Functionalization of inorganic nanoparticles with polymers for stealth biomedical applications
Koon Gee
Neoh
* and
En Tang
Kang
Department of Chemical and Biomolecular Engineering, National University of Singapore, Kent Ridge, Singapore 119260
First published on 29th October 2010
Abstract
Inorganic nanoparticles offer great opportunities in biomedicine, for example, serving as contrast agents for imaging, hyperthermia agents and drug delivery vehicles. A critical factor limiting the applications of nanoparticles for therapeutic and diagnostic purposes is their short circulation time in vivo due to clearance by the body's reticuloendothelial system. One of the most efficient strategies to confer stealthiness to nanoparticles is the use of a polymer “cloak” which inhibits plasma protein adsorption and recognition by the immune system. This review describes some of the polymers which are suitable for these applications, and the strategies which can be employed to functionalize inorganic nanoparticles with such polymers.
 Koon Gee Neoh | K. G. Neoh is currently a Professor at the National University of Singapore. She received her Bachelor's and ScD degrees in Chemical Engineering from MIT in 1976 and 1980, respectively. Her primary research interests are in the areas of nanotechnology, surface and molecular engineering targeted at biomedical applications, in particular, the manipulation of chemical functionalities and nano/micro features of surfaces to achieve selective interactivity with biomolecules and cells. She is a recipient of the National Science Award of Singapore and the National University of Singapore Staff Achievement Award. |
 En Tang Kang | E. T. Kang is a professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore. He received his PhD in chemical engineering from SUNY-Buffalo in 1983. His research interest is in exploring new roles of polymers in nanoscience, molecular electronics biomolecular engineering and environmentally-benign processes. He has published more than 550 papers in international journals and 20 book chapters, and is a co-inventor of 25 patents. He received the National Science Award of Singapore in 1996. He is on the Editorial Advisory Board of half a dozen journals. |
1. Introduction
Nanoparticles possess a number of features which are advantageous for application in biomedicine. They can be administered intravenously and be distributed to organs and tissues. Their dimensions also allow close interaction with cells and the crossing of biological membranes. In recognition of such potential, much research has been carried out to develop nanoparticles as imaging/contrast agents (e.g. magnetic nanoparticles for magnetic resonance imaging (MRI)1,2 and gold nanoparticles for cancer diagnostics3,4), carriers for targeted drug and gene delivery,5–8 hyperthermia agents9,10 or multifunctional agents encompassing diagnostic and therapeutic modalities.11,12 However, the in vivo applications of nanoparticles face a critical challenge. The nanoparticles may be recognized as foreign bodies and rapidly eliminated (possibly within seconds) from the bloodstream by the macrophages and other cells of the reticuloendothelial system (RES).13 This process is thought to be initiated by the adsorption of opsonins from the blood plasma which renders the particles recognizable by the phagocytic cells.Since the initial opsonization of particles is critical to the process of phagocytic recognition and subsequent clearance from the bloodstream, surface engineering of the nanoparticles to reduce opsonization is a logical way to achieve a stealth effect. Much of the earlier work on stealth nanoparticles was focused on the development of liposomes and polymeric nanoparticles for drug delivery.14,15 Research has shown that the physicochemical properties of the nanoparticles such as size, surface charge and hydrophobicity/hydrophilicity play an important role in affecting their uptake by the phagocytic cells. It is generally accepted that nanoparticles with neutral and hydrophilic surface will have a longer half-life.16,17 As such, the coating of the nanoparticles with a hydrophilic polymer is the most commonly used method to modify the nanoparticle surface properties. The majority of such polymers are based on polyethylene glycol (PEG) and its derivatives18 since PEG has been well proven to be non-immunogenic, non-toxic, and protein-resistant.19 It is postulated that these polymer coatings prevent opsonization by shielding the surface charges, increasing surface hydrophilicity, and sterically repulsing the blood components.20 The chain length, density and conformation of the polymer on the nanoparticle surface will determine the effectiveness of the stealth effect. The “mushroom” and “brush” configurations (Fig. 1) have been used to describe PEG coverage at low and high chain density on a nanoparticle surface, respectively. At low surface coverage, the PEG chains have a larger range of motion and will be located closer to the surface of the particle, assuming the “mushroom” configuration. Very low surface coverage can also lead to gaps in the PEG protective layer where opsonin proteins can freely bind to the nanoparticle surface. On the other hand, at high surface coverage the range of motion of the PEG chains will be greatly restricted and they will most often exhibit a semi-linear or “brush” configuration. A high surface density will ensure coverage for the entire nanoparticle surface, but it will also decrease the mobility of the PEG chains and thus decrease the steric hindrance properties of the PEG layer.13
 | ||
| Fig. 1 Schematic diagrams of PEG configurations on the upper hemisphere of a polymeric nanoparticle. In (a), the low surface coverage of PEG chains leads to the “mushroom” configuration where most of the chains are located closer to the particles surface. In (b), the high surface coverage and lack of mobility of the PEG chains leads to the “brush” configuration where most of the chains are extended away from the surface. Reprinted with permission from ref. 13. Copyright 2006 Elsevier. | ||
The incorporation of a polymer coating on the nanoparticles can be achieved either via the “one pot” method where in situ formation of the nanoparticles and the formation of a polymer coating take place concurrently, or the two-step process method where the nanoparticles are first formed and then coated with polymer. The coating process may involve electrostatic interactions or chemical conjugation. This review will cover both the “one pot” and two-step methods.
2. Coating with polymers during nanoparticle synthesis
Magnetic nanoparticles
There has been increasing interest in the use of superparamagnetic iron oxide nanoparticles for biomedical applications since these nanoparticles have low toxicity and are suitable for in vivo applications as they are biodegradable, with the iron product being recycled by cells. Furthermore, with their magnetic properties, manipulation in vivo using an external magnetic field is possible. Polymer coatings have been conferred on these nanoparticles to control the surface properties and functional groups. Coatings with dextran,21 citrate22,23 or aminosilane24,25 prevent particle aggregation in vivo but such coatings are not effective in preventing the particles from being taken up by the RES and accumulating in the liver and spleen shortly after intravenous injection. Much attention is now focused on developing methods to incorporate PEG on the surface of the magnetic nanoparticles surface.Magnetic nanoparticles coated with copolymers of PEG have been prepared in situ during the nucleation and growth of the Fe3O4.26–28 Wan et al. used two different types of PEG-containing polymers: a diblock copolymer, poly(ethylene glycol) monomethyl ether-b-poly(glycerol monoacrylate) (PEG-b-PGA),26 and a graft copolymer, poly(glycerol monoacrylate)-g-poly(PEG methyl ether acrylate) (PGA-g-PEG).27 To prepare the diblock copolymer, PEG-b-poly(solketal acrylate) was first prepared by atom transfer radical polymerization (ATRP) of solketal acrylate using PEG-Br as the macroinitiator, CuBr-1,1,4,7,7-pentamethyldiethylenetriamine as the catalyst and cyclohexanone as the solvent. This was followed by acid hydrolysis of PEG-b-poly(solketal acrylate) to PEG-b-PGA. In a similar manner, PGA-g-PEG was prepared by the acid hydrolysis of poly(solketal acrylate)-g-poly(PEG methyl ether acrylate), which was synthesized via the copolymerization of solketal acrylate and PEG methyl ether acrylate by ATRP. The iron oxide nanoparticle dispersions were prepared by the conventional coprecipitation of Fe2+ and Fe3+ from an aqueous solution by a base in the presence of the copolymer. The stability of the iron oxide dispersion in the presence of the copolymer was attributed to the coordination of the PGA via its 1,2-diols to the Fe atoms on the nanoparticle surface26 (Fig. 2) and the extension of the PEG chains into the aqueous medium. The size of the nanoparticles could be controlled from 4 to 18 nm by changing the graft density of the copolymers.
