DOI:
10.1039/C0PY00420K
(Paper)
Polym. Chem., 2011,
2, 1195-1202
Preparation of poly(2-oxy-6-naphthoyl) and copolymers using reaction-induced phase separation during direct polymerization in the presence of boronic anhydride
Received
23rd December 2010
, Accepted 26th January 2011
First published on 21st February 2011
Abstract
Poly(2-oxy-6-naphthoyl) (PON) was obtained as precipitates by direct polymerization of 2-hydroxy-6-naphthoic acid (HNA) in the presence of boronic anhydrides such as 2,4,6-tris(3,4,5-trifluorophenyl)-1,3,5,2,4,6-trioxatriborinane (TFBA) and 2,4,6-tri(biphenyl-4-yl)-1,3,5,2,4,6-trioxatriborinane (BPBA). The polymerizations were carried out in an aromatic solvent at 300 °C for 24 h. Both morphology and molecular weight of the PON precipitates were considerably influenced by not only the structure of the boronic anhydride but also its concentration (cB). Spheres with needles on their surface were formed in the polymerization with TFBA at cB of 50–70 mol%, and those having smooth surface were obtained at cB of 100 mol%. The Mn increased with the value of cB in the range from 2.7 × 103 to 9.0 × 103. Aggregates of cone-like crystals were prepared in the polymerization with BPBA at cB of 10–30 mol%, and spheres having rugged surface were formed at cB of 50–100 mol%. The Mn also increased with the value of cB in the range from 5.4 × 103 to 12.9 × 103. These PON precipitates possessed high crystallinity in spite of the morphology. The copolymerization of HNA and 4-hydroxybenzoic acid (HBA) was also examined in the presence of BPBA at cB of 100 mol% with varying the content of HBA in feed (rf). Copolymers were obtained as spheres of which the content of 4-oxybenzoyl moiety was lower than the value of rf due to the difference in the solubility of the oligomers.
Introduction
Poly(2-oxy-6-naphthoyl) (PON) possesses many excellent properties such as mechanical strength, thermal stability, chemical resistance and so on, and it has been received great attention as high performance materials.1–3 These properties are influenced by the morphology of the polymers, and therefore the morphology control is of great importance to control the performance. PON does not exhibit solubility and fusibility, and this intractability makes PON difficult to process. Morphology control of PON had been studied by using reaction-induced phase separation of oligomers and PON whisker was prepared by the polymerization of 2-acetoxy-6-naphthoic acid in an inert solvent.2,3Copolymerization is also useful to improve the intractability of the polymers with maintaining the properties, and 2-oxy-6-naphthoyl (ON) moiety has been often used as a component for thermotropic liquid crystalline aromatic polyesters. Various types of copolyesters comprising of ON moiety had been prepared so far.4–11 Among them, copolymers of ON moiety and 4-oxybenzoyl (OB) moiety are industrially important as super engineering plastics, and some of these copolymers are commercially available under the trade name of Vectra.
Recently, high molecular weight poly(4-oxybenzoyl) (POB) was directly synthesized from 4-hydroxybenzoic acid (HBA) in the presence of boronic anhydride with eliminating water, and unique morphologies such as a needle and a sphere were successfully obtained as precipitates by using the reaction-induced phase separation of oligomers.12 In this polymerization, boronic anhydride and boronic acid acted as a catalyst. The morphology of the POB precipitates was influenced by the structure of the boronic anhydride. This polymerization is of interest from the viewpoint of not only the morphology control but also in view of the environmental benign procedure. In this study, the direct polymerization of 2-hydroxy-6-naphthoic acid (HNA) was examined in the presence of two boronic anhydrides such as 2,4,6-tris(3,4,5-trifluorophenyl)-1,3,5,2,4,6-trioxatriborinane (TFBA) and 2,4,6-tri(biphenyl-4-yl)-1,3,5,2,4,6-trioxatriborinane (BPBA) by using reaction-induced phase separation of oligomers to control the morphology of PON. Based on this result, copolymerization of HNA and HBA was also examined as shown in Scheme 1.
Experimental
Materials
HNA and HBA were purchased from TCI (Tokyo, Japan), and recrystallized from a mixture of water and methanol. 3,4,5-Trifluorophenylboronic acid and 4-biphenylboronic acid were purchased from Sigma-Aldrich (St Louis, USA), and recrystallized from toluene. It has been reported that boronic acids undergo dehydration upon simple heating to convert anhydrides.13–15 These two boronic acids were converted into boronic anhydride during the purification by recrystallization confirmed by IR spectroscopy. An isomeric mixture of dibenzyltoluene (DBT) was obtained from Matsumura Oil (Osaka, Japan, Trade name was Barrel Therm 400, Mw: 380, bp: 382 °C) and distilled under reduced pressure (170–175 °C/0.3 mmHg).
