Controlled grafting of polystyrene on silica nanoparticles using NMP: a new route without free initiator to tune the grafted chain length
Chloé
Chevigny†
*a,
Didier
Gigmes
b,
Denis
Bertin
b,
Ralf
Schweins
c,
Jacques
Jestin
*a and
François
Boué
a
aLaboratoire Léon Brillouin, CEA Saclay, 91191, Gif sur Yvette Cedex, France. E-mail: jacques.jestin@cea.fr
bLaboratoire Chimie Provence, UMR 6264, CNRS et Universités d'Aix-Marseille 1,2 et 3, Site de St Jérôme, Av. Esc. Normandie-Niemen case 542, 13393, Marseille Cedex 20, France
cInstitut Laue Langevin DS/LSS 6 rue Jules Horowitz, 38042, Grenoble Cedex 9, France
First published on 18th November 2010
Abstract
We synthesized well-defined polystyrene-grafted silica nanoparticles by adapting our previous synthesis process without using free initiator. We were able to obtain a more versatile system in which we can tune the masses of the grafted chains while controlling the polymerization, the colloidal stability and avoid the formation of free polymer chains. The final grafted objects were characterized in a refined way using SANS and the contrast matching method.
Creating model nano-composites composed of nano-fillers introduced into a polymer matrix is a step towards the understanding of mechanical reinforcement of polymers, by studying the relationship between the filler structure and the obtained mechanical properties. For a better understanding of these properties, controlling the dispersion state is a key step. We are interested in an internal trigger, by grafting polymer chains on the particle surface using refined chemistry. Due to our final project, i.e. using the grafted particles as fillers in nanocomposites, it is particularly important for us to obtain non-aggregated, individual particles and to characterize them with non-destructive methods. Tuning the grafted chain length while keeping control of the polymerization reaction is still a challenge for a clearer view of the polymerization mechanisms, and also for the application in nanocomposites, as the chain length ratio between the grafted and the free matrix chains is a key parameter for particle dispersion.1 One of the main methods for obtaining such hybrid objects is the “grafting from” technique,2 which consists of growing polymer chains from a surface, after grafting a polymerization initiator on it. In order to obtain well-defined objects, reversible-deactivation radical polymerizations like atom-transfer radical polymerization (ATRP)3 or nitroxide-mediated polymerization (NMP)4 are often used. The kinetics of both these polymerization types are governed by the persistent radical effect (PRE),5 which needs a minimal concentration of controller (nitroxide radical in the case of NMP, or Cu II, for example, in the case of ATRP) to set up. This is necessary to control the polymerization. In this article, the term controller refers to the aminoxy moiety necessary to control the radical polymerization. The term initiator refers to the alkyl moiety able to start chain polymerization.
Usually, in solution polymerizations (no surfaces/grafting involved), the PRE is easily set up after a short transitional period of time during which a small proportion of self combination reactions between the initiating radicals or the polymeric radical are unavoidable and creates the necessary amount of free controller in solution.
When it comes to polymerizations from surfaces, difficulties in attaining this amount of free controller may arise from too low a quantity of initiator (small grafting density or low particle concentration), or from spatial confinement of the initiator, leading to uncontrolled polymerizations. The simplest solution to this consists of adding free “sacrificial” initiator in solution.6,7 The main drawback of this method is the formation of untethered chains in solution, due to the presence of free initiator, which implies another purification step. Another way to ensure control which could avoid these unwanted free chains is directly adding free controller in solution, permitting the set up of the PRE without polymerizing in solution. Matyjazewski et al.8 first reported the use of this alternative method by the introduction of free Cu II for ATRP of styrene and acrylates from silicon surfaces, successfully leading to well-controlled polymerizations. A detailed study of the differences in kinetics between the “free initiator” and the “free controller” routes was conducted by Tomlinson et al.9 in the case of multiblock copolymers of acrylates, synthesized by ATRP from silicon wafers.
