Synthesis of heterobifunctional poly(ethylene glycol)s by an acetal protection method†
Zhongyu
Li
and
Ying
Chau
*
Department of Chemical and Biomolecular Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong SAR, China. E-mail: ying.chau@ust.hk; Fax: +852-2358 0054; Tel: +852-2358 8935
First published on 22nd October 2010
Abstract
A versatile synthetic route to prepare well-defined heterobifunctional poly(ethylene glycol) (PEG) derivatives via sequential modification of the hydroxyl ends was established using a newly synthesized initiator for anionic polymerization, 2-(1-ethoxyethoxy)ethanol (EEE), which contains an acid-cleavable acetal protection group. As illustrative examples, two heterobifuntional PEGs, HO–PEG–alkyne and HS–PEG–alkyne, were successfully prepared. This approach is applicable for synthesizing a wide variety of heterobifunctional PEGs.
Polyethylene glycol possesses a variety of physicochemical properties pertinent to biomedical and biotechnological applications.1 Telechelic α,ω-heterobifunctional PEG, with the structure of X–PEG–Y, where X and Y are different functional groups, has been extensively studied and been used as a hydrophilic, flexible, biocompatible, and inert spacer for linking macromolecules to surfaces,2 constructing targeted drug and gene delivery vehicles,3 modifying nanoparticles for bioassays and biorecognition,4 and facilitating liquid phase organic synthesis.5
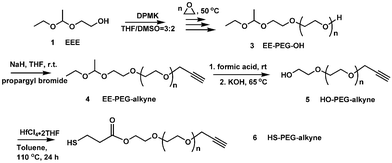
The synthesis of heterobifunctional PEG is key to these applications and has attracted much attention in the last two decades. There are two methods commonly employed for synthesizing heterobifunctional PEG. The direct method involves partial derivatization of PEG diols, followed by isolation of the targeted heterobifunctional oligomers from the reaction mixture. Separating chemically similar polymers that differ only in their end group structures is difficult.6 Usually, expensive and throughput-limited gel permeation chromatography (GPC) was employed. The second and more versatile method is the ring-opening polymerization of ethylene oxide (EO) from a heterobifunctional initiator with one hydroxyl group and one protected or latent group.5a,7 One of the functional groups of the initiator reacts with EO and the other group remains intact. Polymerization was followed by the modification of the hydroxyl group and then the protected (latent) group. Among these heterobifunctional initiators, a protected diol with one active hydroxyl group and one inert, protected hydroxyl group is suitable for synthesizing heterobifunctional PEG due to the versatility of the hydroxyl group.8 We report herein an alternative high-yielding method to synthesize well-defined α,ω-heterobifunctional PEGs using acetals as protecting groups. Acetals are useful as protecting groups of alcohols.9 They are stable in alkaline aqueous or organic solutions and do not interfere with anionic polymerization of ethylene oxide.10To this end, a new initiator, 1-ethoxyethyloxy (EE)-protected ethylene glycol, is designed for the anionic polymerization of ethylene oxide (EO). The feasibility of this approach is demonstrated by the synthesis and characterization of two heterobifunctional PEGs.
Initiation and polymerization of ethylene oxide (EO) from a heterobifunctional initiator are more direct than the partial derivatization of PEG diols for the synthesis of heterobifunctional PEG. Many heterobifunctional PEG polymers have been successfully synthesized using heterobifunctional initiators, such as those with protected amino group,7b protected aldehyde group,7c–e and latent allyl groups.7f,g Initiators with protected hydroxyl groups have also been investigated for the synthesis of heterobifunctional PEG. For example, 2-(benzyloxy) ethanol, 2-(tert-butyldimethylsilyloxy) ethanol, and 2-(tetrahydropyranyloxy) ethanol were used as initiators by Reed and Janda.5a A Pd/C/H2-labile benzyl and two acid-labile monoprotected heterobifunctional PEGs were synthesized. Recently, “click chemistry” compatible heterobifunctional PEGs possessing azide and alkyne functional group respectively were synthesized from a commercial and acid-labile (acetal) protected initiator, 2-(tetrahydro-2H-pyran-2-yloxy)ethanol.7h
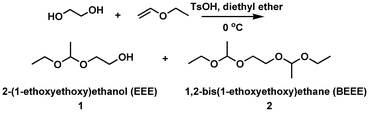
A new initiator with a 1-ethoxyethyloxy (EE)-protected hydroxyl group and a free hydroxyl group was synthesized by the electrophilic addition of ethylene glycol and ethyl vinyl ether using p-toluenesulfonic acid (p-TsOH) as a catalyst (Scheme 1). The successful synthesis was confirmed by NMR and elemental analysis. 1H NMR (ppm) (CDCl3): 1.17 (m, CH3CH2O–), 1.30 (d, –O–CH(CH3)–O–), 3.44 (m, CH3CH2O–), 3.61(m, –CH2CH2OH), 3.79 (m, –CH2CH2OH), 4.68 (m, –O–CH(CH3)–O–) (Fig. 1). Elemental analysis: calculated: C 53.71, H 10.52; found: C 53.65, H 10.56%.