Measurements
Morphology was observed on a HITACHI S-3500N scanning electron microscope (SEM) (Tokyo, Japan). Samples were dried, sputtered with platinum–palladium and observed at 20 kV. Average sizes of the products were determined by taking the average of over 150 observation values. Infrared (IR) spectrum was measured on a JASCO FT/IR-410 spectrometer (Tokyo, Japan). Wide angle X-ray scattering (WAXS) was performed on a Rigaku Gaiger Flex (Matsumoto, Japan) with nickel-filtered CuKα radiation (35 kV, 20 mA). Transition temperatures from crystal phase to anisotropic melt phase (TKN) of copolymers were measured on a Perkin-Elmer DSC 8000 (Waltham, USA) with a scanning rate of 20 °C min−1 in nitrogen atmosphere.
Determination of number-average molecular weight
Number-average molecular weight (Mn) was determined by the end-group analysis. The one end-group of PON molecules was a carboxyl group or a carboxylic boronic anhydridegroup. The content of the carboxylic boronic anhydride end-group was determined by the measurement of the content of the boronic acid on HPLC after the hydrolysis of the samples as follows: samples (2 mg) and 10 wt% KOH methanol solution (5 mL) were placed in a flask, and the mixture was completely hydrolyzed for 12 h at 100 °C. The solution was neutralized with dilute hydrochloric acid. The solution was analyzed by using a HITACHI D-2000 Elite HPLC (Tokyo, Japan) with Nova Pack HR C18 column. The eluent of HPLC was a mixture of water containing 1.0 vol% acetic acid and acetonitrile, and the mixing volume ratio of these two solvents was changed linearly from 90/10 to 73/27 for 8 min. On the other hand, the content of the carboxyl end-group was estimated by calculating the absorbance ratio of carbonyl group of carboxylic acid and that of ester linkage based on an IR spectroscopy. 6-(2-Naphthoyloxy)-2-naphthoic acid and 6-(6-(2-naphthoyloxy)-2-naphthoyloxy)-2-naphthoic acid, which were synthesized according to a previously reported procedure,16 were used to correct the absorbance ratio. On the basis of these analyses, Mn was estimated by the following equation:
| Mn = MON/[(Absacid/1.04 Absester) + (molar fraction of boronic acid residue in the crystals)] |
Absacid: absorbance of carbonyl group in carboxyl end-group; Absester: absorbance of carbonyl group in ester linkage; MON: molar weight of ON moiety; 1.04: calibration coefficient for ON moiety.
M
n of copolymers was estimated by the following equation:
| Mn = [MOBrp/100 + MON(1 − rp)/100] × [1 + 1/{(Absacid/(0.80rp/100 + 1.04(1 − rp/100)Absester) + (molar fraction of boronic acid moiety in the crystals)}] |
MOB: molar weight of OB moiety;
rp (mol%): content of OB moiety in
copolymers; 0.80: calibration coefficient for OB moiety.
Composition analysis of copolymers
Samples were hydrolyzed and the molar ratio of HBA and HNA was determined by HPLC according to the similar procedure for the determination of Mn as described above. Composition of copolymers (rp) was expressed as the content of OB moiety according to the following equation:
| rp (mol%) = [OB] × 100/([OB]+[ON]). |
Polymerization of HNA.
HNA (0.22 g, 1.18 mmol) and 20 mL of DBT were placed into a cylindrical flask equipped with a mechanical stirrer and gas inlet and outlet tubes. Polymerization concentration was 1.0%, defined as (calculated polymer weight/solvent volume) × 100. The reaction mixture was heated under a slow stream of nitrogen up to 300 °C with stirring. HNA was dissolved during heating. BPBA (0.21 g, 0.39 mmol) was added into the solution at 300 °C and stirring was stopped after BPBA was entirely dissolved. Concentration of boronic anhydride (cB) was 100 mol%, defined as (3[boronic anhydride]/[HNA]) × 100 based on the concentration of a boronic acid residue. Temperature was maintained at 300 °C for 24 h. The solution became turbid at an initial stage of the polymerization and then precipitates were formed with time. Precipitated PON crystals were collected by vacuum filtration at 300 °C, and washed with n-hexane and acetone. Polymerizations under other conditions were carried out in a similar manner.