Here we propose to make use of this idea of controlling the polymerization by adding free controller in the frame of a synthesis route using NMP. As we described in a recent paper,10 we set up a very reproducible and controlled method to graft polystyrene chains viaNMP on colloidal silica nanoparticles, while keeping the colloidal stability (checked by small-angle neutron scattering (SANS) measurements) through the whole process. The control of the polymerization was achieved by adding free initiator to the reaction mixture. Similar works can be found in recent publications11–14 which present a series of grafting examples using NMP with free initiator and creation of free polymer chains. These free chains induce a step of purification to get rid of them. The technique we used for this step, ultra-filtration, only permits getting rid of molar masses at most equal to 35000 g mol−1, due to limitations in the cut-off size of the filters. Ultrafiltration is actually not the most used technique to purify the solution of free chains, in most papers dealing with “grafting from” polymerizations, free chains are simply taken away by centrifugation,11,13,14 which does not have any limit on the molar masses. But centrifugation has a huge drawback for our project: it tends to aggregate the particles,14 and the small size of our particles makes them especially sensitive to that. With this free initiator method we cannot then get grafted masses larger than 35000 g mol−1, because we cannot eliminate the free chains. Moreover, to obtain a larger grafted chain mass, we should either reduce the free initiator concentration, which decreases the control of the reaction, or increase the monomer concentration, which destroys the colloidal stability of the particles. By adding free controller SG1 (see Fig. 1) instead of free initiator in solution, we could then polymerize from the nanoparticles in a controlled way, without creating the free chains limiting the reachable mass and thus permitting a tuning of the grafted chain mass. This adds even more versatility to our technique, and is interesting from the fundamental synthesis mechanism point of view. The grafted objects are then characterized by SANS using contrast matching technique.
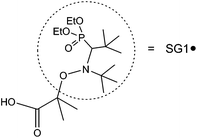 | ||
| Fig. 1 The structure of alkoxyamine MAMA-SG1 with a 1-carboxy-1-methyl ethyl radical as the initiator and N-tert-butyl-N(1-diethylphosphono-2,2-(dimethylpropyl)) nitroxide (SG1) as the controller. | ||
The silica sol is made of Ludox TM-40 silica particles transferred from water to dimethylacetamide (DMAc) according to the procedure described in ref. 10. The initiator-grafting procedure, described in detail in ref. 10, can be briefly summed up as follows: in a first step, the amine-bearing group aminopropyl triethoxy silane (APTES) is grafted to the silica particles via the reactivity of the –OH groups on the surface, and in a second step, the initiation-controlling alkoxyamine moiety is introduced via an over-grafting reaction between the already grafted amino group and the N-hydroxysuccinimide-based MAMA-SG1 activated ester (MAMA-NHS). This process is represented in Fig. 2 (adapted from ref. 10). The initiator grafting density, estimated by TGA with 20% error, is 0.25 molecules nm−2.
![Grafting the initiator in 2 steps: [a] silanization and introduction of the amino group, [b] overgrafting with MAMA-NHS to obtain the initiator-grafted silica particle.](/image/article/2011/PY/c0py00271b/c0py00271b-f2.gif) | ||
| Fig. 2 Grafting the initiator in 2 steps: [a] silanization and introduction of the amino group, [b] overgrafting with MAMA-NHS to obtain the initiator-grafted silica particle. | ||
Previous polymerizations, with free initiator MAMA-SG1 in solution, are used as a reference to set the conditions for the polymerizations without sacrificial initiator here. The solution of initiator-grafted silica particles is diluted to 1.1 wt%, and 20 g are added to a 50 mL three-necked flask. Styrene (from 9 to 1.6 mL) is added dropwise to the solution (feeding rate of 1 drop/s), under constant stirring in order to avoid gelling. The styrene content is adjusted to change the theoretical mass of the grafted polymer. Free nitroxide SG1 (DEPN), about 10% of the quantity of grafted initiator, is added to the solution. The reaction mixture is then deoxygenated by nitrogen bubbling for 30 min. The flask is put in an oil bath heated at 120 °C and the reaction goes on for 4 h. Kinetic samples are taken via sterile syringes to determine conversion by gravimetry: the sample is weighed just after taking, and after complete evaporation of the solvent and the styrene: as styrene is evaporating and polystyrene not, conversion is easily determined.