 | ||
| Fig. 2 Proposed structure in the interaction between the iron oxide surface and PGA. Reprinted with permission from ref. 26. Copyright 2005 The Royal Society of Chemistry. | ||
In another method, poly(oligoethylene glycol methacrylate-co-methacrylic acid) (P(OEGMA-co-MAA)) copolymer was prepared via a two-step procedure: a well-defined precursor poly(oligoethylene glycol methacrylate-co-tert-butyl methacrylate), P(OEGMA-co-tBMA) (Mn = 17300; Mw/Mn = 1.22), was first synthesized by ATRP in the presence of the CuCl/2,2′-bipyridyl catalyst system, and subsequently selectively hydrolyzed in acidic conditions.28 The resultant P(OEGMA-co-MAA) was then utilized as a polymeric stabilizer in the synthesis of the magnetic nanoparticles via the coprecipitation method. The grafting density was estimated to be in the range of 0.2–0.3 chains nm−2. The diameter of the nanoparticles could be tuned in the range 10–25 nm by varying the initial copolymer concentration. The PEGylated nanoparticles exhibited long-term colloidal stability in physiological buffer. Intravenous injection into rats showed no detectable signal in the liver within the first 2 h, and maximum liver accumulation was found after 6 h, providing indirect proof of a prolonged circulation of the nanoparticles in the bloodstream.
Coprecipitation of Fe2+ and Fe3+ has also been carried out in the presence of diblock copolymers based on poly(ethylene glycol) methyl ether methacrylate and 2-(acetoacetoxy)ethyl methacrylate (PEGMAx-b-AEMAy).29 The copolymers were synthesized via reversible addition-fragmentation chain transfer (RAFT) controlled radical polymerization in a two-step procedure (Fig. 3). The first step involved the synthesis of PEGMAx homopolymers with cumyl dithiobenzoate as the chain transfer agent (CTA) and 2,2′-azobisisobutyronitrile (AIBN) as the radical source. In the second step, PEGMAx served as the macro-CTA for the polymerization of AEMA. AEMA was chosen to be the repeating unit for the second block, due to its ability to act as a strong bidentate ligand which can bind effectively onto the inorganic iron oxide surfaces. In aqueous media, the PEGMAx-b-AEMAy diblock copolymers form micelles due to their amphiphilic character, and the insolubility of the AEMAy block in water. The iron oxide formed during coprecipitation was encapsulated in the core of these polymeric micelles stabilized by the AEMA units (Fig. 4). The hybrid micelles did not compromise cell viability in cultures, and in vitro uptake by macrophage cells was found to be significantly lower in comparison to that of the clinically applicable contrast agent, Resovist, suggesting that these systems can evade rapid uptake by the RES.
 | ||
| Fig. 3 A synthetic scheme for the preparation of PEGMAx-b-AEMAy diblock copolymers by RAFT. Reprinted with permission from ref. 29. Copyright 2009 American Chemical Society. | ||
 | ||
| Fig. 4 The strategy followed for the preparation of stabilized magnetic iron oxide nanoparticles in aqueous media: (a) micelle formation of PEGMAx-b-AEMAy diblock copolymers in water; (b) addition of the Fe3+/Fe2+ mixture in the micellar solution, leading to complexation of the iron salts with the β-ketoester ligating units found inside the micellar core; (c) transformation of the iron salt “precursors” into iron oxide nanoparticles inside the micellar core upon addition of ammonium hydroxide aqueous solution. Reprinted with permission from ref. 29. Copyright 2009 American Chemical Society. | ||
In the above-mentioned methods, the magnetic nanoparticles were prepared by the coprecipitation method in the presence of the polymer to achieve a polymeric coating. Magnetic nanoparticles can also be synthesized via the high temperature decomposition of iron acetylacetonate (Fe(acac)3) which gives a narrower size distribution.30 The “one pot” technique to incorporate a polymer coating on the nanoparticles can similarly be applied to this method. Li et al. carried out the high temperature decomposition of Fe(acac)3 in the presence of monocarboxyl-terminated PEG in 2-pyrrolidone.31 The monocarboxyl-terminated PEG was prepared by oxidizing PEG monomethyl ether in the presence of 2,2,6,6-tetramethylpiperidine-1-oxyl as a catalyst. The polymer chains were covalently bound to the surface of the nanoparticles via coordination between the carboxylic acid moieties of the polymer and iron. A higher molar ratio of monocarboxyl-terminated PEG/Fe(acac)3 in the reaction mixture increases the water solubility of the nanoparticles but decreases the size and saturation magnetization of the nanoparticles. A higher molecular weight of the monocarboxyl-terminated PEG (Mn investigated was in the range 550–5000) also increases solubility. From MR images of a rat's liver and kidneys after intravenous injection of the polymer coated nanoparticles, it was concluded that the nanoparticles possess good biocompatibility and long circulation time in blood.
