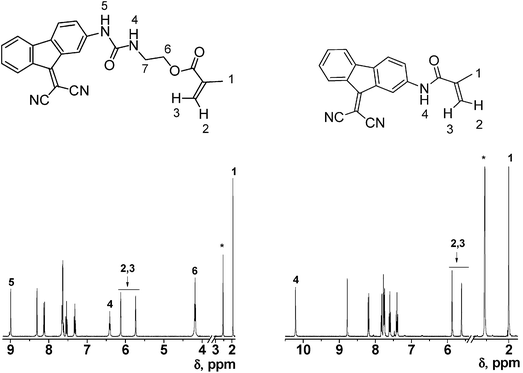 | ||
| Fig. 1 1H NMR of the monomers containing the sensing motifs (DMSO-d6, * = solvent signals, multiplets corresponding to protons 7 of monomer (6) were observed at 3.4 ppm, with water signal). | ||
The hydrophobic and hydrophilic monomers (1) and (2) represented the 98.0% or the 99.8% molar content of the membranes, excluding the crosslinker. Thus, monomer (1) was chosen because it is a commercial chemical and (2) was chosen for reasons including the following: (a) their easy and cheap preparation and purification; (b) their hydrophilicity; (c) their full compatibility with (1), thus permitting the design of a membrane with a la carte hydrophilicity (i.e., the water uptake or gel behaviour); and (d) the good handling of the crosslinked membranes obtained with it, both as dry or water-swelled materials.
Membrane preparation and characterisation
Structural monomers (1) and (2), as well as the crosslinker, were fully miscible and could be mixed in any molar ratio. On the other hand, the monomers containing the sensing motifs (6) and (10) were highly insoluble and could not be solved in common organic solvents other than DMF and DMSO. Thus, preparation of the dense membrane started with a solution of the radical initiator and the mixture of monomers (1) and (2) (600 μL), to which the functional monomers (6) or (10) were added in small quantities of DMF (500 μL). Lowering the amount of DMF gave rise to precipitation or insolubilisation of (6) or (10), and an increase in the DMF content led to excessive plasticisation of the membrane as well as to handling problems immediately after demoulding. The homogeneous polymerisation mixture was then nitrogen bubbled, to eliminate the solved oxygen, and immediately injected in silanised glass moulds, and upon heating, the “almost” bulk polymerisation was conducted smoothly. The silanisation of the glass mould facilitated demoulding of the plasticised material, in which DMF was first air dried and then dried under vacuum (always at rt).The structure of the copolymer network of the membrane, the sensing material, is depicted in Scheme 2 along with a digital picture of one of the membranes, and the composition of each of the 8 membranes and their properties are shown in Table 1. The good optical properties of the membranes can be appreciated in the picture inset of Scheme 2.
The membranes were characterised in the dry solid state by ATR-FTIR showing the absorption bands corresponding to the stretching and bending of the groups presented in the chemical structure, as depicted in Fig. 2 for membrane (d).
 | ||
| Fig. 2 ATR-FTIR of membrane (d) before (black line) and after (red line) hindering in a water cyanide solution. | ||
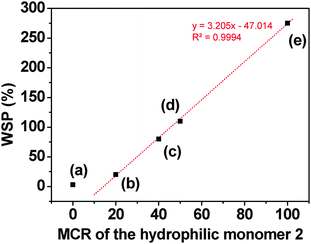 | ||
| Fig. 3 Water-swelling percentage (WSP) of membranes (a) to (e) vs. the molar composition ratio (MCR) of the hydrophilic monomer 2 (the code of each membrane, as shown in Table 1, is depicted close to each symbol). The straight line is a linear fit of the data corresponding to membranes with a molar content of 1 lower than 100%. | ||
Cyanide sensing
Depending on the molar composition ratio (MCR) of the monomers used for preparation of the films, their observed colours were brownish or reddish [films (a) to (f)] or light violet [films (g) and (h)], both when dry and when water-swelled. Upon soaking in aqueous solutions of cyanide, the films turned colourless in less than 72 seconds, which represents a low apparent response time,28 with the lower values corresponding to the higher hydrophilic character of the membrane, depicted in Table 1 and Fig. 2 by the water-swelling percentage (WSP). Fig. 4 depicts the evolution of the colour of a membrane strip upon soaking in an aqueous solution of cyanide. | ||
| Fig. 4 Colour disappearance of a film strip of the membrane (d) upon soaking in an aqueous solution of cyanide. | ||
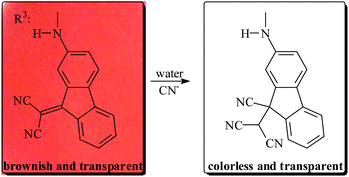 | ||
| Scheme 3 Schematic representation of the proposed sensing mechanism (background: digital picture of film (d)). | ||
Membranes prepared with monomers containing the urea subgroup [monomer (6), Scheme 1] and without it [monomer (10), Scheme 1] showed a similar response toward cyanide, indicating that this group plays a negligible role in the chemodosimeter response of the fluorene derivative motif. Nevertheless, the fluorene derivative with and without the urea unit influenced the UV/Vis spectra of the corresponding membranes, as shown below. The binding characteristics of the ureagroups were probably not observed due to the aqueous media, the hydrogen bonds and water solvation and the neutral conditions.