SANS measurements were performed at the Institut Laue-Langevin (ILL) on the spectrometer D11. Three configurations were used, with one wavelength (10 Å) and three sample-to-detector distances (2 m, 8 m and 34 m), corresponding to a total Q-range of 1 × 10−3 to 0.1 Å−1. Details of data processing and treatment can be found in ref. 10. The incoherent scattering background is subtracted using a blank sample with no silica. In order to measure only the scattering of the polymer coronas we use the contrast matching method. The silica contribution is matched using a 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :43 h-DMAc–d-DMAc mixture as solvent. The total scattered intensity, in our centrosymmetrical system can be written as follows:
:43 h-DMAc–d-DMAc mixture as solvent. The total scattered intensity, in our centrosymmetrical system can be written as follows:
| I(Q) = Φ(Δρ)2VpartP(Q)S(Q) | (1) |
The conditions for the polymerization (particles concentration, temperature and maximum styrene concentration) were optimized to avoid destabilization of the system. The mass of styrene monomer is varied in order to change the grafted mass Mntheoretical (mass of the chains for complete conversion), from the formula
| Mntheoretical = mmonomer/ninitiator | (2) |
The initiator quantity ninitiator is fixed by the grafting density (0.25 initiator/nm2) and by the quantity of introduced particles, unchanged for all polymerizations.
For all the reactions we followed the conversion during time and thus extracted the parameters allowing checking of a good control of the polymerization, represented in Fig. 3.
 | ||
Fig. 3 Monomer conversion as a function of reaction time and semi-logarithmic evolution of the conversion (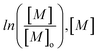 being the monomer concentration) as a function of t2/3 for the four initial monomer concentrations: 28% ( being the monomer concentration) as a function of t2/3 for the four initial monomer concentrations: 28% ( ), 22% ( ), 22% ( ), 15% ( ), 15% ( ) and 6.5% ( ) and 6.5% ( ). ). | ||
We obtain good enough conversions (between 11 and 18%), which increase along with the initial monomer concentration.
The polymerization seems well-controlled for the three highest monomer concentrations (linear progression of  as a function of t2/3, meaning constant radical concentration, which is a first hint of control), but the conversion reaches a plateau before the end of the reaction for the lowest concentration (lowest theoretical mass). This could be due to a competition between recombination of the growing radical with nitroxide and addition on monomer. As styrene quantity is relatively low and recombination between nitroxide and the growing radical is very fast, it might be that above a certain conversion this recombination becomes as important as the growing of the chains.
as a function of t2/3, meaning constant radical concentration, which is a first hint of control), but the conversion reaches a plateau before the end of the reaction for the lowest concentration (lowest theoretical mass). This could be due to a competition between recombination of the growing radical with nitroxide and addition on monomer. As styrene quantity is relatively low and recombination between nitroxide and the growing radical is very fast, it might be that above a certain conversion this recombination becomes as important as the growing of the chains.
After polymerization, the solution of grafted particles is dried and the dry particles characterized by thermogravimetric analysis (TGA) in order to get an estimate of the chains grafting density, which varies from 0.15 to 0.2 chains nm−2. We use the following formula to determine the grafting density (from ref. 10):
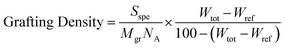 | (3) |
000 g mol−1. All these characteristics of the grafted particles are summed up in Table 1.
| Name | Initial styrene concentration (%v/v) | Conversion (%v/v) | Estimated Mn/g mol−1 | Chains nm−2 | Chains/particle | Initiation efficiency (%) |
|---|---|---|---|---|---|---|
| CC-122 | 28 | 18 | 50000 |
0.15 | 400 | 0.60 |
| CC-125 | 22 | 16 | 32000 |
0.20 | 550 | 0.80 |
| CC-124 | 15 | 15 | 20000 |
0.20 | 550 | 0.80 |
| CC-123 | 6,5 | 11 | 5300 | 0.18 | 490 | 0.72 |
No direct measurement of chain length (like SEC) was performed on the grafted chains, this would require dissolution of the silica core by HF, hence a destruction of the grafted particles. We do not want to use this technique, as we need all the particles afterwards to form composites,15 not to mention the high dangerousness of HF. However, as previously demonstrated,10SANS characterization has proven to be a powerful technique for grafted and free chain mass determination, in perfect accordance with SEC experiments, and is then here essential (and sufficient) to check the good agreement between the experimental and theoretical molecular weight, and to characterize the particles.