Gold nanoparticles
Gold nanoparticles have potential applications as drug- and gene-delivery carriers, optical imaging probes, and for photothermal therapy. The “one-pot” method has also been used to prepare PEGylated gold nanoparticles.32,33 “Double-hydrophilic” poly(ethylenimine)-poly(ethylene glycol) block copolymer (PEI-b-PEG) was prepared by the coupling of semi-methylated poly(ethylene oxide) (PEO) with PEI.30 PEG-stabilized gold colloids were obtained by mixing AuCl3 with this copolymer. The amino group in the PEI segment of the PEI-b-PEG copolymer reduced the auric cations without additional reducing reagent. Neither the pure PEO (Mw = 2000) nor the pure PEI (Mw = 700) separately can stabilize the gold nanoparticles, and only the copolymer provides the appropriate conditions for the self-reduction and stabilization of the gold nanoparticles. In this method, the distal end of PEG has no reactive group for further functionalization. To introduce a functional group at the end of the PEG chain, another copolymer, acetal-PEG-b-poly(2-(N,N-dimethylamino) ethyl methacrylate) (acetal-PEG-b-PAMA), can be used.33 This copolymer was synthesized via potassium 3,3-diethoxypropyl alcoholate-initiated ring opening anionic polymerization of ethylene oxide in THF, followed by the block copolymerization of 2-(N,N-dimethylamino)-ethyl methacrylate (AMA) to form acetal-PEG-b-PAMA. The α-acetal group of this copolymer was quantitatively converted into an aldehyde group using acetic acid, and biocytin hydrazide was then reacted with this group to form α-biotinyl-PEG-b-PAMA (Fig. 5).33 The block copolymer catalyzed the autoreduction of the HAuCl4 at room temperature and the PAMA segment in the block copolymer coordinates with the gold surface, allowing the PEG segment to extend from the surface into the aqueous exterior and shielding the cationic charge of the PAMA segment. The interaction of the PAMA block with the gold surface will be discussed in greater detail in a later section. The biotin molecule at the end of the tethered PEG chain offers the possibility of further functionalization. | ||
| Fig. 5 A schematic representation of the functionalization of gold nanoparticles with α-biotinyl-PEG-b-PAMA in a “one pot” method. Reprinted with permission from ref. 33. Copyright 2004 American Chemical Society. | ||
Silica nanoparticles
Silica nanoparticles is another class of nanoparticles which can offer favorable advantages for use in biomedical applications since their synthesis is relatively straightforward and low cost, and surface modification for bioconjugation can be readily carried out. Organo-silica nanoparticles possessing an organo-silica core and monolayer PEG shell were synthesized by the co-hydrolysis and copolycondensation reactions of ω-methoxy(polyethyleneoxy)propyltrimethoxysilane (Mw = 460–590) and hydroxymethyltriethoxysilane mixtures.34 The reactions were catalyzed by sodium hydroxide in the presence of a surfactant, benzethonium chloride. The nanoparticles ranged from 2 to 10 nm in diameter, depending on the ratio of the reactants. Another method of preparing PEG-coated silica nanoparticles involved the hydrolysis of tetramethylorthosilicate in a mixed solution of PEG monomethyl ether, ammonium hydroxide and methanol.35 The PEG chains attached to the particle surface via the reaction between the silanol groups on the particle surface and the alcohol end groups on the PEG chain. The diameter of these particles ranged from about 50 to 350 nm. The amount of bovine serum albumin (BSA) adsorbed on the silica nanoparticles decreased with increase in the molecular weight of the PEG in the range of 750 to 6000, and the amount of adsorbed protein on the PEG-6000-coated particles was ∼3 times lower than that on the bare silica particles3. Coating of preformed nanoparticles with polymer
A polymer coating may be grafted on preformed nanoparticles by either the “grafting to” or “grafting from” method. In the former, preformed polymer chains are grafted to the surfaces of the nanoparticles via electrostatic interaction or coordination between groups on the polymer chains and the nanoparticles. In the latter, an initiator is first immobilized on the nanoparticle surface, after which polymerization of the selected monomer(s) proceeds from the nanoparticle surface. While the “grafting to” technique is more easily carried out, the grafting density achieved is often low since the chains which are already attached present a steric barrier to the approaching polymer molecules. The film thickness is also limited by the molecular weight of the polymer chains in solution.3.1 “Grafting to” method
 | ||
| Fig. 6 Groups that can be used to anchor polymers on iron oxides nanoparticles. Reprinted with permission from ref. 36. Copyright 2010 NPG Asia Materials. | ||
In applications of stealth nanoparticles for active targeting of tumors, specific targeting ligands are required in addition to the protein-resistant polymer coating. Silane-PEG-trifluoroethylester was synthesized and used to confer a PEG coating with functional groups for attaching the targeting ligands on magnetic nanoparticles.40,41 The treatment of the PEG-trifluoroethylester with ethylenediamine converted the terminal group of the PEG chain to amine which was then used for conjugation with folic acid (FA), a tumor targeting ligand. An alternative protocol for preparing magnetic nanoparticles coated with PEG functionalized with FA involved first the reaction of FA with tert-butyloxycarbonyl (t-boc) and N-hydroxysuccinimide (NHS), followed by the reaction of (t-boc)folate-NHS with amino-PEG-carboxyl.42 The resultant (t-boc)folate-PEG-carboxyl was then reacted with the magnetic nanoparticles which had been conferred with amine groups by reaction with (3-aminopropyl)-trimethoxysilane. Finally, the t-boc protective group was removed.
Dopamine (DPA) has been proposed as an alternative to silane due to its high affinity for the iron oxide nanoparticle surface and the possibility of functionalization with other molecules through amide bonds.43 The PEGylation of hydrophobic nanoparticles produced from the high-temperature decomposition of Fe(acac)3via a DPA anchor has been reported.44 DPA was first linked to a COOH group in PEG diacid (HOOC-PEG-COOH) via N,N′-dicyclohexylcarbodiimide (DCC)/NHS chemistry to produce HOOC-PEG-DPA. This was then used to replace the oleate/oleylamine coating on the particles thereby converting the particle surface from hydrophobic to hydrophilic. PEG diacid molecules of Mw of 600, 3000, 6000, and 20000 were used and the uptake of these PEG-coated nanoparticles by macrophages was compared with that of dextran-coated nanoparticles. At a concentration of 0.01 mg Fe mL−1 in the incubation medium, the dextran-coated nanoparticles gave the highest uptake of >5 pg Fe/cell whereas the uptakes of the PEG-3000-, PEG-6000-, and PEG-20000-coated magnetic nanoparticles were < 0.5 pg/cell. However, when the concentration was increased to 0.1 mg Fe mL−1, the uptake of both dextran- and PEG-coated magnetic nanoparticles increased about 20-fold. The use of DPA as an anchor for iron oxide nanoparticles may have some potential problems since another group has pointed out that DPA reacts with Fe3+ which facilitated a rapid degradation of the nanoparticles.45
Phosphonic acid is another group which can be used for anchoring polymers on iron oxide nanoparticles. Phosphonic acid-terminated poly(oligoethylene glycol acrylate) (poly(OEG-A)) was synthesized using a trithiocarbonate RAFT agent bearing dimethyl phosphonate group.46 Following the polymerization process, the dimethylphosphonate was cleaved into phosphonic acid by bromosilane, and the trithiocarbonate was transformed into pyridyl disulfide group via aminolysis and reaction with dithiopyridine. The α-phosphonic acid, ω-dithiopyridine functionalized poly(OEG-A) was then grafted onto the iron oxide nanoparticles. The phosphonic acid end group provides strong iron oxide chelation properties for stabilization of the particles, while the pyridyl disulfide end group enables the conjugation of biomolecules to the nanoparticles via selective thiol–pyridyldisulfide exchange reaction. The poly(OEG-A)-stabilized iron oxide nanoparticles exhibited low protein adsorption (<30 μg g−1 nanoparticles) over a wide range of BSA concentration (0.05 to 10 g L−1). The same strategy was used to prepare α-phosphonic acid terminal poly(dimethylaminoethyl acrylate) (poly(DMAEA)).47 The poly(OEG-A) and poly(DMAEA) homopolymers can be simultaneously assembled on the surfaces of iron oxide nanoparticles, and the polymer composition on the surface of the nanoparticles could be controlled by manipulating the feed ratio of the polymers used. By tailoring the relative density and chain length of the two homopolymers on the nanoparticle surface, the poly(OEG-A) chains can effectively mask the positive charges originating from poly(DMAEA) to limit protein adsorption on these particles. The poly(DMAEA) chains can be exploited for the complexation of siRNA, thereby generating iron oxide siRNA nano-carriers with antifouling poly(OEG-A) shells.