Copolymerization of HNA and HBA.
Copolymerizations of HNA and HBA were carried out with varying the content of HBA in feed (rf) in DBT at 300 °C for 24 h in the presence of BPBA at cB of 100 mol% according to the procedure similar to the polymerization of HNA. Concentration of polymerization was 1.0%.
| rf (mol%) = [HBA] × 100/([HBA]+[HNA]). |
Results and discussion
Polymerizations of HNA were carried out in DBT at a concentration of 1.0% at 300 °C for 24 h in the presence of two different boronic anhydrides with varying the value of cB. HNA was insoluble in DBT at 25 °C but it became dissolved during heating. Table 1 presents results of the polymerization. In the polymerization with TFBA, precipitates were obtained with the yields of 34 to 80%. The precipitates were insoluble in common organic solvents, and therefore their chemical structures were analyzed by the IR spectroscopy. Fig. 1(a) shows an IR spectrum of the precipitates formed with TFBA at cB of 50 mol% (run no. 2) as a representative. Bands of carbonyl groups and ethergroups in ester linkages are clearly observed at 1736 and 1180 cm−1, and this spectrum is identical with that of PON. Further, bands of hydroxyl end-groups and carboxyl end-groups are hardly visible, suggesting high molecular weight. The Mn determined by the end-group analysis increased with the value of cB and it ranged from 2.7 × 103 to 9.0 × 103. The precipitates did not exhibit clear morphology at cB of 10 mol%, whereas those prepared at cB of 50–100 mol% exhibited clear morphology as shown in Fig. 2(a) and (b). Spheres with needles on their surface were formed at cB of 50–70 mol%. The average diameters of the spheres prepared at cB of 50 and 70 mol% were 2.22 and 1.94 μm, respectively. The average length and width of the needles on the surface prepared at cB of 50 mol% were 540 and 130 nm, respectively. And those at cB of 70 mol% were 290 and 140 nm. In contrast to these, spheres having smooth surface were formed at cB of 100 mol%. The average diameter and its coefficient of variation (cv) of the spheres were 1.76 μm and 18%, respectively. Fig. 3(a) and (b) show WAXS intensity profiles of the spheres with needles on their surface (run no. 2) and the spheres having smooth surface (run no. 4), respectively. In spite of the morphology, diffuse halo attributed to amorphous region was hardly detected in these profiles, indicating that these precipitates possessed high crystallinity. Diffraction peaks were observed at 2θ of 10.4, 15.4, 18.7, 23.0, 27.8, 38.2 and 42.6°, which could be assigned according to the orthorhombic unit cell of the PON crystal with a = 0.766, b = 0.598, and c = 1.712 nm.17
| Run no. |
Polymerization conditiona |
Polymer yield (%) |
M
n (×103) |
Morphology |
|
Boronic anhydride
|
c
B
(mol%) |
|
Polymerizations were carried out in DBT at 300 °C at a concentration of 1.0% at 300 °C for 24 h.
c
B (mol%) = (3[boronic anhydride]/[HNA]) × 100.
SN stands for spheres with needles on their surface.