The samples are in silica-matching conditions, we observe only the polymer corona. The scattered intensities for each of the four grafted masses are shown in Fig. 4, after subtracting the background and normalizing to obtain the absolute intensity.
 | ||
| Fig. 4 The scattering curves of diluted solutions of silica Ludox particles grafted with PS chains under contrast conditions for which the scattering contribution of the silica core is matched with a mixture of hydrogenated and deuterated solvent (DMAc). The estimated different grafted masses are: 50000 g mol−1 (), 32000 g mol−1 (), 19000 g mol−1 () and 5300 g mol−1 (). | ||
At the lowest Q-values, the intensity reaches a plateau, indicating finite-size objects, which confirms there is no aggregation of the particles. Moreover, the extrapolated intensity at zero Q limit, directly depending on the scattering volume of the objects, increases when the estimated grafted mass increases. This is the first insight that we succeeded in producing grafted particles with increasing total mass of grafted chains. At intermediate Q-values, an oscillation is present for all the samples and its position is characteristic of the corona size: the thicker the corona, the smaller the value of Q. Here again, it supports the fact that increasing the Mnestimated leads to larger coronas. At the highest Q-values, we observe a slope in Q−2 representative of the scattering of Gaussian chains. The four whole scattering curves are then in good agreement with well-dispersed polymer coronas of different masses.
In order to extract the relevant parameters from the scattering data, we fit the intensity curves with a non-interacting Gaussian-chains model,16 which we have previously proven to be well-adapted to these h-PS grafted particles.10 The best fit parameters are plotted in Fig. 5.
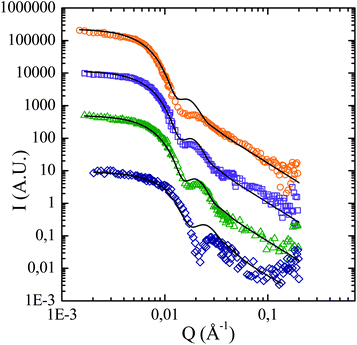 | ||
| Fig. 5 Scattering curves of the polymer coronas (open symbols) and the corresponding fit results with a Pedersen model (solid lines). For the sake of clarity the intensities are shifted by a decade. The estimated different grafted masses are: 50000 g mol−1 (), × 103; 32000 g mol−1 (), × 102; 19000 g mol−1 (), × 10; and 5300 g mol−1 (). | ||
Several parameters of this model are already known from previous measurements on silica particles (particles radius Rcore = 134 Å, log-normal polydispersity index σ = 0.18). The only unknown parameters are the volume fraction in scattering objects Φ, the number of grafted chains per particle (N) and the radius of gyration of the grafted chains (Rg). After this process the polydispersity is re-adjusted in order to take into account the polydispersity of the grafted chains. Table 2 presents the fit results for all the masses. With increasing Mnestimated, the number of grafted chains remains roughly constant, between 410 and 470, and the gyration radius increases from 24 to 87 Å.