Block copolymer micelles have been explored as long circulating vehicles of hydrophobic drugs, with the hydrophobic micelle core serving as a reservoir for the drugs, while the corona shell shields the hydrophobic core and prevents nonspecific interactions with plasma proteins in the biological environment.48,49 Nasongkla et al. have developed amphiphilic block copolymers of maleimide-terminated poly(ethylene glycol)-block-poly(D,L-lactide) (MAL-PEG-PLA) for the encapsulation of iron oxide nanoparticles and doxorubicin.50 The MAL-PEG-PLA was synthesized by ring opening polymerization of D,L-lactide at 110 °C. Poly(ethylene glycol) monoethyl ether maleimide (Mn = 3210) was used as a macro-initiator and stannous (II) octoate was the catalyst. Multiple hydrophobic iron oxide nanoparticles (synthesized from high temperature decomposition of Fe(acac)3) together with doxorubicin can be encapsulated in the micelles of ∼45–50 nm in diameter. Cancer targeting capability was achieved by attaching cRGD to the micelle surface through a covalent thiol-maleimide linkage.
Gold nanoparticles
Thiol groups have been widely used as anchors for polymer chains on gold surface, and PEG with terminal thiol group (PEG-SH) is commercially available. A number of investigators have prepared PEG-coated gold nanoparticles via the reaction of the thiol group of PEG-SH with the gold surface in an aqueous solution.51–53 Akiyama et al. grafted PEG onto the surface of gold nanorods stabilized by hexadecyltrimethylammonium bromide (CTAB), and since CTAB is cytotoxic, it was removed by dialysis against water. The effects of grafting level and injection dose on the biodistribution of the nanorods in the tumor-bearing mice after intravenous injection were investigated.54 Higher PEG grafting levels were found to be advantageous for RES avoidance and for suppression of aggregation of the gold nanorods during in vivo circulation. A PEG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :gold molar ratio of 1.5 was sufficient to show both prolonged circulation and the EPR effect. When the injection dose was increased above 19.5 μg of gold, the RES uptake in the liver was saturated and surplus gold nanorods were distributed to other tissues, especially the spleen and the tumor. The same group also evaluated the effect of the length of the tethered PEG chain on the stability of the gold nanorods in aqueous solution and blood circulation.53 PEG-SH of four molecular weights (2000, 5000, 10000, 20000) were used, and the thickness of the polymer shells was found to increase with increasing length of PEG chain. The coating with PEG-2000 was unable to stabilize the gold nanorods in physiological conditions for a long period even though it was sufficient to electrostatically shield the surface of the gold nanorods. From the analysis of the gold content in the blood and organs of mice 30 min after the intravenous injection of the nanorods, it was concluded that the PEG-5000- and PEG-10000-modified gold nanorods showed higher stability in blood circulation compared with PEG-2000- and PEG-20000-modified gold nanorods, with >60% of the injected dose of the former present in the blood. The lack of effectiveness of the PEG-2000 chains has also been shown in earlier works and this has been attributed to the lack of flexibility of the shorter chains.13,55,56 Although the authors did not give a reason for the lack of effectiveness for the PEG-20000 coating, it is likely that excessively long chains do not offer an optimum conformation as discussed below.
:gold molar ratio of 1.5 was sufficient to show both prolonged circulation and the EPR effect. When the injection dose was increased above 19.5 μg of gold, the RES uptake in the liver was saturated and surplus gold nanorods were distributed to other tissues, especially the spleen and the tumor. The same group also evaluated the effect of the length of the tethered PEG chain on the stability of the gold nanorods in aqueous solution and blood circulation.53 PEG-SH of four molecular weights (2000, 5000, 10000, 20000) were used, and the thickness of the polymer shells was found to increase with increasing length of PEG chain. The coating with PEG-2000 was unable to stabilize the gold nanorods in physiological conditions for a long period even though it was sufficient to electrostatically shield the surface of the gold nanorods. From the analysis of the gold content in the blood and organs of mice 30 min after the intravenous injection of the nanorods, it was concluded that the PEG-5000- and PEG-10000-modified gold nanorods showed higher stability in blood circulation compared with PEG-2000- and PEG-20000-modified gold nanorods, with >60% of the injected dose of the former present in the blood. The lack of effectiveness of the PEG-2000 chains has also been shown in earlier works and this has been attributed to the lack of flexibility of the shorter chains.13,55,56 Although the authors did not give a reason for the lack of effectiveness for the PEG-20000 coating, it is likely that excessively long chains do not offer an optimum conformation as discussed below.
In another study on the cellular level biodistribution of gold nanorods, Tong et al. compared nanoparticles coated with linear and branched PEG.57 The nanorods coated with linear PEG were prepared by replacing the surface CTAB on the nanorods with commercial methyl-PEG-5000-SH or methyl-PEG-2000-SH. For coating with branched PEG, the nanorods were conjugated with SH-PEG-5000-NH2 3HCl. After dialysis, (methyl-PEO12)3-PEO4-NHS ester was added in the presence of triethylamine. Acetic anhydride was then added to block unreacted NH2 groups. The PEGylated gold nanorods injected into the tail vein of mice were found to exhibit a biphasic clearance mode, with a significantly prolonged blood residence time for branched PEG as compared to the linear PEG. It was postulated that one reason for the biphasic decay might be the surface heterogeneity among PEGylated nanoparticles, with a population bearing insufficient protection. The poor steric shielding allows opsonic binding, which enhances phagocytosis by RES. The binding of serum components to the nanorods coated with linear PEG was found to be higher than those coated with the branched PEG which is consistent with the prolonged blood circulation of the latter.
Gold nanoshells, synthesized by the deposition of gold on silica nanoparticle core, were PEGylated using methoxy-PEG-thiol (mPEG-thiol) and subjected to an in vitro macrophage assay to examine the effect of surface density and chain length of the PEG, and size of the gold nanoshells on their uptake.58 The results showed that a relatively larger amount of phagocytosis takes place for gold nanoshells PEGylated with both the shortest (750 Da) and longest (20000 Da) chains used (Fig. 7a). The finding that intermediate PEG chain length is optimum is consistent with those of Niidome et al.53 although the two studies differ on the effectiveness of PEG of 2000 Da. The higher uptake observed with nanoshell PEGylated with PEG chain of 750 Da was attributed to its lack of flexibility. On the other hand, PEG chains that are too long have a high tendency to fold into coils or bend into a mushroom-like configuration on the surface of the gold nanoshell. The difference in effectiveness of PEG chains of the same length in preventing macrophage uptake of nanoparticles as reported by different investigations may be the result of the different surface PEG density, as illustrated in Fig. 7b.58 This study reported that when 2.5μmol of mPEG-thiol was used in the PEGylation of 2 × 1013 gold nanoshells (of 227 nm in diameter), the surface will be saturated with PEG. This would correspond to about 75000 molecules of mPEG-thiol on each gold nanoshell particle with each molecule having a footprint of 2.2 nm2, which is comparable to the hydrodynamic size of the mPEG-thiol molecule (∼ 2 nm).59 Thus, at saturation, the PEG molecules would be so tightly packed that no further binding of PEG is possible, and opsonins will also be prevented from approaching the nanoshell.