|
| 1 |
TFBA
|
10 |
34 |
2.7 |
Unclear |
| 2 |
TFBA
|
50 |
80 |
4.9 |
SNc |
| 3 |
TFBA
|
70 |
77 |
6.6 |
SN |
| 4 |
TFBA
|
100 |
62 |
9.0 |
Sphere |
| 5 |
BPBA
|
10 |
40 |
5.4 |
Cone |
| 6 |
BPBA
|
30 |
71 |
8.3 |
Cone |
| 7 |
BPBA
|
50 |
55 |
9.9 |
Sphere |
| 8 |
BPBA
|
70 |
66 |
12.1 |
Sphere |
| 9 |
BPBA
|
100 |
58 |
12.9 |
Sphere |
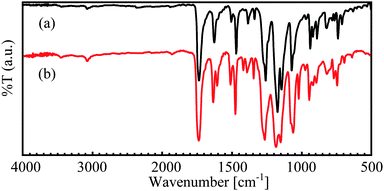 |
| | Fig. 1
IR spectra of (a) PON prepared in the presence of TFBA at cB of 50 mol% and (b) copolymer of HBA and HNA (rf = 50 mol%) prepared in the presence of BPBA at cB of 100 mol%. | |
 |
| | Fig. 2
PON precipitates prepared in the presence of TFBA at cB of (a) 50 mol% and (b) 100 mol%, and those prepared in the presence of BPBA at cB of (c) 30 mol% and (d) 100 mol%. | |
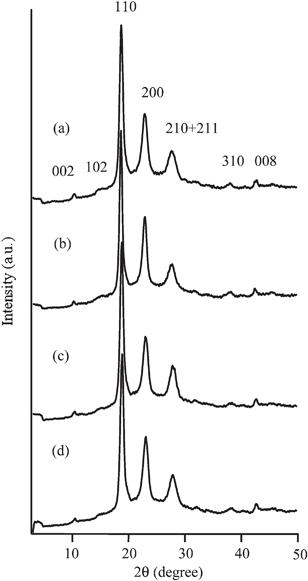 |
| | Fig. 3
WAXS intensity profiles of the PON prepared in the presence of TFPBA at cB of (a) 50 mol% and (b) 100 mol%, and those prepared in the presence of BPBA at cB of (c) 30 mol% and (d) 100 mol%. | |
With respect to the polymerization with BPBA, the PON precipitates were formed with the yields of 40 to 71%. The Mn also increased with the value of cB in the range from 5.4 × 103 to 12.9 × 103. The Mn prepared with BPBA was higher than that with TFBA at the same cB value. The morphology of the precipitates changed drastically with the value of cB. Aggregates of cone-like crystals were formed at cB of 10 and 30 mol% as shown in Fig. 2(c). The cone-like crystals grew radially from the centre part. The average length from the centre part and the width of the cone-like crystals prepared at cB of 10 mol% were 690 and 280 nm, respectively. And those at cB of 30 mol% were 610 and 170 nm, respectively. The size of the corn-like crystals prepared at cB of 30 mol% was slightly smaller than that at cB of 10 mol%. Morphology was drastically changed at cB of 50–100 mol% from cone-like crystal to spheres as shown in Fig. 2(d). The average diameter and the cv value of these spheres were 620–670 nm and 13–16%, respectively. These spheres did not possess smooth surface and many protuberances were observed on the surface. The spheres have rugged surface which are different from those prepared with TFBA. Fig. 3(c) and (d) show WAXS intensity profiles of the aggregates of the cone-like crystals (run no. 6) and the spheres (run no. 9). They also possessed high crystallinity as well as the precipitates obtained with TFBA.
The polymerization mechanism is explainable as follows: the boronic anhydride reacts with phenolic hydroxylgroup of HNA to form a boronate. Then the carboxylic acid in HNA is activated by an isomerization of the boronate to form a carboxylic boronic anhydride. Then phenolic hydroxylgroup of HNA attacks the carboxylic boronic anhydride to form an ester linkage with eliminating boronic acid. This boronic acid reacts with carboxylic acid directly to form the carboxylic boronic anhydride or with another boronic acid to form boronic anhydride with elimination of water. This reaction continues to form the oligomers. When the molecular weight of the oligomers exceeds a critical value, the oligomers are precipitated to form the products via super-saturated state. If the crystallization is induced, the oligomer crystals having clear habit are formed. The polymerization between the oligomers occurs in or on the crystals. On the other hand, if the liquid–liquid phase separation is induced, the spheres are formed via the formation of droplets. In the droplets, the concentration of the oligomers becomes high and the polymerization proceeds to increase the Mn, leading the solidification with maintaining the spherical morphology. The morphology of the precipitates is significantly influenced by the value of cB. The crystals are formed at a low value of cB and the spheres are done at a high value of cB. The oligomers having carboxyl end-group are precipitated at a low value of cB and those having carboxylic boronic anhydride end-group are precipitated at a high value of cB. The oligomers having carboxylic boronic anhydride end-group exhibit lower freezing point than those having carboxyl end-group, bringing about the liquid–liquid phase separation rather than crystallization. Further the oligomers having carboxylic boronic anhydride end-group possess lower crystallizability due to the bulkiness of end-group. Due to these effects, the spheres are naturally formed at a high value of cB. In contrast, the crystals having clear habit are formed at a low value of cB.