| Name | Φ (%v/v) | R core/Å | σ | N (chains/particle) | R g/Å |
|---|---|---|---|---|---|
| CC-122 | 2 | 134 | 0,24 | 440 | 87 |
| CC-125 | 1,7 | 0,23 | 410 | 74 | |
| CC-124 | 1,2 | 0,20 | 440 | 59 | |
| CC-123 | 0,7 | 0,17 | 470 | 24 |
While our model of non-interacting Gaussian-chains fits perfectly the two intermediate masses (samples CC-124 and CC-125), slight differences are found for the lowest and highest grafted chains masses. For the lowest mass (CC-123), they occur at high Q-range (2.10−2–0.2 Å−1), where intensity is below 0.1 cm−1, and can be attributed to the low precision due to this low intensity. For the highest mass, the difference is localized on the oscillation (Q = 2 × 10−2 Å−1). Here, the deviation may come from a slight change in chain conformation (more stretched, or beginning to interact), but our simple model still allows a good estimation of the parameters, as discussed below. It is also noteworthy that this model always fits the intensity at the largest Q-values well. If there were any chains in solution at a significant concentration, the model would under-estimate the scattered intensity in this Q-range, because of the extra scattering of free chains (not included in the model, which is written for polymer coronas only). This is a strong evidence that thermal polymerization is negligible here. The idea here is to show a robust and precise way to characterize grafted objects using SANS, using a previously set procedure.10
From the estimated molecular mass we can also evaluate a magnitude order of the gyration radius from the relation Rg = 0.275 × Mw0.5, by assuming the PDI of the chains is 1.5. This gives respectively Rg = 75.3 Å, Rg = 60.2 Å, Rg = 47.6 Å and Rg = 24.5 Å for CC-122, CC-125, CC-124 and CC-123. Finally a basic analysis of the curves, using the Guinier approximation, gives a global radius of gyration of the scattering objects Rgglobal without any geometrical approximations. Another value of the radius of gyration Rg of the grafted chains can then be simply obtained by subtracting the radius of gyration of the silica core, equal to  , from Rgglobal which gives 89.5, 72.5, 60.5 and 42.0 Å for Rg of grafted chains for CC-122, CC-125, CC-124 and CC-123, respectively. All these values of Rg of the grafted chains are compared in Fig. 6 (a) with the ones obtained from the Pedersen model (see Table 2) and with the values extracted from the estimation of Mn through conversion. There is a good agreement between the different methods of determination and also between the different SANS analyses, indicating the independence of Rg relative to the fitting model. Similarly, Fig. 6 (b) compares the number of chains per particle N obtained by SANS and by TGA. The agreement is excellent for the highest (CC-122) and smallest (CC-123) masses, while not so good for the two intermediate masses (CC-125, CC-124). However, the mean values obtained from the two methods, respectively N = 497.5 for TGA and N = 435 for SANS, are consistent. Finally, Fig. 6 (c) shows a similar correlation, this time for determination of particle volume fraction (SANS/TGA), which is always good. To conclude on these different methods of characterization, we should keep in mind that the modelling of the grafted particle form factor is carried out with the hypothesis that the solutions are diluted enough (typically 0.5% v/v) to eliminate the interactions between the particles. Such interactions, when repulsive, are characterized by a maximum of the scattered intensity in the small-Q region, which is not visible in our present curves. Effective interactions between particles can, however, exist at larger distances, for smaller Q-values not accessible here, and influence the form factor parameters, and especially its pre-factor parameters like N and Φ.
, from Rgglobal which gives 89.5, 72.5, 60.5 and 42.0 Å for Rg of grafted chains for CC-122, CC-125, CC-124 and CC-123, respectively. All these values of Rg of the grafted chains are compared in Fig. 6 (a) with the ones obtained from the Pedersen model (see Table 2) and with the values extracted from the estimation of Mn through conversion. There is a good agreement between the different methods of determination and also between the different SANS analyses, indicating the independence of Rg relative to the fitting model. Similarly, Fig. 6 (b) compares the number of chains per particle N obtained by SANS and by TGA. The agreement is excellent for the highest (CC-122) and smallest (CC-123) masses, while not so good for the two intermediate masses (CC-125, CC-124). However, the mean values obtained from the two methods, respectively N = 497.5 for TGA and N = 435 for SANS, are consistent. Finally, Fig. 6 (c) shows a similar correlation, this time for determination of particle volume fraction (SANS/TGA), which is always good. To conclude on these different methods of characterization, we should keep in mind that the modelling of the grafted particle form factor is carried out with the hypothesis that the solutions are diluted enough (typically 0.5% v/v) to eliminate the interactions between the particles. Such interactions, when repulsive, are characterized by a maximum of the scattered intensity in the small-Q region, which is not visible in our present curves. Effective interactions between particles can, however, exist at larger distances, for smaller Q-values not accessible here, and influence the form factor parameters, and especially its pre-factor parameters like N and Φ.