 | ||
| Fig. 7 (a) The percentage of the gold nanoshells PEGylated with different chain lengths of mPEG (at 2.5 μmol mL−1) which was phagocytosed, (b) the percentage of the gold nanoshells PEGylated with different amounts of mPEG (Mw = 2000 Da) which was phagocytosed. Reprinted with permission from ref. 58. Copyright 2009 Taylor & Francis. | ||
PEG-b-PAMA has been grafted on gold nanoparticles to render them dispersible under physiological conditions.60 The PEG macroinitiator, methoxy-poly(ethylene glycol)-2-bromoisobutyrate, was synthesized by the anionic ring-opening polymerization of ethylene oxide, followed by acylation with 2-bromo-2-methylpropionyl bromide to introduce the initiating moiety at the ω-chain end (Fig. 8). Using this initiator, the block copolymerization of AMA was carried out by ATRP in the presence of CuBr and 2,2′-bipyridine in methanol. By varying the polymerization time, PEG-b-PAMA was synthesized with the degree of polymerization (DP) of AMA ranging from 3 to 43. Gold nanoparticles were modified with PEG-b-PAMA of different PAMA chain lengths under different PEGylation conditions (pH, polymer concentration). After 18 h, the PEGylated nanoparticles prepared under acidic conditions showed signs of aggregation while those prepared at pH > 10 showed constant size. It is postulated that above pH of 10, the amino groups of the PAMA segment of the block copolymers were completely deprotonated since the pKa of the amino groups of PEG-b-PAMA is 7.0. Thus, it is likely that the PEG-b-PAMA interacted with the gold nanoparticle surface via multipoint coordination of the tertiary amino groups of PAMA, and not via electrostatic interactions. The gold nanoparticles modified with PEG-b-PAMA having a short PAMA chain length showed excellent dispersion stability, and the charge on the PEGylated nanoparticle surface was effectively shielded by the PEG brush over a wide pH range from 2 to 12. These nanoparticles showed excellent dispersion stability when kept in 95% human serum for 4 days. The increased PEG chain density resulting from PEG-b-PAMA with a short PAMA chain length is one of the factors that improve the dispersion stability of gold nanoparticles, indicating that three amino nitrogen-bearing side chains in the PAMA segment are sufficient to stabilize the PEG brushes on the gold colloid surface via the polyvalency effect.
 | ||
| Fig. 8 A schematic representation of PEG-b-PAMA synthesis. Reprinted with permission from ref. 60. Copyright 2008 American Chemical Society. | ||
Gold nanoparticles have been grafted with thermosensitive copoly(oligoethylene oxide) acrylates to achieve dual thermosensitive and antifouling properties.61,62 The synthesis of different copolymers based on di(ethylene glycol) ethyl ether acrylate (DEG-A) and OEG-A was achieved by RAFT polymerization using 3-(benzylsulfanylthiocarbonylsulfanyl) propionic acid as the RAFT agent and AIBN as the initiator (Fig. 9). The feed ratios and the compositions of the copolymers were proximate; indicating that these two monomers have similar reactivity in the RAFT mediated copolymerization reaction. Depending on the ratio of the two monomers, the lower critical solution temperature (LCST) of the copolymer can be tuned between 15 °C (LCST for poly(DEG-A)) and 92 °C (LCST for poly(OEG-A)). The RAFT end group was preserved after purification, and the strong affinity of the trithiocarbonate functionality for the gold surface resulted in the assembly of the polymers onto the gold surface. The zeta potential of the gold nanoparticles increased from about −40 mV before grafting (i.e. citrate-stabilized) to ∼0 after grafting with the copolymer. In the presence of the copolymer stabilizing layer below the LCST, less than 0.10 mg m−2 (9 × 1014 molecules m−2) of BSA was adsorbed. In comparison, 2.8 mg m−2 (or 2.5 × 1016 BSA m−2) of the protein was adsorbed on the gold nanoparticles in the absence of the polymer coating due to the strong charge interactions between the citrate stabilizing groups and the protein. In an extension of this technique, the co-assembly of two different polymers, neutral and thermosensitive (e.g. poly(OEG-A-co-DEG-A)) and charged (cationic or anionic) polymers, onto gold nanoparticles surfaces has been carried out.62 The surface charge of such nanoparticles can then be modulated by two different stimuli, pH and temperature.
 | ||
| Fig. 9 Copolymerization of oligo(ethylene glycol) acrylate (1) and di(ethylene glycol) acrylate (2) using 3-(benzylsulfanylthiocarbonylsulfanyl) propionic acid (3). Reprinted with permission from ref. 61. Copyright 2009 American Chemical Society. | ||
A layer-by-layer (LBL) approach has been used to build up a hydrophilic polymer layer on gold nanoparticles.63 In this work, the gold nanoparticles synthesized via the citrate reduction method have negative surface charges which were exploited for the deposition of the cationic polyethyleneimine (PEI). This was followed by a layer of poly(acrylic acid)-g- poly(oligoethylene glycol methylether acrylate) (POEGMEA) (PAA-g-POEGMEA). The gold nanoparticles coated with PAA-g-POEGMEA were found to be stable in phosphate buffer for 14 h. Schneider et al. reported an electrostatic and covalent LBL assembly strategy for the synthesis of stealth nanoparticles possessing a high stability in physiological media and a covalently attached pro-drug for release after endocytosis and enzymatic cleavage.64 The colloidal core of gold nanoparticle was first coated with five internal primer layers of poly(allylamine) (PAH) and poly(styrenesulfonate) (PSS) to create a defined polyamine surface that is independent of the core material. A terpolymer (F-HPMA) was attached by covalent LBL deposition on the functionalized nanoparticle to yield the final core/shell multifunctional nanoparticles (MFNP) (Fig. 10). The terpolymer (structure presented in Fig. 10B; Mw = 56400, Mw/Mn = 1.9) bears three different monomer repeat units, the majority (∼90%) being N-(2-hydroxypropyl) methacrylamide which provides a highly water-solvated corona layer to minimize opsonization of MFNP by serum proteins. The second monomer unit, N-methacryloyl-glycyl-glycyl thiazolidine-2-thione (∼8%), enables the covalent attachment of the terpolymer to the PSS/PAH functionalized nanoparticles through the aminolysis of the thiazolidine-2-thione reactive groups by the primary amino groups present on the surface of the PAH corona layer. The third comonomer is N-methacryloyl-glycyl-DL-phenylalanyl-leucyl-glycyl doxorubicin (∼2%) which would release the active drug after endocytosis through the enzymatic degradation of the oligopeptide spacer (Y in Fig. 10). While the (PSS/PAH) multilayer-coated particles are highly stable in pure water, such dispersions aggregate when the ionic strength approaches physiological concentrations. The attachment of the terpolymer further stabilizes the particles and prevents their aggregation in PBS at pH 7.4 or in PBS containing 0.5 mg mL−1 human serum albumin (Fig. 10C and inset). TPA-differentiated THP-1 monocytes were used as model macrophages for testing the stealthiness of the PSS/PAH functionalized nanoparticles with and without the terpolymer coating. Optical microscopy observation of the macrophages after incubation with the PSS/PAH functionalized nanoparticles for 72 h indicated significant association of these nanoparticles with the cells (either internalized or adsorbed). On the other hand, the corresponding experiments with the MFNP did not show a significant degree of adsorption on or internalization into the macrophages.