The Mn increased with cB and it became the highest at cB of 100 mol%. Further, BPBA gave the higher Mn than TFBA as described. When the value of cB is 100 mol%, all carboxylic end-groups of the precipitated oligomers must be converted to carboxylic boronic anhydride end-groups. Phenolic hydroxyl end-group attacks the activated end-group of the oligomer in or on the crystals, and the polymerization proceeds efficiently to yield the highest Mn. On the other hand, when the value of cB is lower than 100 mol%, some oligomers having carboxyl end-group are precipitated to form the crystals and the carboxyl end-group terminates the polymerization. With respect to the structure of boronic anhydride, the tendency that BPBA afforded the higher molecular weight was also observed in the polymerization of HBA due to the size effect of carboxylic and boronic anhydride end-group as previously reported.12 The yield and the Mn of the PON precipitates prepared in the presence of TFBA and BPBA at cB of 100 mol% were plotted as a function of time in Fig. 4. In both polymerizations, they increased with time in the initial stage of the polymerization. The Mn increased continuously after the yield was levelled off. This fact suggests the efficient polymerization in the precipitates. The linearity between the Mn and the time after the yield was levelled off indicates that the polymerization in the precipitates apparently obeys second-order kinetics. The polymerization rate in the precipitates could not be accurately determined because the initial concentration of the end-group was not determined. Therefore, the polymerization rate was compared using the slope (R). The values of R of the polymerization with TFBA and BPBA were calculated as 3.92 and 5.06 min−1, respectively. The value of R in the polymerization with BPBA is slightly larger, but there is not so large difference between TFBA and BPBA. The PON did not exhibit a melting temperature under the decomposition and the polymerization proceeded in the solid state.2,3,7,18 It had been reported that a axis of the PON crystal unit cell slowly increased with temperature up to the transition at 330 °C, and b and c remained almost constant.18 The volume of the crystal unit cell increased ca. 10% at 330 °C. The crystal structure changed from an orthorhombic structure to a new orthorhombic one which was almost pseudohexagonal. The polymerization temperature 300 °C was closed to the transition temperature, suggesting that the polymerization in the precipitates might occur in a new orthorhombic structure rather than an orthorhombic structure. This structural change is one possibility for the efficient polymerization in the precipitates. The polymerization reaction proceeded more rapidly at the initial stage of the polymerization until the yield was levelled off than that at the middle stage of polymerization in the both polymerization systems. When the oligomers precipitate to form the crystals, they polymerized not only in the crystals but also on the crystallization site. The contribution of the polymerization on the crystals makes the total polymerization rate higher. At the initial stage of the polymerization, the yield increased more rapidly in the polymerization with TFBA than BPBA, but oppositely the Mn increased more slowly. In the polymerization with TFBA, the rapid precipitation of the oligomers in the polymerization with TFBA makes the polymerization on the crystals slower than that with BPBA.
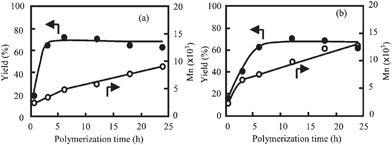 |
| | Fig. 4 Plots of yield (●) and Mn (○) of PON precipitates prepared in the presence of (a) TFBA and (b) BPBA at cB of 100 mol% as a function of time. | |
According to the reaction mechanism, by-produced boronic acid activates a carboxyl group directly or converts to anhydride with elimination of water. The cB value of 100 mol% is desirable to prepare high molecular weight PON precipitates. If the value of cB in the solution keeps more than 100 mol% in the solution, the polymerization will proceed efficiently to afford high molecular weight PON precipitates even after the addition of HNA. In order to prove this, HNA was added after 24 and 48 h into the polymerization system. The results were presented in Table 2. The concentration of HNA for the addition was fixed as 1.0% to maintain the cB value of 100 mol% in the solution phase. The Mn and the yield of the PON precipitates after adding HNA once were 3.5 × 104 and 65%, respectively. Those after adding twice were 2.5 × 104 and 75%, respectively. The total cB value became lower to 50 mol% and 33.3 mol% after adding HNA once and twice. The Mn increased naturally with the polymerization time and those prepared without the addition of HNA for 24 h and 48 h were 1.3 × 104 and 2.7 × 104. Although molecular weight distribution could not be measured due to the insolubility of PON, the Mn prepared with the addition of HNA was not lower than that without the addition. This result reveals that the boronic anhydride acts as a catalyst even in the heterogeneous polymerization system.
Table 2 Results of polymerization of HNA in the presence of BPBA with additions of HNA
| Run no. |
Polymerization conditiona |
Polymer yield |
M
n (×103) |
| Initial conc. (%) |
Time for monomer addition/h |
Total conc. (%) |
Total polymerization time/h |
Calc./g |
Obs./g |
Yield (%) |
|
Polymerizations were carried out in DBT at 300 °C in the presence of BPBA at cB of 100 mol% for initial HNA at 300 °C.
HNA of 1.0% was added once after 24 h and the polymerization was carried out for another 24 h.
HNA of 1.0 wt% was added twice after 24 h and 48 h, and the polymerization was carried out for another 24 h.
|
| 10 |
1.0 |
No |
1.0 |
24 |
0.20 |
0.12 |
60 |
12.9 |
| 11 |
1.0 |
No |
1.0 |
48 |
0.20 |
0.15 |
75 |
26.5 |
| 12 |
1.0 |
24b |
2.0 |
48 |
0.40 |
0.26 |
65 |
35.1 |
| 13 |
1.0 |
24 and 48c |
3.0 |
72 |
0.60 |
0.45 |
75 |
24.6 |
| 14 |
3.0 |
No |
3.0 |
24 |
0.60 |
0.45 |
75 |
17.0 |
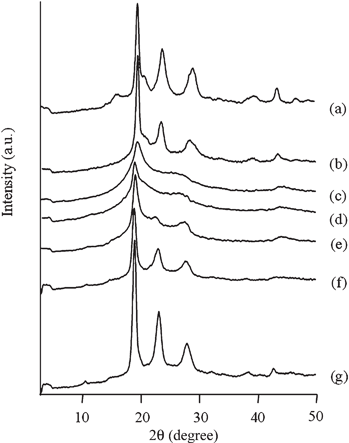
Poly(4-oxybenzoyl-co-2-oxy-6-naphthoyl) is usually prepared by the melt polymerization of 4-acetoxybenzoic acid and 2-acetoxy-6-naphthoic acid. The viscosity of the melt polycondensation limits the molecular weight, and the efficient elimination of acetic acid from the polymerization phase is of importance to gain high molecular weight copolymers. Further, copolymers usually show high melting temperature and this makes difficult to extrude the polymers from a reaction vessel. To overcome these problems, one procedure has been to prepare oligomers by the melt polycondensation, followed by a subsequent solid-state polymerization.19 Another procedure has been to prepare the copolymers in an inert solvent. The polymerization proceeded in dispersion and coarse granules consisting of small particles are ultimately obtained in this procedure.20,21 In this polymerization, the small particle size and high surface area of droplets allow rapid removal of the by-product, leading to higher molecular weight than that prepared by the melt polycondensation.22 Based on these previous studies, the copolymerization of HBA and HNA was also examined in the presence of BPBA at cB of 100 mol% with varying the value of rf. The results are presented in Fig. 5 and 6. The value of rp was determined by the HPLC analysis after hydrolysis. Their chemical structures were also analyzed by the IR spectroscopy. Fig. 1(b) shows an IR spectrum of the precipitates formed with BPBA at cB of 100 mol% at rf of 50 mol% as a representative. Carbonyl and ether bands of ester linkage are clearly observed at 1734 and 1179 cm−1. In addition, bands of 1,4-phenylene group and 2,6-naphthylenegroup are visible at 1599 cm−1 and 1629 cm−1, respectively. Additionally, the bands corresponding to the end-groups are hardly visible. This spectrum indicates the formation of a high molecular weight copolymer. In all copolymerizations, the value of rp was lower than that of rf, indicating that the oligomers rich in ON moiety were precipitated more rapidly than those rich in OB moiety due to lower solubility. The spheres were formed as precipitates, as shown in Fig. 6. Although the spheres having rugged surface were formed at rf of 90 mol%, those prepared at rf of 10–70 mol% possessed smooth surface. The diameter of the spheres was approximately 0.4–1.7 μm. TKN of copolymers highly relates to the value of rp, and it becomes lower than 300 °C when the value of rp is in the range from 30 to 78 mol%.7,18 The copolymerization makes the freezing point of the oligomers lower, leading to the liquid–liquid phase separation rather than crystallization. The spherical morphology was formed via the liquid–liquid phase separation at rf of 50–90 mol%, and the polymerization occurred in the discontinuous dense phase. Further, the copolymerization generally lowers the crystallizability of the oligomers bringing about to extinguish clear crystal habit. These tendencies caused the formation of the spherical morphology in copolymerization system. The yield of the precipitates decreased with the decrease in the value of rf until 50 mol% and then increased. The rf of 50 mol% gave the minimum yield of 23%. The solubility of the oligomers also relates to the value of rf and generally it becomes maximum at the middle range of the composition, leading to the minimum yield of the precipitates at rf of 50 mol%. The Mn also became lower with the copolymerization. Fig. 7 shows the WAXS intensity profiles of the spheres of the copolymers. Crystalline peaks became broader and disappeared in the middle range of rf and the crystallinity of the spheres prepared in the middle range of rf became lower. The spheres prepared at rf of 90 mol% possessed relatively high crystallinity, being attributed to the rugged surface.