The UV/Vis calculated spectra, by means of molecular modelling of the reaction of model compounds of copolymers, supported the proposed mechanism. The model compounds used to model the behaviour of the sensing motifs within the membrane had a similar structure to (6) and (10), replacing the methacrylate and the methacrylamidegroup by a 2-methylpropanoate or 2-methylpropanamidegroups; thus, mimicking the polymer network backbone in the membrane. Modelling of (6) in aqueous media gave rise to absorption in the visible region that disappeared upon reaction with cyanide, as depicted in Fig. 5. The experimental spectra of membrane (d) showed comparable behaviour.
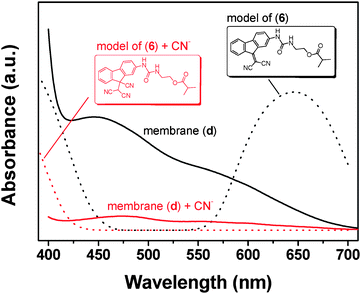 | ||
| Fig. 5 Theoretical calculation of UV/Vis spectra of a model molecule of monomer 6 before and after reaction with CN− (UV-Vis DFT calculations at B3LYP/6-31G*, in water), along with the spectra corresponding with membrane (d) before and after immersing it in water solution of CN− (10−1 M), solid lines. | ||
This mechanism was fully confirmed by means of the preparation and characterisation of 2-(9H-fluoren-9-ylidene)malononitrile and its reaction with cyanide to give 2-(9-cyano-9H-fluoren-9-yl)malononitrile, as depicted in Scheme 4. The former reacted with cyanide in a selective and quantitative manner though a Michael reaction.
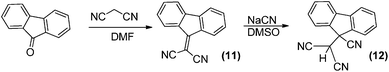 | ||
| Scheme 4 Synthesis of the model 2-(9-cyano-9H-fluoren-9-yl)malononitrile. | ||
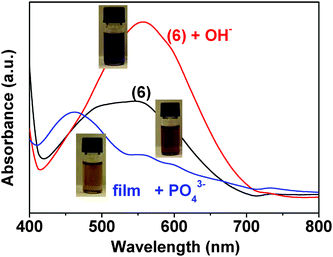 | ||
| Fig. 6 Solution of monomer (6) in DMSO/water (90:10, v/v), 2 × 10−3 M, before and after addition of hydroxide or phosphate anions 2 × 10−2 M). | ||
 | ||
| Fig. 7 Selected UV/Vistitration curve of film (d) with cyanide in buffered water (pH = 7.5) (inset: cyanide concentration vs. I/I0 at 445 nm). | ||
Upon lowering the MCR of the copolymer from 2% (films (a)–(f)) to 0.2% [films (g) and (h)], the absorption bands of the UV region were observed {a maximum with shoulders at 351 nm [film (g)], and two maxima were observed at 290 and 354 nm [film (h)]}. The progressive addition of cyanide to the cell containing the copolymer films gave rise to subsequent diminishment of the absorbance intensity of these bands, thus permitting titration and giving rise to a detection limit lower than 13 ppb for film (h), which is much lower than the MCL (Fig. 8), and 260 ppb for film (g) (Fig. 9).
 | ||
| Fig. 8 Selected UV/Vistitration curve of film (h) with cyanide in buffered water (pH = 7.5) (inset: absorbance intensity ratio at 290 and 354 nm (I290/I354) vs.cyanide concentration). | ||
 | ||
| Fig. 9 Selected UV/Vistitration curve of film (g) with cyanide in buffered water (pH = 7.5) (inset: cyanide concentration vs. I/I0 at 351 nm). | ||
Conclusion
In summary, water-insoluble organic molecules (6) and (10), with a chemodosimeter behaviour toward the cyanide anion, were chemically incorporated in the lateral chain of a methacrylic copolymer to give different, dense membranes. The design of the membrane composition allowed for the preparation of a solid material with ‘a la carte’ hydrophilic character. Upon swelling with water, the membrane permitted the colorimetric detection ofcyanide with an extremely low detection limit. Thus, through diffusion into the swelled membrane, the solvated ions reached the highly hydrophobic sensing organic motifs, giving rise to a recognition phenomenon. Following this procedure, this supramolecular sensing phenomenon can be easily performed by means of anchoring organic sensing molecules to appropriate polymeric nets. Using this approach, we are now successfully developing membranes with the ability of detecting biologically and medically important molecules. Moreover, we are extending our studies to the preparation of linear copolymers for the water solubilisation of all types of chemicals, with an eye on the development of new drugs.Acknowledgements
The authors gratefully acknowledge financial support provided by the Spanish Ministerio de Educación y Ciencia—Feder (MAT2008-00946/MAT) and by the Junta de Castilla y León (BU001A10-2).References
- N. M. Bergmann and N. Peppas, Prog. Polym. Sci., 2008, 33, 271 CrossRef CAS.