 | ||
| Scheme 1 Synthesis of initiator. | ||
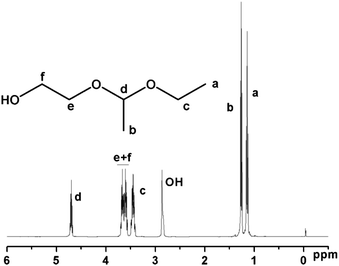 | ||
| Fig. 1 1H NMR spectrum of 2-(1-ethoxyethoxy) ethanol (EEE) (CDCl3 at 20 °C). | ||
The reaction was conducted in diethyl ether solution in an ice-water bath. Initially, diols (ethylene glycol) were not dissolved well in diethyl ether and two phases were observed. The addition of p-TsOH to the solution under vigorous stirring enabled the reaction to take place and the phase separation disappeared. To ensure that all alcohol groups are protected, ethyl vinyl ether in slight excess over hydroxyl is needed. Low temperature is necessary because the boiling points of both diethyl ether and ethyl vinyl ether are about 36 °C and the reaction is exothermic. Since heat plus acid may cause acetal interchange to happen, which will result in the formation of side products (acetaldehyde acetals),9 TsOH must be removed completely by washing with saturated NaHCO3 solution prior to distillation.
We used diphenylmethyl potassium (DPMK) as a base for generating alkoxide initiators.10c Lowering the ratio of DPMK to hydroxyl and using a DMSO-containing co-solvent system ensure the complete solubility of alkoxide and more importantly, a reduced rate of polymerization. This helps to decrease the polydispersity of the polymer. The current setup produces in one typical batch about 41 g of 1-ethoxyethyl-protected PEG (EE–PEG–OH) with low polydispersity (PDI). This polymerization reaction is conceptually simple but technically demanding. Avoiding moisture is pivotal throughout the entire synthesis procedure. All solvents and reagents must be rigorously dried and degassed to obtain consistent results. The purity of the potassium is critical, since KOH contamination will lead to the formation of PEG diol.
Fig. 2A shows a typical 1H NMR spectrum of EE–PEG–OH. The signals of the protons of the 1-ethoxyethyl group are detected at δ (ppm) 1.17 (m, CH3CH2O–), 1.30 (d, –O–CH(CH3)–O–), and 4.68 (m, –O–CH(CH3)–O–). Table 1 lists the properties of two batches of EE–PEG–OH. The number average molecular weight (Mn) calculated by 1H NMR is in close agreement with the value obtained by SEC. This molecular weight of batch 1 was reconfirmed by MALDI-TOF mass spectroscopy (Fig. 3 and Table 1). The major series of the molecular masses of the product is expressed in the following equation: Mw = 44.03n (EO) + 134.09 (EEE) + 22.99 (sodium), where n is an integer that confirms the initiator is the new synthetic EEE.
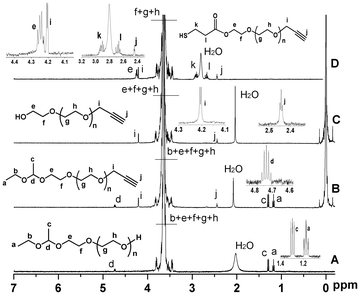 | ||
| Fig. 2 1H NMR spectra of EE–PEG–OH (A), EE–PEG–alkyne (B), HO–PEG–alkyne (C) and HS–PEG–alkyne (D), respectively (CDCl3 at 20 °C). | ||
| [EO]0/[EEE]0 | 10−3 × Mnb/g mol−1 | Polydispersity Mw/Mng | |||||
|---|---|---|---|---|---|---|---|
| Calcdc | NMRd | SECe | MSf | SECg | MSh | ||
| a Yield of products 1 and 2 are 98% and 99% respectively. b M n denotes the number average molecular weight. c Determined from the following equation: Mn(calcd) = Mw(EO)[EO]0/[EEE]0 + Mw(EEE) = 44.03 [EO]0/[EEE]0 + 134.09.2 d The number average molecular weight (Mn) of the polymers was determined by 1H NMR spectrum on the basis of end group analysis using the equation: Mn = 44.03 × (3AEO − 2Am/3)/4Am + 134.09,3 where AEO and Am are the peak areas of sum of protons in the PEG main chain at δ = 3.45–3.80 ppm and methyl protons at δ = 1.17 ppm, respectively; 134.09 and 44.03 are the molecular weight of EEE and EO respectively. e Determined by SEC. f Determined by MALDI-TOF mass spectroscopy. g Polydispersity index (PDI) = Mw/Mn, Mw denotes the weight average molecular weight. h The number average molecular weight according to MALDI-TOF MS. | |||||||
| 1a | 85 | 3.87 | 3.81 | 3.82 | 3.78 | 1.09 | 1.04 |
| 2a | 170 | 7.62 | 7.56 | 7.51 | ND | 1.07 | ND |
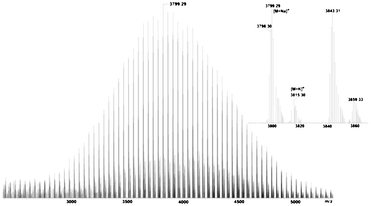 | ||
| Fig. 3 MALDI-TOF mass spectrum of EE–PEG–OH. | ||
The MS spectrum is consistent with the SEC results also in other regards: the polymer distribution is unimodal, the polydispersity is narrow (Mw/Mn = 1.04) and side reaction products are absent.