 | ||
| Fig. 10 (A) From left to right: as-synthesized 13 nm gold nanoparticles (AuNPs) stabilized by adsorbed sodium citrate; AuNPs coated with five primer layers of PAH and PSS (i.e., Au5+:Au/(PAH/PSS)2/PAH); Au5+ further coated with a external layer of F-HPMA (yielding MFNP). The red circles represent doxorubicin moieties (Dox) and are at scale with respect to the size of Dox molecules and the density of Dox moieties on the nanoparticle surface. (B) Chemical structures of the terpolymer and the polyelectrolytes used for the layer-by-layer deposition. (C) (i) Transmission electron micrographs of Au5+ dispersed in pure water, (ii) MFNP dispersed in pure water, (iii) MFNP dispersed in PBS of pH 7.4. The bottom right inset represents Au5+ and MFNP, respectively, dispersed in PBS in the presence of 0.5 mg mL−1 human serum albumin (HSA). Reprinted with permission from ref. 64. Copyright 2009 American Chemical Society. | ||
The grafting methods described above rely on either chemical or electrostatic interactions between the polymer chains and the surface of the gold nanoparticles. In contrast, Prencipe et al. synthesized PEG-grafted branched polymers based on poly(γ-glutamic acid) (γPGA) which has abundant free carboxylic acid groups for attaching lipophilic species such as pyrene or phospholipid, which bind to gold nanoparticles and nanorods via robust physisorption.65 In the first synthetic step of PGA-Py-mPEG, the free carboxylic acid groups of γPGA (Mn ≈ 430000) were used to couple 1-methylaminopyrene via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) amidation. In the second step, the remaining carboxylic acid groups of γPGA were used to attach amine-terminated poly(ethylene glycol) methyl ethers (mPEG-NH2, Mw ≈ 5000). The average number of pyrene moieties grafted to γPGA chains was estimated to be ∼30%, and the remaining ∼70% of the γPGA backbone was loaded with PEG. The preparation of PGA-DSPE-mPEG involved the PEGylation of γPGA followed by the grafting of the lipophilic phospholipid moiety, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) via EDC. The average numbers of DSPE and PEG moieties grafted to γPGA chains were determined to be ∼10 and ∼60%, respectively. Functionalization of gold nanoparticles was achieved by exchanging the capping ligand (citrate) with either PGA-Py-mPEG or PGA-DSPE-mPEG. These functionalized nanoparticles showed no significant changes in suspension dispersity or absorbance after a 48 h incubation in 50% fetal calf serum.
Silica nanoparticles
PEG-grafted silica nanoparticles have been prepared by a two-step method involving the direct facile esterification condensation between alkyl–hydroxyl modified silica nanoparticles and carboxyl-terminated PEG under ambient conditions.66 Epoxy groups modified silica nanoparticles were first prepared by grafting 3-glycidoxypropyl trimethoxysilane onto the surface of silica nanoparticles. The epoxy groups were converted into alkyl–hydroxyl units by refluxing in an ethanol-HCl mixture. Carboxyl-terminated PEG (PEG–COOH) was then grafted onto the silica surface via direct esterification condensation using DCC as an activating agent and 4-(dimethylamino)pyridinium-p-toluenesulfonate as a catalyst.PEG-silanes of different chain lengths have also been covalently attached to 150 nm mesoporous silica nanoparticles (MSNs) to study the nonspecific binding of serum proteins and cellular responses.67 The polymers were synthesized via the hydrogen-transfer nucleophilic addition reaction between the end hydroxyl group of the PEG and the isocyanate group of 3-(triethoxysilyl) propylisocyanate. The results showed that the optimal molecular weight of the PEG for inhibiting nonspecific binding of PEGylated MSNs to human serum protein (HSA) should not be less than 10000 and the corresponding optimal chain densities (defined as wt% of the coating on the MSNs) for PEG-10000–MSNs and PEG-20000–MSNs were 0.75 wt% and 0.075 wt%, respectively. At these conditions, the HSA adsorption was ∼2.5% compared to 18.7% on MSNs without PEGylation. It was postulated that the increase in HSA adsorption observed when the PEG chain density is increased beyond the optimal is due to the loss in chain flexibility which depressed the steric hindrance effect against the nonspecific binding to HSA.
Another method of conferring a PEG coating on silica nanoparticles is via electrostatic self-assembly of polyethyleneimine-polyethylene glycol (PEI-PEG) copolymer. This results from the strong interactions between the polyamino backbone of the copolymer and the negatively charged silica surface.68 MethoxyPEG (Mw of 3400 and 5000) was converted into methoxyPEG-acid via a 3-step process: reaction with sodium hydride, followed by methyl bromoacetate, and finally hydrolysis of the obtained ester in aqueous sodium hydroxide. The carboxylic acid groups introduced on the methoxyPEG were activated with DCC and NHS in dry DMSO before being reacted with primary amines of the PEI in the presence of triethylamine. The ratio of PEG to primary amines in the PEI backbone was about 1:4.5, which represents about 34 grafted PEG chains (Mw = 5000) per PEI molecule. The efficiency of the PEGylated nanoparticle surfaces in preventing biologically nonspecific adsorption events was investigated in vitro using human serum albumin and lysozyme in PBS, and 5% fetal calf serum. Protein fouling was minimal (at the limit of sensitivity of XPS) for the nanoparticles coated with the two PEI-PEG copolymers investigated, and no aggregation was observed for the coated nanoparticles.
3.2 “Grafting from” method
In recent years, surface-initiated graft polymerization based on living polymerization techniques such as ATRP and RAFT polymerization have gained popularity. A dense polymer layer with controlled structures can be achieved with such “grafting from” approaches as these techniques allow for good control over molecular weight and monodispersity. ATRP has recently been successfully employed to form a PEGMA shell around magnetic iron oxide nanoparticles, using a “grafting from” approach.72,73 In the first method, a silane initiator, 4-(chloromethyl)phenyl trichlorosilane, was first immobilized onto the surface of the nanoparticles synthesized by the high-temperature decomposition of Fe(acac)3, after which PEGMA was graft polymerized from the nanoparticle surface via copper-mediated ATRP in water (Fig. 11).72 The grafted poly(PEGMA) chains are stable and enable the modified magnetic nanoparticles to disperse well in aqueous solutions. The morphology and viability of macrophages cultured in a medium containing 0.2 mg mL−1 of poly(PEGMA)-immobilized magnetic nanoparticles were similar to those of cells in the control experiment without any nanoparticles. The uptake of nanoparticles by macrophages was greatly reduced, from 158 pg per cell to <2 pg per cell after grafting with the poly(PEGMA) layer. The second method is a solvent-free ATRP method where the macroinitiators on the surface of the magnetic iron oxide nanoparticles were introduced through effective ligand exchange of the long alkane chain surfactant (oleic acid) on the nanoparticle surface by 3-chloropropionic acid.73 This process rendered the magnetic nanoparticles soluble in the PEGMA monomer. After the solvent-free ATRP of PEGMA, the nanoparticles have a hydrodynamic diameter of 36 nm and possess good solubility and stability in water. In the solvent-free method, the polymerization rate and resultant molecular weight are easier to control than ATRP in aqueous solution. The poly(PEGMA)-modified nanoparticles prepared by this method are just as effective in evading macrophage uptake as the nanoparticles grafted with poly(PEGMA) via surface-initiated ATRP in water.