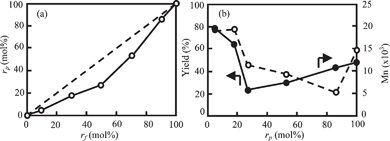 |
| | Fig. 5 Plots of (a) relation between rf and rp, and (b) yield (●) and Mn (○) as a function of rp of precipitates prepared in the presence of BPBA at cB of 100 mol% for 24 h. | |
 |
| | Fig. 6
Copolymer precipitates prepared in the presence of BPBA at cB of 100 mol% at rf of (a) 10, (b) 50, (c) 70 and (d) 90 mol%. | |
 |
| | Fig. 7
WAXS intensity profiles of copolymers at cB of 100 mol% at rf of (a) 100, (b) 90, (c) 70, (d) 50, (e) 30, (f) 10 and (g) 0 mol%. | |
Changes in the yield, the Mn and the value of rp were examined during the polymerization at rf of 70 mol%. Fig. 8 plots the results as a function of polymerization time. The yield increased very slowly with time until 18 h and then it became constant. The Mn increased linearly with time and it reached 13.5 × 103 after 36 h. In the polymerization of HNA, the Mn also increased with time as aforesaid. However, the Mn did not increase linearly throughout the polymerization. It increased gradually after the yield was levelled off, being attributed to the solid-state polymerization. In the copolymerization at rf of 70 mol%, the polymerization occurred in the dense phase and the oligomers were supplied very slowly from the dilute solution phase to the dense phase. Because of these, the polymerization rate apparently obeys the second-order kinetics throughout the polymerization. With respect to the composition, the value of rp was not constant throughout the polymerization. The value of rp was 66 mol% for 6 h, and afterward it decreased gradually to 54 mol% with the increase in the yield until 18 h. In the initial stage of the polymerization, the oligomers rich in OB moiety were precipitated and then the oligomers rich in ON moiety started to be gradually precipitated with time. This fact provides a possibility that the copolymers have sequence gradations or the spheres are comprised of a mixture of copolymers having different composition. The sequence of the prepared copolymers could not be directly analyzed by NMR because of insolubility. It is well known that TKN is significantly influenced by not only the composition but also the sequence of copolymer even at the same composition.23TKNs of the copolymers measured on a DSC were plotted as a function of the content of ON moiety (100-rp mol%) on the phase-diagram previously prepared with the copolymers produced by the melt polymerization.17Fig. 9 shows the plot. TKNs of the copolymers prepared in this study are on the phase line between the crystal and the anisotropic phase, and they are in good agreement with the previous results. This good conformity indicates that the copolymers prepared in this study have not sequence gradation and they are random copolymers. The reshuffling reaction such as trans-esterification reaction takes place in the dense droplets at high temperature, leading to the randomization of the sequence.
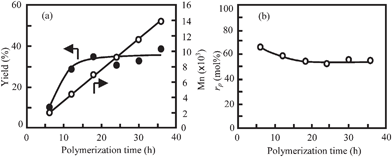 |
| | Fig. 8 Plots of (a) yield (●) and Mn (○), and (b) rp of copolymer prepared rf of 70 mol% as a function of polymerization time. Polymerizations were carried out in the presence of BPBA at cB of 100 mol%. | |
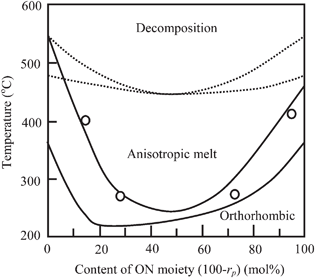 |
| | Fig. 9 Plot of TKN (○) as a function of content of ON moiety on a phase-diagram previously reported in ref. 18. | |
Conclusions
The morphology of the PON precipitates was influenced by not only the structure of the boronic anhydride but also the value of cB. Spheres with needles on their surface were obtained in the polymerization with TFBA at cB of 50–70 mol%, and those having smooth surface were formed at cB of 100 mol%. These precipitates possessed high crystallinity and the Mn increased with the value of cB in the range from 2.7 × 103 to 9.0 × 103. With respect to the polymerization with BPBA, the aggregates of cone-like crystals were formed at cB of 10–30 mol% and spheres having rugged surface were formed at cB of 50–100 mol%. The Mn also increased with the value of cB in the range from 5.4 × 103 to 12.9 × 103 and it became the highest at cB of 100 mol%. The Mn prepared with BPBA was higher than that with TFBA at the same cB value. When the value of cB was 100 mol%, all carboxylic end-groups of the precipitated oligomers were converted to carboxylic boronic anhydride end-groups which reacted with the phenolic hydroxyl end-group in or on the crystals to afford high molecular weight. The copolymerization of HBA and HNA was also examined in the presence of BPBA at cB of 100 mol% with varying the value of rf. Polymerization proceeded in the droplet formed by the liquid–liquid phase separation in the middle range of rf. Spheres were formed with high molecular weight, of which the value of rp was lower than that of rf. This indicates that the oligomers rich in ON moiety were precipitated more rapidly than those rich in OB moiety due to lower solubility.
References
- A. Mühlebach, J. Lyerla and J. Economy, Macromolecules, 1989, 22, 3741 CrossRef.
- G. Schwarz and H. R. Kricheldor, Macromolecules, 1991, 24, 2829 CrossRef CAS.
- K. Kimura, S. Endo, Y. Kato and Y. Yamashita, Polymer, 1993, 34, 1054 CrossRef CAS.
- M. G. Dobb and J. E. McIntyre, Adv. Polym. Sci., 1984, 60/61, 61.
- J. B. Stamatoff, Mol. Cryst. Liq. Cryst., 1984, 110, 75 CrossRef CAS.
- G. D. Butzbach, J. Wendorff and H. J. Zimmerman, Makromol. Chem. Rapid Commun., 1985, 6, 821 CrossRef CAS.
- M.-Y. Cao and B. Wunderlich, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 521 CrossRef CAS.
-
G. H. Ba and E. F. Cluff, in Polymeric Liquid Crystals, ed. A. Blumstein, Plenum, New York, 1985, p. 217 Search PubMed.
-
G. W. Calundann, in High Performance Polymers: their Origin and Development, ed. R. B. Seymour and G. S. Krishenbaum, Elsevier, New York, 1986, p. 235 Search PubMed.
-
J. Blackwell and A. Biswas, in Developments in Oriented Polymers-2, ed. I. M. Ward, Elsevier, Barking, UK, 1987, ch. 5 Search PubMed.
-
A. M. Donald, A. H. Windle and S. Hanna, Liquid Crystalline Polymers, Cambridge University Press, 2nd edn, 2006, p. 82 Search PubMed.
- M. Kihara, S. Kohama, S. Umezono, K. Wakabayashi, S. Yamazaki and K. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1088 CrossRef CAS.
- H. Gilman, L. Santucci, D. R. Swayampati and R. O. Ranck, J. Am. Chem. Soc., 1957, 79, 3077 CrossRef CAS.
- H. R. Snyder, M. S. Konecky and W. J. Lennarz, J. Am. Chem. Soc., 1958, 80, 3611 CrossRef CAS.
- T. M. Bulbul Islam, K. Yoshino, H. Nomura, T. Mizuno and A. Sasane, Anal. Sci., 2002, 18, 363 CAS.
- K. Kimura, T. Horii, S. Kohama and Y. Yamashita, Polym. Adv. Technol., 2005, 16, 693 CrossRef CAS.
- P. Iannelli, D. Y. Yoon and W. Parrish, Macromolecules, 1994, 27, 3295 CrossRef CAS.
- A. Habenschuss, M. Varma-Nair, Y. K. Kwon, J. Ma and B. Wunderlich, Polymer, 2006, 47, 2369 CrossRef CAS.
-
J. Economy and W. Volksen, in Strength and Stiffness of Polymers, ed. A. E. Zachariades and R. S. Porter, Dekker, New York, 1983, p. 299 Search PubMed.
-
S. G. Cottis, J. Economy and B. E. Nowak, US Pat., 3975486, 1973.
-
G. W. Calundann, US Pat., 4067852, 1978.
- A. Brunn, B. P. Griffin, W. A. MacDonald and D. G. Rance, Polymer, 1992, 33, 3066 CrossRef.
- C. Ober, W. R. Lenz, G. Galli and E. Chiellini, Macromolecules, 1983, 16, 1034 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.