These results support that 1-ethoxyethyl-protected PEG (EE–PEG–OH) (3) has been successfully synthesized using 2-(1-ethoxyethoxy)ethanol (EEE) as the initiator.
To demonstrate that this polymer is suitable for preparing heterofunctional derivatives, we modified EE–PEG–OH with propargyl bromide (Scheme 2). Fig. 2B shows the 1H NMR spectrum of EE–PEG–alkyne. New signals at δ (ppm) 2.40 (s, –CCH) and 4.17 (s, –CH2–CCH), along with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 peak area ratio of alkyne groups to methyl groups (δ = 1.17 ppm), show that the hydroxyl terminals were completely changed to alkyne groups. Alkyne groups are amenable for “click chemistry”, forming a stable triazole with azido groups under mild conditions.11
:1 peak area ratio of alkyne groups to methyl groups (δ = 1.17 ppm), show that the hydroxyl terminals were completely changed to alkyne groups. Alkyne groups are amenable for “click chemistry”, forming a stable triazole with azido groups under mild conditions.11
 | ||
| Scheme 2 Synthesis of α,ω-heterobifunctional PEG. | ||
The protecting group on EE–PEG–alkyne (4) was successfully cleaved by hydrolysis to restore the hydroxyl end. Fig. 2C shows the 1H NMR spectrum of HO–PEG–alkyne. The signals assigned to 1-ethoxyethyl group at δ (ppm) 1.17 (m, CH3CH2O–), 1.30 (d, –O–CH(CH3)–O–) and 4.68 (m, –O–CH(CH3)–O–) all disappeared, supporting that complete cleavage occurred. We tried but failed to synthesize HO–PEG–alkyne (5) by an alternative method employing alkynyl alcohol as the initiator. When DPMK solution was added into the THF/DMSO = 3/2 solution containing anhydrous alkynyl alcohol, the color turned black (instead of orange-red) immediately, meaning that carbanion (alkynyl potassium) was produced. The pKa of the terminal acetylene proton is 25, much lower than that of diphenylmethane (DPM) (pKa = 34), causing the proton abstraction from the acetylene moieties at the initial stage of the polymerization.12
The recovered hydroxyl group on HO–PEG–alkyne enables further modification to introduce different reactivity.8 Here, we used HS–PEG–alkyne (6) as an example. HS–PEG–alkyne was synthesized and characterized using 1H NMR (Fig. 2D). New signals at 2.67 (m, –HS–CH2CH2–), 2.91 (m, –HS–CH2CH2–), and 4.28 (m, –COOCH2–), along with the 1:1 molar ratio of CH2 at 4.28 (m, –COOCH2–) to CH at 4.68 (m, –O–CH(CH3)–O–), support that the hydroxyl ends on HO–PEG–alkyne were completely converted to –SH groups. Thiol-containing PEGs are widely used in the modification of gold nanoparticles and quantum dots (QDs) to improve water solubility, reduce toxicity, and link to targeting moities.4b The alkyne end group of HS–PEG–alkyne can react with azido-containing materials by the mild and efficient “click chemisty”.11
The α,ω-heterobifunctional PEGs shown in this report serve as examples to illustrate a new synthesis approach. It should be pointed out that a large variety of heterobifunctional PEGs can be designed and synthesized by the sequential modification of hydroxyl groups enabled by the cleavable acetal protecting group.8,13
In summary, a new initiator, 2-(1-ethoxyethoxy)ethanol (EEE), was synthesized and the anionic polymerization of ethylene oxide (EO) yielded well-defined PEGs containing cleavable acetals as protecting groups. Two α,ω-heterobifunctional PEGs, HO–PEG–alkyne and HS–PEG–alkyne, were successfully prepared by the deprotection and sequential modification of the hydroxyl ends. This approach is suitable for synthesizing a wide variety of heterobifunctional PEGs.