- B. N. Chen, S. Piletsky and A. P. F. Turner, Comb. Chem. High Throughput Screening, 2002, 5, 409 CAS.
- A. Tulinsky, Semin. Thromb. Hemostasis, 1996, 22, 117 Search PubMed.
- M. Britschgi, S. von Greyerz, C. Burkhart and W. J. Pichler, Curr. Drug Targets, 2003, 4, 1 Search PubMed.
- P. Cudic, D. C. Behenna, J. K. Kranz, R. G. Kruger, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Chem. Biol., 2002, 9, 897 CrossRef CAS.
- E. J. Sundberg and R. A. Mariuzza, J. Protein Chem., 2002, 61, 119.
- R. Jimenez, G. Salazar, K. K. Baldridge and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 92 CrossRef CAS.
- S. A. Hofstadler and R. H. Griffery, Chem. Rev., 2001, 101, 377 CrossRef CAS.
- R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef CAS.
- F. García, J. M. García, B. García-Acosta, R. Martínez-Mañez, F. Sancenón, N. San-José and J. Soto, Chem. Commun., 2005, 2790 RSC.
- B. García-Acosta, F. García, J. M. García, R. Martínez-Mañez, F. Sancenón, N. San-José and J. Soto, Org. Lett., 2007, 9, 2429 CrossRef CAS.
- J. M. García, F. C. García, F. Sena and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623 CrossRef CAS.
- K. C. Persaud, Mater. Today, 2005, 8, 38 CrossRef CAS.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79 RSC.
- K. W. Kulig, in Cyanide Toxicity, US Department of Health and Human Services, Atlanta, GA, 1991 Search PubMed; Guidelines for Drinking-Water Quality, World Health Organization, Geneva, 1996 Search PubMed.
- For a recent review see: Z. Xu, X. Chen and J. Yoon, Chem. Soc. Rev., 2010, 39, 127 Search PubMed.
- S. Vallejos, P. Estévez, F. C. García, F. Serna, J. L. de la Peña and J. M. Garacía, Chem. Commun., 2010, 46, 7951 RSC.
- T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, J. Organomet. Chem., 2009, 694, 3833 CrossRef CAS; V. Kumar, M. P. Kaushika, A. K. Srivastava, A. Pratap, V. Thiruvenkatam and T. N. Row, Anal. Chim. Acta, 2010, 663, 77 CrossRef CAS; J. Wang and C.-S. Ha, Tetrahedron, 2010, 66, 1846 CrossRef CAS.
- See, for instance: T. Ábalos, S. Royo, R. Martínez-Máñez, F. Sancenón, J. Soto, A. M. Costero, S. Gil and M. Parra, New J. Chem., 2009, 33, 1641 Search PubMed; H.-T. Niu, D. Su, X. Jiang, W. Yang, Z. Yin, J. He and J.-P. Cheng, Org. Biomol. Chem., 2008, 6, 3038 RSC; Y. Sun, G. Wang and W. Guo, Tetrahedron, 2009, 65, 3480 RSC; S. K. Kwon, S. Kou, H. N. Kim, X. Chen, H. Hwang, S.-W. Nam, S. H. Kim, K. M. K. Swamy, S. Park and J. Yoon, Tetrahedron Lett., 2008, 49, 4102 CrossRef CAS; D.-G. Cho, J. H. Kim and J. L. Sessler, J. Am. Chem. Soc., 2008, 130, 12163 CrossRef CAS; J. L. Sessler and D.-G. Cho, Org. Lett., 2008, 10, 73 CrossRef CAS; J. Ren, W. Zhu and H. Tian, Talanta, 2008, 75, 760 CrossRef CAS; S.-J. Hong, J. Yoo, S.-H. Kim, J. S. Kim, J. Yoon and C.-H. Lee, Chem. Commun., 2009, 189 CrossRef CAS; Y.-K. Yang and J. Tae, Org. Lett., 2006, 8, 5721 RSC; G. Qian, X. Li and Z. Y. Wang, J. Mater. Chem., 2009, 19, 522 CrossRef CAS; J. O. Huh, Y. Do and M. H. Lee, Organometallics, 2008, 27, 1022 RSC; T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, Chem.–Eur. J., 2009, 15, 5056 CrossRef CAS; M. Jamkratoke, V. Ruangpornvisuti, G. Tumcharen, T. Tuntulani and B. Tomapatanaget, J. Org. Chem., 2009, 74, 3919 CrossRef CAS.