 | ||
| Fig. 11 A schematic representation of the preparation of PEGMA-coated magnetic nanoparticle by surface-initiated ATRP. Reprinted with permission from ref. 72. Copyright 2006 American Chemical Society. | ||
Hyperbranched polyglycerol (HPG) has recently been shown to be resistant to protein adsorption, and its branched nature may be more efficient in reducing protein interactions than a linear polymer.74,75 Polyglycerol has also been found to be as biocompatible as PEG.76,77 HPG-grafted magnetic iron oxide nanoparticles have been prepared by the surface-initiated polymerization of glycidol via two different methods.78–80 In the first method, the magnetic nanoparticles (prepared by the coprecipitation method) were first treated with 3-mercaptopropyltrimethoxysilane followed by deprotonation with sodium methoxide solution in methanol for 5 h at 60–70 °C. The surface-initiated anionic ring-opening polymerization of glycidol was then carried out in toluene at 95 °C (Fig. 12).78 The polymerization mechanism has been described79 as follows: the deprotonated group attacks the glycidol monomer on the unsubstituted side to produce a secondary alkoxide. The alkoxide can subsequently react with another glycidol monomer or exchange a proton with a neighboring primary alcohol group. With further reaction of the monomer with the alkoxides, a hyperbranched polymer is obtained. The content of HPG on the nanoparticle surface was calculated to be 13.4% (by wt). The adsorption of three proteins, fibrinogen (340 kDa, isoelectric point = 5.5), lysozyme (14.6 kDa, isoelectric point = 11.1), γ-globin (150 kDa, isoelectric point = 7.1) on the nanoparticles in PBS for 24 h at pH 7.4 and 4 °C was investigated. The results indicated that the HPG-grafted nanoparticles resisted the adsorption of all three proteins. The efficacy of the HPG-grafted nanoparticles in resisting proteins adsorption was found to be comparable with that of nanoparticles grafted with silanated methyloxypoly(ethylene glycol) (Mw = 750) at a similar grafting density.
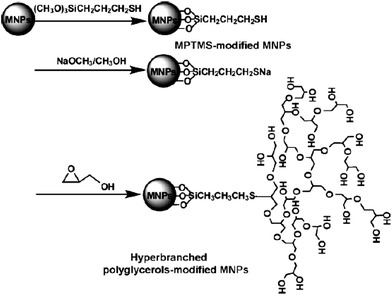 | ||
| Fig. 12 A schematic representation of the preparation of hyperbranched polyglycerol-modified magnetic nanoparticle via anionic ring-opening polymerization of glycidol. Reprinted with permission from ref. 78. Copyright 2008 Elsevier. | ||
In the second method, the magnetic iron oxide nanoparticles were synthesized via the high temperature decomposition of Fe(acac)3 in the presence of oleic acid and oleylamine. The oleic acid-stabilized nanoparticles were subjected to a two-phase ligand exchange reaction with 6-hydroxy caproic acid. The hydroxyl groups on the nanoparticle surface after treatment with aluminium isopropoxide formed the initiators for the subsequent surface-initiated polymerization of glycidol.80 The HPG-grafted nanoparticles have an average hydrodynamic diameter of about 24 nm, and possessed excellent solubility and stability in water, PBS and cell culture medium. The HPG-grafted nanoparticles did not exhibit significant cytotoxicity towards macrophages and fibroblast cells. The uptake of the HPG-grafted nanoparticles by macrophages was very low (<3 pg Fe/cell), even when cultured with a relatively high nanoparticle concentration (1 mg nanoparticles/mL) over 3 days.
:1 to 61.7:1. The HPG shell protects the QDs from biological media, and hence the HPG-grafted QDs can still emit strong fluorescence in cells, while the fluorescence of pristine QDs was almost quenched.
4. Conclusions
Inorganic nanoparticles are promising candidates for biomedical applications but their use in vivo will be very restricted if they are rapidly cleared from blood circulation by the RES. To address this issue, a number of strategies have been developed to coat nanoparticles with polymers to inhibit protein adsorption and uptake by macrophages. In general, these polymer coatings require a component which serves to anchor them to the nanoparticle surface (e.g. through electrostatic interactions or covalent bonds) while another component comprising hydrophilic chains will extend outwards from the surface. In view of the variety of nanoparticles with high potential for biomedical applications, many different designs of polymer coating have been developed as summarized in this review. The intended application of the nanoparticles will determine whether additional functionality is required beyond the macrophage-evading capability. For example, to achieve active targeting of tumors by nanoparticles, targeting ligands are needed, and hence, functional groups in the coating would be necessary to facilitate conjugation of these targeting ligands. The development of controlled/“living” radical polymerization techniques like ATRP and RAFT polymerization over the past decade has enabled the design of polymer coatings with well-controlled structure, composition and a multitude of incorporated functional groups. Undoubtedly, with further advances in this field, stealth polymer coatings can be designed to serve as a multifunctional platform acting in synergy with the nanoparticles to provide new opportunities for therapeutic and diagnostic applications.References
- S. Mornet, S. Vasseur, F. Grasset, P. Veverka, G. Goglio, A. Demourgues, J. Portier, E. Pollert and E. Duguet, Prog. Solid State Chem., 2006, 34, 237–47 CrossRef CAS.
- L. J. D. Thorek, A. K. Chen, J. Czupryna and A. Tsourkas, Ann. Biomed. Eng., 2006, 34, 23–38 CrossRef.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS.
- Y. F. Huang, Y. W. Lin, Z. H. Lin and H. T. Chang, J. Nanopart. Res., 2009, 11, 775–783 CrossRef CAS.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. V. Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- R. Singh and J. W. Lillard Jr., Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS.
- B. Pan, D. Cui, Y. Sheng, C. Ozkan, F. Gao, R. He, Q. Li, P. Xu and T. Huang, Cancer Res., 2007, 67, 8156–8163 CrossRef CAS.
- S. C. McBain, H. H. P. Yiu and J. Dobson, Int. J. Nanomed., 2008, 3, 169–180 Search PubMed.
- C. C. Berry, J. Phys. D: Appl. Phys., 2009, 42, 224003 CrossRef.
- F. Gazeau, M. Lévy and C. Wilhelm, Nanomedicine, 2008, 3, 831–844 CrossRef CAS.
- O. C. Farokhzad and R. Langer, Adv. Drug Delivery Rev., 2006, 58, 1456–1459 CrossRef CAS.
- V. I. Shubayev, T. R. Pisanic II and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- C. Monfardini and F. M. Veronese, Bioconjugate Chem., 1998, 9, 418–50 CrossRef CAS.
- S. M. Moghimi and J. Szebeni, Prog. Lipid Res., 2003, 42, 463–478 CrossRef CAS.
- A. S. Zahr, C. A. Davis and M. V. Pishko, Langmuir, 2006, 22, 8178–8185 CrossRef CAS.
- A. Vonarbourg, C. Passirani, P. Saulnier and J. P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- M. Q. Zhang, T. Desai and M. Ferrari, Biomaterials, 1998, 19, 953–960 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS.
- M. Ohgushi, K. Nagayama and A. Wada, J. Magn. Reson., 1978, 29, 599–601 CAS.
- M. Racuciu, D. E. Creanga and A. Airinei, Eur. Phys. J. E, 2006, 21, 117–121 CrossRef CAS.
- M. Taupitz, S. Wagner, J. Schnorr, I. Kravec, H. Pilgrimm, H. Bergmann-Fritsch and B. Hamm, Invest. Radiol., 2004, 39, 394–405 CrossRef CAS.
- M. Ma, Y. Zhang, W. Yu, H.-Y. Shen, H.-Q. Zhang and N. Gu, Colloids Surf., A, 2003, 212, 219–226 CrossRef CAS.
- Y. J. Ma and H. C. Gu, J. Mater. Sci.: Mater. Med., 2007, 18, 2145–2149 CrossRef CAS.