Acknowledgements
The authors gratefully acknowledge the financial support from the Hong Kong Research Grant Council (General Research Fund 600207).Notes and references
- (a) J. M. Harris, Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Publishing Corporation, New York, 1992 Search PubMed; (b) J. M. Harris and S. Zalipsky, Poly(ethylene glycol) Chemistry and Biological Applications, American Chemical Society, Washington, 1997 Search PubMed; (c) J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- H. Chen, Y. Chen, H. Sheardown and M. A. Brook, Biomaterials, 2005, 26, 7418–7424 CrossRef CAS.
- (a) A. Warnecke and F. Kratz, Bioconjugate Chem., 2003, 14, 377–387 CrossRef CAS; (b) G. Pasut, F. Canal, L. D. Via, S. Arpicco, F. A. Veronese and O. J. Schiavon, J. Controlled Release, 2008, 127, 239–248 CrossRef CAS; (c) J. Nguyen, X. L. Xie, M. Neu, R. Dumitrascu, R. Reul, J. Sitterberg, U. Bakowsky, R. Schermuly, L. Fink, T. Schmehl, T. Gessler, W. Seeger and T. J. Kissel, J. Gene Med., 2008, 10, 1236–1246 CrossRef CAS; (d) E. Kleemann, M. Neu, N. Jekel, L. Fink, T. Schmehl, T. Gessler, W. Seeger and T. J. Kissel, J. Controlled Release, 2005, 109, 299–316 CrossRef CAS.
- (a) J. W. Bae, E. Lee, K. M. Park and K. D. Park, Macromolecules, 2009, 42, 3437–3442 CrossRef CAS; (b) S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314–320 Search PubMed.
- (a) N. N. Reed and K. D. Janda, J. Org. Chem., 2000, 65, 5843–5845 CrossRef CAS; (b) P. Wentworth and K. D. Janda, Chem. Commun., 1999, 1917–1924 RSC; (c) D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489–509 CrossRef CAS.
- J. Li and W. J. Kao, Biomacromolecules, 2003, 4, 1055–1067 CrossRef CAS.
- (a) M. S. Thompson, T. P. Vadala, M. L. Vadala, Y. Lin and J. S. Riffle, Polymer, 2008, 49, 345–373 CrossRef CAS; (b) M. Yokoyama, T. Okano, Y. Sakurai, A. Kikuchi, N. Ohsako, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 1992, 3, 275–276 CrossRef CAS; (c) Y. Nagasaki, T. Kutsuna, M. Iijima, M. Kato, K. Kataoka, S. Kitano and Y. Kadoma, Bioconjugate Chem., 1995, 6, 231–233 CrossRef CAS; (d) Y. Akiyama, H. Otsuka, Y. Nagasaki, M. Kato and K. Kataoka, Bioconjugate Chem., 2000, 11, 947–950 CrossRef CAS; (e) T. Nakamura, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 1998, 9, 300–303 CrossRef CAS; (f) S. Cammas, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 1995, 6, 226–230 CrossRef CAS; (g) S. Hiki and K. Kataoka, Bioconjugate Chem., 2007, 18, 2191–2196 CrossRef CAS; (h) S. Hiki and K. Kataoka, Bioconjugate Chem., 2009, 21, 248–254.
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150–165 CrossRef CAS.
- B. C. Barot and H. W. Pinnick, J. Org. Chem., 1981, 46, 2981–2983 CrossRef CAS.
- (a) D. Taton, A. Leborgne, M. Sepulchre and N. Spassky, Macromol. Chem. Phys., 1994, 195, 139–148 CrossRef CAS; (b) S. Peleshanko, J. Jeong, V. V. Shevchenko, K. L. Genson, Y. Pikus, M. Ornatska, S. Petrash and V. V. Tsukruk, Macromolecules, 2004, 37, 7497–7506 CrossRef CAS; (c) Z. Y. Li, P. P. Li and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4361–4371 CrossRef CAS.
- J. F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS.
- T. Ishizone, G. Uehara, A. Hirao, S. Nakahama and K. Tsuda, Macromol. Chem. Phys., 1998, 199, 1827–1834 CrossRef CAS.
- Z. Y. Li and Y. Chau, Bioconjugate Chem., 2009, 20, 780–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods and experimental details. see DOI: 10.1039/c0py00310g |
| This journal is © The Royal Society of Chemistry 2010 |