- L. Peng, M. Wang, G. Zhang, D. Zhang and D. Zhu, Org. Lett., 2009, 11, 1943 CrossRef CAS; Y. Sun, Y. Liu and W. Guo, Sens. Actuators, B, 2009, 143, 171 CrossRef; J. Jo and D. Lee, J. Am. Chem. Soc., 2009, 131, 16283 CrossRef CAS; Y. Sun, Y. Liu, M. Chen and W. Guo, Talanta, 2009, 80, 996 CrossRef; H.-T. Niu, X. Jiang, J. He and J.-P. Cheng, Tetrahedron Lett., 2009, 50, 6668 CrossRef CAS; K.-S. Lee, H.-J. Kim, G.-H. Kim, I. Shin and J.-I. Hong, Org. Lett., 2008, 10, 49 CrossRef CAS.
- J. H. Lee, A. R. Jeong, I.-S. Shin, H.-J. Kim and J.-I. Hong, Org. Lett., 2010, 12, 764 CrossRef CAS; R. Guliyev, O. Buyukcakir, F. Sozmen and O. A. Bozdemir, Tetrahedron Lett., 2009, 50, 5139 CrossRef CAS; L. Shang, L. Zhang and S. Dong, Analyst, 2009, 134, 107 RSC; F. H. Zelder, Inorg. Chem., 2008, 47, 1264 CrossRef CAS; C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272 CrossRef CAS.
- X. Lou, L. Zhang, J. Qin and Z. Li, Chem. Commun., 2008, 5848 RSC.
- X. Chen, S. W. Nam, G. H. Kim, N. Song, Y. Jeong, I. Shin, S. K. Kim, J. Kim, S. Park and J. Yoon, Chem. Commun., 2010, 46, 8953 RSC.
- Z. Xu, J. Pan, D. R. Spring, J. Cui and J. Yoon, Tetrahedron, 2010, 66, 1678 CrossRef CAS; L. Shang, L. Jin and S. Dong, Chem. Commun., 2009, 3077 RSC; P. Kaur, S. Kaur and K. Singh, Inorg. Chem. Commun., 2009, 12, 978 CrossRef CAS; Y. Liu, K. Ai, X. Cheng, L. Huo and L. Lu, Adv. Funct. Mater., 2010, 20, 951 CrossRef CAS; X. Lou, J. Qin and Z. Li, Analyst, 2009, 134, 2071 RSC; C. Männel-Croisé, B. Probst and F. Zelder, Anal. Chem., 2009, 81, 9493 CrossRef CAS; S.-Y. Chung, S.-W. Nam, J. Lim, S. Park and J. Yoon, Chem. Commun., 2009, 2866 RSC.
- N. Gimeno, X. Li, J. R. Durrant and R. Vilar, Chem.–Eur. J., 2008, 14, 3006 CrossRef CAS.
- Q. Zeng, P. Cai, Z. Li, J. Qina and B. Z. Tang, Chem. Commun., 2008, 1094 RSC; Z. Li, X. Lou, H. Yu, Z. Li and J. Qin, Macromolecules, 2008, 41, 7433 CrossRef CAS; Z. Ekmekci, M. D. Yilmaz and E. U. Akkaya, Org. Lett., 2008, 10, 461 CrossRef CAS.
- A. Touceda-Varela, E. I. Stevenson, J. A. Galve-Gasión, D. T. F. Dryden and J. C. Mareque-Rivas, Chem. Commun., 2008, 1998 RSC.
- P. Kaur, D. Sareen, S. Kaur and K. Singh, Inorg. Chem. Commun., 2009, 12, 272 CrossRef CAS.
- V. Compañ, P. Tiemblo, F. García, J. M. García, J. Guzmán and E. Riande, Biomaterials, 2005, 26, 3783 CrossRef CAS.
- H. D. Hartzler, J. Org. Chem., 1966, 31, 2654 CrossRef CAS.
- D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Intermediates and monomer NMR and IR-FT spectra, a TGA curve, as well as a movie showing the colorimetric response of a film (d) toward cyanide. See DOI: 10.1039/c1py00013f |
| This journal is © The Royal Society of Chemistry 2011 |