 | ||
Fig. 6 Correlation between (a) the gyration radiuses estimated from grafted mass, from Pedersen fit (●) and from Guinier approximation ( ) of the SANS data; (b) the number of chains per particle N obtained from TGA (●) and SANS (); and (c) the particles volume fraction Φ obtained from TGA and SANS. ) of the SANS data; (b) the number of chains per particle N obtained from TGA (●) and SANS (); and (c) the particles volume fraction Φ obtained from TGA and SANS. | ||
In summary, the key point of this communication is to describe a way to perform controlled NMP on particles without using free initiator in solution. The route presented here is based on the use of free controller SG1 instead of free initiator, which forces the conditions of the persistent radical effect (leading to the control of the polymerization) without creating free polymer chains in solution (occurring due to the presence of free initiator). The second interesting point is the use of SANS to characterize the particles. Following our former full article10 detailing the refined procedure of fitting the scattering data with the most adequate model, here we take advantage of these previous results by directly using the best model for our particles. The SANS measurements prove here unambiguously the success of this synthesis method to obtain particles grafted with chains of different masses. However, some open questions remain. The quantity of free nitroxide to add was not optimized, and more experiments on this would be necessary in order to better understand the kinetic mechanisms behind this new procedure.
To conclude, we have presented in this communication the successful adaptation of an existing synthesis route10 towards more versatility. By simply changing the initial monomer concentration in the reaction mixture, we can tune the grafted mass of the chains, and obtain very well-defined objects. The use of free nitroxide in solution is then a very valuable way to control the polymerization. This result is interesting both from a chemistry point of view, as it is a first attempt to control polymerization with free nitroxide, but also from physics point of view, as we succeeded in creating particles grafted with longer chains as previously.10 Indeed, creating grafted nanoparticles with a wide accessible range of grafted chains length is of a great interest for many applications and especially in the field of nanocomposites. We will detail in a further paper how these different grafted particles can be used as fillers in nanocomposites and how changing the grafted mass is important to control the dispersion.
References
- P. G. De Gennes, Macromolecules, 1981, 35, 3523.
- (a) O. Prücker and J. Ruhe, Macromolecules, 1998, 31, 592–601 CrossRef; (b) O. Prücker and J. Ruhe, Macromolecules, 1998, 31, 602–613 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS.
- M. Husseman, E. E. Malmstrom, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef.
- K. Matyjaszewski, P. J. Miller, N. Shukla, I. Immaraporn, A. Gelman, B. B. Luokala, T. M. Siclovan, G. Kickelbick, T. Vallant, H. Hoffmann and T. Pakula, Macromolecules, 1999, 32, 8716–8724 CrossRef CAS.
- M. R. Tomlinson, K. Efimenko and J. Genzer, Macromolecules, 2006, 39, 9049–9056 CrossRef CAS.
- C. Chevigny, D. Gigmes, D. Bertin, J. Jestin and F. Boué, Soft Matter, 2009, 5, 3741–3753 RSC.
- V. Ladmiral, T. Morinaga, K. Ohno, T. Fukuda and Y. Tsujii, Eur. Polym. J., 2009, 45, 2788–2796 CrossRef CAS.
- M. K. Brinks and A. Studer, Macromol. Rapid Commun., 2009, 30, 1043–1057 CrossRef CAS.
- C. Bartholome, E. Beyou, E. Bourgeat-Lami, P. Chaumont, F. Lefebvre and N. Zydowicz, Macromolecules, 2005, 38, 1099–1106 CrossRef CAS.
- R. Inoubli, S. Dagreou, A. Khoukh, F. Roby, J. Peyrelasse and L. Billon, Polymer, 2005, 46, 2486–2496 CrossRef CAS.
- C. Chevigny, J. Jestin, D. Gigmes, R. Schweins, E. Di Cola, F. Dalmas, D. Bertin and F. Boué, Macromolecules, 2010, 43, 4833–4837 CrossRef CAS.
- J. S. Pedersen and M. C. Gerstenberg, Macromolecules, 1996, 29, 1363–1365 CrossRef CAS.
Footnote |
| † Present address: Stranski-Lab., Institut für Chemie, TU Berlin, Sekr. TC 9, Strasse des 17. Juni 124, 10623 Berlin, Germany, E-mail: mailto:chloe.chevigny@tu-berlin.de |
| This journal is © The Royal Society of Chemistry 2011 |