- S. R. Wan, Y. Zheng, Y. Liu, H. S. Yan and K. L. Liu, J. Mater. Chem., 2005, 15, 3424–3430 RSC.
- S. R. Wan, J. S. Huang, H. S. Yan and K. L. Liu, J. Mater. Chem., 2006, 16, 298–303 RSC.
- J. F. Lutz, S. Stiller, A. Hoth, L. Kaufner, U. Pison and R. Cartier, Biomacromolecules, 2006, 7, 3132–3128 CrossRef CAS.
- P. Papaphilippou, L. Loizou, N. C. Popa, A. Han, L. Vekas, A. Odysseos and T. Krasia-Christoforou, Biomacromolecules, 2009, 10, 2662–2671 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- Z. Li, L. Wei, M. Y. Gao and H. Lei, Adv. Mater., 2005, 17, 1001–1005 CrossRef CAS.
- L. M. Bronstein, S. N. Gourkova, A. Y. Sidorov, P. M. Valetsky, J. Hartmann, M. Breulmann, H. Colfen and M. Antonietti, Inorg. Chim. Acta, 1998, 280, 348–354 CrossRef CAS.
- T. Ishii, H. Otsuka, K. Kataoka and Y. Nagasaki, Langmuir, 2004, 20, 561–564 CrossRef CAS.
- H. Du, P. D. Hamilton, M. A. Reilly, A. d'Avignon, P. Biswas and N. Ravi, J. Colloid Interface Sci., 2009, 340, 202–208 CrossRef CAS.
- H. Xu, F. Yan, E. E. Monson and R. Kopelman, J. Biomed. Mater. Res., 2003, 66A, 870–879 Search PubMed.
- C. Boyer, M. R. Whittaker, V. Bulmus, J. Liu and T. P. Davis, NPG Asia Mater., 2010, 2, 23–30 Search PubMed.
- Y. Zhang, N. Kohler and M. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS.
- S. Jon, J. Seong, A. Khademhosseini, T. T. Tran, P. E. Laibinis and R. Langer, Langmuir, 2003, 19, 9989–9993 CrossRef CAS.
- H. Lee, E. Lee, D. K. Kim, N. K. Jang, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2006, 128, 7383–7389 CrossRef CAS.
- C. Sun, R. Sze and M. Zhang, J. Biomed. Mater. Res., Part A, 2006, 78A, 550–557 CrossRef CAS.
- N. Kohler, G. E. Fryxell and M. Zhang, J. Am. Chem. Soc., 2004, 126, 7206–7211 CrossRef CAS.
- Y. Zhang and J. Zhang, J. Colloid Interface Sci., 2005, 283, 352–357 CrossRef CAS.
- C. J. Xu, K. M. Xu, H. W. Gu, R. K. Zheng, H. Liu, X. X. Zhang, Z. H. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–6 CrossRef CAS.
- M. D. Shultz, J. U. Reveles, S. N. Khanna and E. E. Carpenter, J. Am. Chem. Soc., 2007, 129, 2482–2487 CrossRef CAS.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111–123 RSC.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Carroll, R. Woodward and T. St Pierre, J. Mater. Chem., 2010, 20, 255–265 RSC.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS.
- T. Niidome, Y. Akiyama, M. Yamagata, T. Kawano, T. Mori, Y. Niidome and Y. Katayama, J. Biomater. Sci., Polym. Ed., 2009, 20, 1203–1215 CrossRef CAS.
- Y. Akiyama, T. Mori, Y. Katayama and T. Niidome, J. Controlled Release, 2009, 139, 81–84 CrossRef CAS.
- M. T. Peracchia, STP Pharma Sci., 2003, 13, 155–161 Search PubMed.
- M. T. Peracchia, C. Vauthier, C. Passirani, P. Couvreur and D. Labarre, Life Sci., 1997, 61, 749–761 CrossRef CAS.
- L. Tong, W. He, Y. Zhang, W. Zheng and J. X. Cheng, Langmuir, 2009, 25, 12454–12459 CrossRef CAS.
- J. C. Y. Kah, K. Y. Wong, K. G. Neoh, J. H. Song, J. W. P. Fu, S. Mhaisalkar, M. Olivo and C. J. R. Sheppard, J. Drug Targeting, 2009, 17, 181–193 CrossRef CAS.
- M. Nagahama, S. Hayashi, S. Morimitsu and J. Sakurai, J. Biol. Chem., 2003, 278, 36934–36941 CrossRef CAS.
- D. Miyamoto, M. Oishi, K. K. Keitaro and Y. Nagasaki, Langmuir, 2008, 24, 5010–5017 CrossRef CAS.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- C. Boyer, M. R. Whittaker, K. Chuah, J. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS.
- A. Bousquet, C. Boyer, T. P. Davis and M. H. Stenzel, Polym. Chem., 2010, 1, 1186–1195 RSC.
- G. F. Schneider, V. Subr, K. Ulbrich and G. Decher, Nano Lett., 2009, 9, 636–642 CrossRef CAS.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, J. Am. Chem. Soc., 2009, 131, 4783–4787 CrossRef CAS.
- L. Feng, Y. Wang, N. Wang and Y. Ma, Polym. Bull., 2009, 63, 313–327 CrossRef CAS.
- Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS.
- B. Thierry, L. Zimmer, S. McNiven, K. Finnie, C. Barbé and H. J. Griesser, Langmuir, 2008, 24, 8143–8150 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS.
- O. Carion, B. Mahler, T. Pons and B. Dubertret, Nat. Protoc., 2007, 2, 2383–2390 Search PubMed.
- C. Flesch, Y. Unterfinger, E. Bourgeat-Lami, E. Duguet, C. Delaite and P. Dumas, Macromol. Rapid Commun., 2005, 26, 1494–1498 CrossRef CAS.
- F. X. Hu, K. G. Neoh, L. Cen and E. T. Kang, Biomacromolecules, 2006, 7, 809–816 CrossRef CAS.
- Q. L. Fan, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Biomaterials, 2007, 28, 5426–5436 CrossRef CAS.
- C. Siegers, M. Biesalski and R. Haag, Chem.–Eur. J., 2004, 10, 2831–2838 CrossRef CAS.
- P. Y. Yeh, R. K. Kainthan, Y. Zou, M. Chiao and J. N. Kizhakkedathu, Langmuir, 2008, 24, 4907–4916 CrossRef CAS.
- R. K. Kainthan, S. R. Hester, E. Levin, D. V. Devine and D. E. Brooks, Biomaterials, 2007, 28, 4581–4590 CrossRef CAS.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS.
- S. Wang, Y. Zhou, S. Yang and B. Ding, Colloids Surf., B, 2008, 67, 122–126 CrossRef CAS.
- M. Khan and W. T. S. Huck, Macromolecules, 2003, 36, 5088–5093 CrossRef CAS.
- L. Wang, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Adv. Funct. Mater., 2009, 19, 2615–2622 CrossRef CAS.
- L. Zhou, C. Gao, W. Xu, X. Wang and Y. Xu, Biomacromolecules, 2009, 10, 1865–1874 CrossRef CAS.
- M. Joubert, C. Delaite, E. Bourgeat-Lami and P. Dumas, Macromol. Rapid Commun., 2005, 26, 602–607 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |