Bioinspired conformational changes: an adaptable mechanism for bio-responsive protein delivery
William J.
King
PhD
a and
William L.
Murphy
PhD
*abc
aBiomedical Engineering Department, 1550 Engineering Drive, Madison, WI, USA. E-mail: wlmurphy@wisc.edu; Fax: +608-265-9239; Tel: +608-262-2224
bDepartment of Orthopaedics and Rehabilitation, 1111 Highland Avenue, Madison, WI, USA. E-mail: wlmurphy@wisc.edu; Tel: +608-265-9978
cDepartment of Pharmacology, 1300 University Avenue, Madison, WI, USA. E-mail: wlmurphy@wisc.edu; Tel: +608-262-3753
First published on 29th October 2010
Abstract
Hydrogels that respond to their environment have been extensively used as controlled drug delivery devices. An emerging trend is to form these “dynamic” hydrogels from polymers that undergo conformational changes. Indeed, nanometer scale polymer conformational changes have translated to macroscopic changes in hydrogel properties and controlled the release of encapsulated drugs. This review will focus on the mechanisms that control protein release from dynamic hydrogels. Specifically, we will highlight emerging mechanisms to form dynamic hydrogels, whose functional nature is derived from nature-inspired polymer conformational changes. Pertinent results from the literature will be examined to illustrate how these nanometer scale polymer conformational changes influence therapeutic protein release from dynamic hydrogels.
 William J. King | William King is a graduate student in the department of Biomedical Engineering at the University of Wisconsin, where he has been since 2005. His research interests include the development of dynamic materials and combinatorial systems for regenerative medicine applications. He has published 10 manuscripts. |
 William L. Murphy | William Murphy is an Associate Professor of Biomedical Engineering, Pharmacology, and Orthopedics/Rehabilitation at the University of Wisconsin, where he has been since 2004. He received his Ph.D. in Biomedical Engineering from the University of Michigan in 2002, and was a postdoctoral fellow in Chemistry at the University of Chicago from 2002–2004. Murphy's research interests focus on designing “bioinspired” materials that mimic and exploit biological systems. Examples of mimicking biology include biomaterials that translate nature's molecular dynamics into controllable drug delivery. Examples of exploiting biology include biomaterials that bind and regulate specific molecules and stem cells already present in the body to engineer new tissues. He has published over 50 manuscripts and filed 15 patents. |
1. Introduction
Hydrogels, which are crosslinked networks of hydrophilic polymers, have been extensively characterized for drug delivery applications. Hydrogels are particularly well suited for drug delivery, as they are formed using chemistries that do not degrade drugs, they can be processed into clinically relevant geometries, and their network properties can be varied to modulate drug release. Drug release from hydrogel networks is normally a function of the drug's diffusivity (De) in the hydrogel network, which is tuned by the hydrogel network mesh size and non-covalent interactions between the hydrogel network and the encapsulated drug. Also, in the presence of an external force, convective fluid flow out of the hydrogel network can modulate drug release (Fig. 1).1 Together, the adaptability of hydrogel networks gives them tremendous potential for controlled drug delivery applications, and they are a particularly relevant platform for delivery of protein-based drugs. | ||
| Fig. 1 Schematic representation of the network properties that control protein release from hydrogels. A) The small mesh size of highly crosslinked hydrogel networks decreases protein diffusivity (De) and slows protein release out of the hydrogel network. B) Electrostatic attraction between the hydrogel network and encapsulated proteins decreases protein De and slows protein release out of the hydrogel network. C) The larger mesh size of lightly crosslinked hydrogel networks has less of a pronounced attenuation of protein De in hydrogel networks than highly crosslinked hydrogels. D) Mechanical strain increases the hydrogel's internal fluid pressure, produces fluid exclusion, and induces protein release from hydrogel networks. | ||
Protein-based drugs have emerged as a particularly important class of therapeutics, and an important cargo in emerging drug delivery systems. There are over 20 FDA approved therapeutic proteins and more than 200 proteins are currently in development for therapeutic applications.2 Furthermore, the annual growth rate for the therapeutic protein market is four times that of the small molecule drug market, and proteins have comparable development timescales and costs to bring to market.2 Also, proteins are particularly attractive drug candidates because many of their biological roles and intermolecular interactions have been well-characterized. Therapeutic proteins have a typical half-life of tens of minutes in vivo due to their rapid proteolytic degradation and clearance from the body. Therefore, investigators have attempted to increase protein stability by encapsulating them in hydrogels which have acted as a barrier to proteolytic degradation and influenced their release rate. Temporal control over protein release will be necessary for the success of many protein therapeutics, as the timing of protein delivery is a critical regulator of in vivo physiological processes including tissue development,3 wound healing,4 and tissue regeneration.5 The timing of protein delivery is also a key regulator of stem cell differentiation in vitro, which could have significant consequences in basic research, tissue engineering, and drug screening applications.6 Dynamic hydrogels, which can change their porosity over time in a controllable manner, may be especially valuable for controlling the timing of protein delivery.
A wide range of design approaches have been used to generate dynamic hydrogels and a subset of these can be directly applied to protein delivery applications (reviewed by Qiu and Park).7 Many of these design approaches have used polymer components that undergo changes in their ordered structure, herein termed “conformational changes”, which mimic or directly harness biomolecule conformational changes. Significantly, these bio-inspired conformational changes have translated to changes in material properties, ranging from molecular transport to mechanical properties. Therefore, the fundamental principles that control natural conformational changes could give insights into the mechanisms that control dynamic hydrogels' network properties and drug release characteristics.
The purpose of this review is to highlight emerging mechanisms by which polymer conformational changes modulate protein release from dynamic hydrogels. Specifically, we will start by discussing the mechanisms that control three types of dynamic processes in polymer chains: 1) helix assembly, 2) β-sheet assembly, and 3) tertiary protein conformational changes. Then, we will discuss how the underlying fundamental chemical and physical principles that govern these conformational changes control dynamic hydrogel properties. Finally, we will examine the mechanisms that influence protein release from these dynamic hydrogels. Illustrative examples from the literature will be highlighted in each section to interpret how nanometer-scale polymer conformational changes have translated to macroscopic alterations in hydrogel properties and protein release.
This review will not cover alternative mechanisms to control protein release from hydrogels, including forming hydrogels with interpenetrating polymeric networks, encapsulating sustained protein release devices inside hydrogels, forming macropores in hydrogel networks, and forming hydrogel networks in which a fraction of the crosslinks comprise reversible receptor-ligand bonds. The reader is encouraged to read recent reviews for a more detailed discussion of these alternative mechanisms.7,8
2. Forming hydrogel networks from dynamic building blocks
Polymer conformational changes have the potential to provide nanometer scale control over hydrogel network structure. Understanding the mechanisms that control polymer conformational changes and how these dynamic polymers are incorporated into hydrogel networks will be critical for tuning hydrogel properties and protein release.2.1 Helix-crosslinked hydrogels
Investigators have used specific interpolymer interactions to form helix-based crosslinks in hydrogel networks. Helix-crosslinked hydrogels have often been based on ABA block copolymers in which the “A” blocks from separate polymers interacted to form multi-polymer helices and reversible crosslinks in hydrogel networks. In general, helix assembly has been promoted by either changing the pH to increase the interpolymer interactions, decreasing the ionic strength to lessen charge shielding between polymers, increasing the polymer concentration in the pre-polymer solution, or maintaining sub-physiologic temperature. Helix disassociation has been promoted by reversing the physiochemical stimuli that promoted helix assembly or increasing the rate of polymer degradation.Stereocomplexation and dissociation between L- and D-polymer stereoisomers has extensively been used to form helix-crosslinked hydrogels with tunable dynamic properties. Stereocomplexation between D- and L-stereoisomers of poly(lactic acid) (PDLA and PLLA) has been particularly well characterized.9 PDLA has interacted with PLLA to form parallel packed 31 left-handed helices in aqueous solutions via van der Waals interactions. Although this example of a 31 helical-crosslink is from a synthetic polymer, 31 helices have been commonly observed in natural poly(amino acid)-based polymers like globular proteins. 31 helices have been characterized by their left-handed coils that readily progress to β-sheets, right handed α-helices, or 310 helices.10 31 helix formation has been dependent on the polymer concentration and temperature of the pre-polymer solution prior to crosslinking. Increasing the polymer concentration in the pre-polymer solution increased the probability of two stereoisomer chains encountering each other and forming a 31 helix, while increasing the temperature disrupted the van der Waals interactions between stereoisomers and decreased the 31 helical content of the hydrogels.11–13 In contrast, solutions that contained a single PDLA or PLLA stereoisomer did not form 31 helices.14
Vert and colleagues have formed helix-crosslinked hydrogels from pre-polymer solutions that contained mixtures of PDLA-PEG-PDLA with PLLA-PEG-PLLA15–17 and multiblock copolymers of PEG-PLLA and PEG-PDLA.11 Other investigators have formed 31 helix-crosslinked hydrogels by combining eight-arm block copolymers of PEG8-PDLA with PEG8-PLLA18 and linear block copolymers of PLA stereoisomers with eight-arm stereoisomer block copolymers.19 In these studies, the PEG and PLA components had to be of comparable molecular weights in order to form hydrogel networks. The mesh size (ξ) of these 31 helix-crosslinked hydrogels was tuned by varying the molecular weight of the PEG component of the block copolymer, the solubility of the block copolymers, and the degradation of the PLA segments.18 Together, these components have given investigators significant control over the resulting hydrogel network structure. Other block copolymers, including PLA-Dextran-PLA20 and PEG-poly(hydroxybutyrate)-PEG, have also formed dynamic hydrogels.21 Recently, Slager et al. reported that L-stereoisomer peptides may have formed complexes with PDLA13 but this finding has been controversial.22 31 helix-crosslinked hydrogels may find broad applicability in the biomedical applications, as their constituent monomers are produced on industrial scales. However, precise control over their hydrogel network properties may be difficult to achieve due to their polydispersity when synthesized using standard approaches.
Peptide-based polymers have been extensively used to form helix-crosslinked hydrogels with tightly controlled network properties. There are many well characterized examples of natural helix forming peptides, which have been used to crosslink dynamic hydrogels including collagen based peptides26 and leucine zipper peptide domains.27,28 Investigators have formed superhelical coiled-coil crosslinks in hydrogels with precise control over the polymer chain sequence, length, and the number of polymer chains that emanate from a crosslink.29 Together, these variables have given investigators precise control over hydrogel network properties. Peptide based coiled-coils have formed from 2 to 5 parallel helices that cross at a 20° angle. Their formation has been driven by cooperative non-covalent interactions, including van der Waals forces, hydrophobic interactions, and charge-charge interactions.30 Peptide-based left-handed coiled-coil helices have typically contained a heptapeptide repeating sequence in which positions “a” and “d” were hydrophobic residues, and every other position contained a hydrophilic residue.31 Right-handed coiled-coil helices have typically contained an eleven amino acid repeating sequence.32 Combining left-handed and right-handed coiled-coil forming peptides into block copolymers could produce hydrogels with high levels of control over their network structure and stability due to their selective interpolymer interactions.
The assembly and stability of coiled-coils has been modulated by the pH, ionic strength, and the temperature of their environment.33 One of the most extensively characterized coiled-coil forming peptides has been the leucine zipper domain.34 Tirrell and colleagues have formed leucine zipper-based hydrogels whose macroscopic properties have been tuned using the fundamental principles that govern coiled-coil formation (Fig. 2A).23,35–38 Varying the amino acid content and sequence in leucine zipper domains has given investigators control over helix self assembly,39 the number of interacting peptides,40 helix stability,41 and the overall complex conformation.42 These studies highlight the powerful adaptability of peptides, whose composition can be readily varied using standard peptide synthesis techniques.
 | ||
| Fig. 2 Schematic representations of conformation changing polymers and their assembly into hydrogels. A) Schematic representation of coiled-coil peptides that self-assemble to form crosslinks in dynamic hydrogels.23 B) Schematic representation a peptide that self-assembles to form β-sheets, fibrils, and ultimately branched hydrogel networks.24 C) Schematic representation of mutant CaM(T34C, T110C) undergoing a Michael-type addition reaction with PEGDA to form PEG-CaM-PEG conjugates that can be photocrosslinked to form hydrogel networks.25 | ||
In a separate approach, Kiick and colleagues have formed hydrogels based on the coiled-coil conformational change of the heparin-binding peptide PF4ZIP.43–46 Heparin has been characterized as a glycosaminoglycan that is found on the cell surface and in the extracellular matrix.47 The viscoelastic properties of PF4ZIP hydrogels have been tuned by varying the ratio of PF4ZIP to heparin, the temperature of the hydrogels' environment, and the peptides' affinity for heparin.44 These studies provided an important example that coiled-coil peptides with varying affinities for their target polymer could be used to tune hydrogel network properties. Also, many glycosaminoglycan-peptide interactions have been well-characterized and could be used to form dynamic hydrogels. Therefore, this approach might be broadly adaptable.48
Taken together, these initial examples clearly demonstrate that peptides can form coiled-coils, and that these interactions can be used for hydrogel assembly. The readers are directed to a review by Mart et al. for a more comprehensive list of 14 peptides that have been used to form helices or helix-derived coiled-coils in biomaterials applications.49 Only a fraction of these peptides have been used to form crosslinks in dynamic hydrogels, which suggests an opportunity to form several new dynamic hydrogels via coiled-coil crosslinks.
2.2 β-sheet- and β-hairpin-derived hydrogels
β-sheet and β-hairpin peptide conformational changes have also been used to assemble dynamic hydrogels. Peptides have formed β-sheets and β-hairpins via cooperative non-covalent interactions including electrostatic interactions, hydrogen bonding, and hydrophobic interactions.36,49 β-sheet forming peptides have typically contained alternating cationic, hydrophobic, and anionic amino acid residues, and the resulting sheets have interacted with each other to form curved peptide surfaces or fibrils. Some fibrils have formed with defects, which produced branching and, in turn, hydrogel network formation (Fig. 2B). β-sheet and β-hairpin hydrogels have possessed unique properties that have been a product of from their cooperative intermolecular interactions. In particular, β-sheet derived hydrogels have remained stable even after exposure to high salt concentrations, high temperatures, and denaturing agents.50 This hydrogel stability has been attractive for applications in wound environments, which often contain high concentrations of proteases.51 Also, these hydrogels have not typically engendered a strong immune response when they have been implanted in vivo.52 This finding has been critical for many biomedical applications, as the function of implanted hydrogels has been restricted if they were encapsulated by the body's foreign body response.53Among the first peptides that formed β-sheet derived hydrogels was the EAK16 peptide (AEAEAKAKAEAEAKAK), which formed β-sheets in the presence of monovalent cations.54 Zhang et al. assembled β-sheet derived hydrogels by systematically mutating the EAK16 peptide to form the RAD16 peptide (RADARADARADARADA), which formed unusually stable hydrogels that have been resistant to proteolysis, heat, and denaturing chemicals due to cooperative non-covalent interactions.54,55 Therefore, the RAD16 peptide has been used in a wide variety of biomedical applications including use as a drug delivery device and extracellular matrices for cell encapsulation.24 Also, the material properties of RAD16 peptide hydrogels have served as benchmarks for new β-sheet derived hydrogels formed for specific applications.
The rate of β-sheet assembly has been tuned by the precursor peptides' physiochemical environment. For example, the rate of β-sheet assembly from RAD16 peptides dramatically increased when the pH was adjusted to neutral, or upon the addition of physiologically relevant salt concentrations.24,54,55 Significantly, the RAD16 peptide formed β-sheet derived hydrogels in less than 30 min when injected into an in vitro skin wound model.56 This result suggested that β-sheet derived hydrogel assembly could occur over time scales that may allow its use in medical applications. In a separate approach, Yu and colleagues used oppositely charged decapeptides that rapidly self assembled to form β-sheet derived hydrogels over the course of minutes upon physical mixing.57–59 These hydrogels could be useful in applications where accelerated hydrogel formation is necessary for clinical success, such as in induction of hemostasis.
A similar approach to forming β-sheet derived hydrogels has used small, oppositely charged peptides with an N-(fluorenylmethoxycarbonyl) (fmoc) group attached to peptides' N-termini to facilitate interpeptide π-stacking.60 These hydrogels formed over the course of 12 h, which was much slower the than oppositely charged β-sheet forming peptides characterized by Yu and colleagues. The decreased hydrogel assembly rate could have been the product of the fmoc moiety sterically hindering electrostatic interactions between the charged peptides. These results suggested that the rate of β-sheet derived hydrogel formation could be tuned by covalently modifying the β-sheet forming peptides with aromatic molecules that undergo π-stacking. Many important biological molecules include aromatic moieties, therefore this approach may provide a new way to incorporate biological molecules into β-sheet derived hydrogels.61
Alternatively, Gazit and colleagues and Ulijn and colleagues have formed β-sheet derived hydrogels from fmoc-FF dipeptides,62,63 which formed 3 nm β-sheet fibrils via π-stacking of the fluorenyl groups and phenyl rings.64 These short peptides self-assembled to form rigid hydrogels that were resistant to changes in heat, pH, or chemical induced degradation. The crosslinking density of the hydrogels was increased by increasing the concentration of fmoc-FF peptides in the pre-polymer solution. Significantly, these fmoc-FF derived hydrogels were 200 times more rigid than RAD16 derived hydrogels, which could result in a better match to the mechanical environment of stiffer tissues and thereby minimize tissue or hydrogel damage.65 Ulijn and colleagues also developed a technique in which hydrogel crosslinking was controlled by enzyme-mediated amide bond formation between a dipeptide and a fmoc-conjugated amino acid.63 This process could provide an additional level of control over the rate of hydrogel assembly.
Separately, Xu and colleagues have formed β-sheet crosslinked hydrogels based on the reversible assembly of naphthalene-FFGEY.66 They chose to use the naphthalene moiety because several drugs have been used in clinical trials that contain a naphthalene moiety including propanolol,67 naphazoline,68 and nafronyl.69 These peptides formed hydrogels when the pre-polymer solution pH was adjusted from 7.8 to 7.5. Then, the formation and disassociation of naphthalene-FFGEY hydrogels was enzymatically controlled by adjusting the solubility of the peptides. Specifically, the tyrosine amino acid of the naphthalene-FFGEY peptide was phosphorylated by a tyrosine kinase in the presence of adenosine triphosphate (ATP), which increased the peptides' solubility, dissociated the β-sheet crosslinks in the hydrogel network, and dissolved the hydrogels. To induce the hydrogels to form again, the phosphorylated peptides were exposed to alkali phosphatase which removed the phosphate groups from the naphthalene-FFGEY peptide, induced β-sheet formation, and produced a hydrogel network.66 Xu and colleagues have also formed enzyme sensitive hydrogels that responded to light,70 integrated the anti-cancer drug taxol as a functional unit,71 integrated protease resistant D-amino acids,72 and facilitated enzyme immobilization.73 Various types of kinase and phosphatase enzymes have been extensively characterized,74 and therefore this approach may provide a broadly adaptable approach to controlling β–sheet peptide hydrogel formation and disassociation.
In a distinct approach, Schneider and colleagues assembled hydrogels from peptides that formed β-hairpins in the presence of physiologically relevant concentrations of NaCl.75–79 The parent peptide of this family, MAX1, was a 20 amino acid long peptide which contained eight lysine residues. The addition of physiologically relevant NaCl concentrations shielded the lysine residues' charge, which induced the peptides' conformational change into a β-hairpin. This hairpin was composed of two β-strands that spontaneously self-assembled to form β-sheets and β-sheet derived hydrogels. This mechanism to assemble hydrogels may allow for them to be injected through a syringe and form in situ for a wide variety of biomedical applications.
The kinetics of β-hairpin derived hydrogel assembly have been key regulators of the mesh size (ξ) of the hydrogel networks. In general, increasing the rate of β-hairpin assembly in the pre-polymer solution has induced greater crosslinking rates and smaller hydrogel ξ.76,78 The rate of β-hairpin assembly has been tuned by varying the components in the pre-polymer solution, including the pH, ionic strength, temperature, peptide concentration, amino acid composition, and even the order of amino acids.80 Peptides with lower overall charge densities underwent faster conformational changes than peptides with higher charge densities. This observation was attributed to the decreased number of charges that had to be screened to permit inter-peptide interactions. Also, alterations to the MAX peptides that minimized the positive charge density in the beta-sheet region of the hairpin had the greatest effect on the pH responsiveness of these hydrogels.78 The results of these studies suggest that the network properties of β-hairpin derived hydrogels can be finely tuned with an understanding of the fundamental principles that govern β-hairpin assembly.
In an alternative approach, Stupp and colleagues have formed β-sheet derived “peptide amphiphiles” based on peptide-palmitic acid conjugates. Peptide amphiphiles formed β-sheet crosslinked hydrogels via salt bridges in neutral pH solutions, and therefore constituted an attractive mechanism to incorporate hydrophilic peptides into stable, β-sheet derived hydrogel networks.81,82 Collectively, the diversity of peptides that form β-sheet derived hydrogels allows investigators to choose from a wide variety of physiochemical stimuli to control the formation of stable hydrogels.
2.3 Dynamic, protein based hydrogels
Proteins that undergo tertiary conformational changes have recently been incorporated as integral components in hydrogel networks. During folding, proteins rearrange their hierarchical structures to assume minimum free energy states. Ligand binding alters a protein's energy landscape which can induce a protein to fold into a new tertiary structure with rearranged helices, β-sheets, β-hairpins, and/or unstructured regions.83 A wide variety of protein families undergo conformational changes, including motor proteins,84 riboswitches,85 enzymes,85 cytokine receptors,85 intrinsically disordered proteins,84 and calcium sensing proteins.86 Indeed, there are hundreds of proteins that undergo well characterized protein conformational changes.87,88Protein conformational changes have been classified into distinct protein motions with unique characteristics. Conformational changes referred to as “hinge motions” are a common class of protein motions, and they can occur at the terminal regions of α-helices or β-sheets that are connected with a metastable linker region.89 Another subset of proteins undergo “shear motions” of up to 100 degrees in rotation at the interface between pairs of perpendicular helices in proteins with layered domains.90 From these examples and others, it is clear that the organization of secondary structures has a critical role in controlling proteins' potential tertiary motions.
A particularly advantageous property of dynamic hydrogels whose dynamic properties are derived from protein conformational changes is that they respond to specific biochemical ligands. Traditional dynamic hydrogels have responded to physiochemical changes in their environment, such as temperature and pH changes. These physiochemical changes are not ideal inputs for many potential biological or biomedical applications, as these changes have affected cellular gene expression, function, and survival.91 Also, the in vivo environment is tightly regulated which has made it difficult to vary these physiochemical properties.92 Therefore, protein conformational changes may allow for more specific “bio-responsive” dynamic properties in a broader range of physiological environments.
Currently, proteins in dynamic hydrogels have not possessed the ability to self-assemble into hydrogel networks, and therefore have been incorporated as functional units in hydrogel networks using polymer chemistry techniques. For example, we have used two distinct approaches to form hydrogels based on the dynamic protein calmodulin (CaM), which undergoes a pronounced hinge motion upon binding of specific biochemical ligands.93 In the first approach, the sulfhydryl groups of a mutant version of CaM(T34C, T110C) were conjugated with 4-arm PEG4-acrylate via a Michael-type addition reaction. Then, solutions containing polymer-CaM-polymer conjugates were crosslinked with dithiothreitol (DTT) via a second Michael-type addition reaction. This approach facilitated efficient formation of polymer-CaM-polymer conjugates, and tunable incorporation of CaM into hydrogels.94 In a separate approach, we reacted the mutant CaM(T34C, T110C) with PEG-diacrylate (PEGDA) to form PEG-CaM-PEG conjugates with terminal acrylate groups. PEG-CaM-PEG conjugates were photocrosslinked to form hydrogels with greater than 90% CaM incorporation efficiencies (Fig. 2C). Significantly, this approach to forming dynamic hydrogels enabled the use of photolithography techniques to form hybrid hydrogels with distinct polymer regions25 and injectable microspheres for drug delivery applications.95 Also, the concentration of PEG-CaM-PEG in the pre-polymer solution and the molecular weight of the PEG chains attached to CaM were tuned to control PEG-CaM-PEG hydrogels' Qm.96,97 These results suggest that photocrosslinking may enable the formation of multifunctional materials with tunable dynamic properties. Recent studies have demonstrated the utility of this class of materials in biosensing,97 bio-actuation,8 and triggered protein delivery.95,96 Therefore, this general approach can be applied to a broad range of hinge motion proteins for significant biomedical applications.
In a separate example, Kopeček and colleagues have incorporated the hinge motion protein adenylate kinase (AKe) into hydrogels using thiol-maleimide chemistry. The sulfhydryl groups of mutant AKe(C77S, A55C, V169C) were reacted with pendant maleimide groups incorporated into N-(2-hydroxypropyl)methacrylamide (HPMA). Significantly, the Qm of AKe-based hydrogels was tuned by varying the concentration of AKe in the pre-polymer solution and the chemistry used to form the hydrogel networks.98 This result suggests that proteins in the presence of a radical initiator to form hydrogels.99 There are many proteins with surface exposed cysteines that could be incorporated into hydrogel networks using these approaches.100 Together, these studies highlight the potential of using sulfhydryl groups to form protein-based hydrogels.
Francis and colleagues have formed dynamic hydrogels from a hinge motion protein that binds heavy metal ions. Specifically, they used recombinant DNA techniques to form mutant metallothionein with a C-terminal intein segment and a chitin binding domain. After purifying mutant metallothionein, one ketone group was conjugated to a functionalized cysteine using a native chemical ligation,101 and another ketone was conjugated to the N-terminus of the mutant protein by reacting the protein with pyridoxal 5′-phosphate. The dual-ketone labeled metallothionein was then polymerized with alkoxyamino-functionalized HPMA to form hydrogels.102 This approach could be broadly adaptable, as the native chemical ligation and N-terminal ketone conjugation techniques are not specific to any protein's native residues. Significantly, this approach was insensitive to native cysteines and therefore could be used to incorporate proteins into hydrogels that cannot tolerate cysteine mutations or covalent modification.
3. From polymer conformational changes to hydrogel network dynamics
It is clear that conformation changing polymers can be incorporated into hydrogels with unique network properties. We will now focus on how polymer conformational changes affect hydrogel network dynamics, with a focus on the network properties that may be particularly relevant to drug delivery applications.3.1 Dynamics of helix-derived hydrogel assembly and disassociation
Using fundamental principles as a guide, investigators have tuned the rate of helix assembly and disassociation to control dynamic hydrogel properties. The degradation of 31 helix-crosslinked PLA-PEO-PLA hydrogels has been particularly well characterized. Vert and colleagues demonstrated that helix-derived crosslinks in PLA-PEO-PLA hydrogels underwent rapid unfolding when exposed to physiological salt concentrations and temperatures. In buffered solutions these hydrogels lost more than 50% of their swollen mass within 10 days, then slowly degraded for another 20 days. The decreased degradation rate after 10 days could have been attributed to the increased PLA/PEO ratio, due to PEO release from the hydrogel networks upon partial degradation of PLA crosslinks. The increased PLA/PEO ratio increased the hydrophobicity of the overall hydrogel network and slowed its hydrolytic degradation.15 Also, proteinase K, an enzyme which catalyzes PLA degradation, accelerated the degradation of 31 helices. After 80 h in solution, PLA-PEO-PLA hydrogels exposed to proteinase K lost 56.9% of their weight, whereas hydrogels undergoing only hydrolytic degradation lost 5% of their weight.103 Therefore, proteinase K could be used to trigger rapid degradation of an otherwise slowly degrading hydrogel network.The disassociation rate of 31 helices in PLA-PEO-PLA hydrogels has also influenced by the symmetry of the PLA blocks. Hydrogels formed from PLA-PEO-PLA polymers with asymmetric terminal PLA blocks degraded faster than hydrogels formed from symmetric polymers, because the shorter PLA blocks did not participate in crosslinking as efficiently as longer PLA blocks.104 A common technique to impart stability on 31 helix-crosslinked systems has been to derivatize the polymer chains with reactive groups that participate in photocrosslinking.105 These crosslinks have not been reversible, which could limit their dynamic properties. However, without covalent crosslinks these hydrogels have eroded rather than undergoing reversible volume changes. Therefore, there may be a tradeoff between the stability of 31 helix-crosslinked hydrogels and their ability to undergo reversible volume changes.
Erosion has been an important mediator of dynamic properties in coiled-coil crosslinked hydrogels.31 These hydrogels have typically rapidly eroded in physiological conditions, via diffusion of polymer chains away from the hydrogel matrix after coiled-coil disassociation. For example, unmodified AC10A hydrogels, in which the “A” domain was derived from an acidic zipper domain and the “C” domain was an unstructured linker region, eroded completely in 2.9 h in 100 mM, pH 7.6 phosphate buffer due to the reversibility of coiled-coil helices and their propensity to form looped structures rather than bridges. Tirrell and colleagues quantified the properties that controlled the erosion of helix-crosslinked hydrogels by measuring the stress relaxation time and the leucine zipper strand exchange time in solution.35 In AC10A hydrogels, the strand exchange time of the leucine zipper domains was 3–4 times greater than the stress relaxation time of the hydrogel network (Fig. 3A Left). The increase in strand exchange time was a product of the leucine zipper peptides' propensity to form “looped” configurations rather than “bridges” that crosslinked the hydrogel network.37 The looped structures played an integral role in hydrogel erosion, as they diffused away from the hydrogel network without reintegrated into the hydrogel network as a bridge.
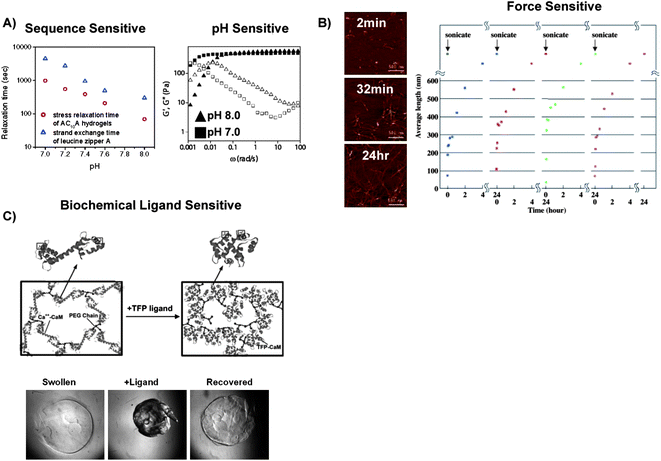 | ||
| Fig. 3 Results of studies that characterized how polymer conformational changes affect hydrogel network dynamics. A) The stress relaxation time of AC10A coiled-coil-crosslinked hydrogels was 3–4 times greater than the strand exchange time of leucine zipper A coiled-coils in solution, which indicated that the AC10A peptide formed extensive “loops” rather than “bridges.” This result was attributed to their peptide sequence which permitted intramolecular interactions (Left). Dynamic moduli (G′ open symbols, G′′ closed symbols) of AC10A peptide coiled-coil crosslinked hydrogels at pH 7.0 (■) and pH 8.0(▲) demonstrated the pH sensitivity of coiled-coil-crosslinks (Right). B) After exposure to sonic forces, RAD16 peptide fibrils elongated and formed networks over time (Left), and repeatedly self-assembled to form hydrogel networks after cycles of exposure to ultrasonic forces (Right).106 C) Schematic representation of CaM's ligand-induced conformational change in a hydrogel network (Top)25 and photomicrographs of a CaM-based hydrogel that underwent a reversible volume change in response to a specific biochemical ligand (Bottom).96 | ||
To slow the hydrogels' erosion rate, Tirrell and colleagues formed block copolymers that contained the previously employed “A” and “C” domains from the AC10A peptide, with a “P” domain derived from rat cartilage oligomeric matrix protein. The “A” and “P” domains did not interact to form coiled-coils. Interestingly, although PC10A monomers underwent more rapid domain swapping and helix assembly than AC10A monomers, the resulting hydrogels degraded 500 times slower in 100mM phosphate buffer, 200 times slower in Dulbecco's phosphate buffered saline (DPBS), and maintained 85% of their initial mass after 96 h in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum.38 These results suggest that the stability of future coiled-coil derived hydrogels can be significantly increased by forming them with ABC block copolymers in which the A and C domains do not interact to form coiled-coils. Also, these studies highlight the importance of using different, physiologically-relevant environments to test the stability of dynamic hydrogels, which will be crucial for their utility in many biomedical applications.
Physiochemical stimuli like heat, pH, and ionic strength have been used to tune coiled-coil assembly and dissociation in hydrogel networks (Fig. 3A Right).23,37 For example, the leucine zipper exchange rate of the AC10A peptide increased more than 10-fold when the pH was decreased from 8.0 to 7.0. The increased exchange rate was a product of electrostatic repulsion between glutamic acid residues in the AC10A peptide. However, pH control over the exchange rate was masked by increasing the ionic strength of the hydrogels' environment to physiologically relevant salt concentrations.35 Importantly, these observations suggest that investigators can tune the kinetics of hydrogel assembly and disassociation by controlling the amino acid composition in helical peptide domains.
However, coiled-coil peptide interactions have not always been reversible, which has affected the reversibility of macroscopic hydrogel volume changes. For example, Kopeček and colleagues formed helix-crosslinked hydrogels based on a leucine zipper forming peptide from the stalk region of kinesin. The coiled-coil crosslinks unfolded at temperatures above 35 °C and did not reform to make a hydrogel network.75 Recently, these crosslinks were made reversible by disassociating the coiled-coil with a chemical denaturing agent, guanidine hydrochloride (GdnHCL), then their reassembly was induced by removing GdnHCL.107 However, this approach is not ideal for biological environments, in which GdnHCL could denature other proteins as well.
3.2 Dynamics of β-sheet and β-hairpin derived hydrogel assembly and disassociation
The rate of β-sheet derived hydrogels' assembly and disassociation has been controlled by their physiochemical environment. The kinetics of RAD16 β-sheet derived hydrogel formation and disassociation has been particularly well characterized. RAD16 peptide hydrogels have undergone repeated cycles of disassociation when exposed to ultrasonic forces, and then they spontaneously reassembled in the absence of these forces. Specifically, after exposure to ultrasonic forces, short β-sheet fibers elongated from 20nm to several μms in length, and the β-sheet assembly rate was controlled by the concentration of short peptide fragments produced. The short peptides diffused in solution and laterally along peptide fibrils to form new crosslinks over the course of hours. This crosslinking phenomena was not observed from larger peptide fragments (Fig. 3B).106 The size distribution of the peptide fragments was tuned by the magnitude and duration of the ultrasonic forces. Therefore, ultrasonic forces may serve as a broadly applicable mechanism to rapidly dissociate β-sheet derived hydrogels,108 and could be used in combination with image-guided ultrasound therapies.109In a separate approach, Yu and colleagues formed β-sheet derived hydrogels that dissociated when exposed to shear forces and recovered when the force was removed. These hydrogels were formed from WK(VK)4 and EW(EV)4 peptides, which were mutually attracted to each other but underwent self-repulsion. Assembly of β-sheet derived crosslinks in these hydrogels was monitored by measuring the hydrogel shear modulus. In less than one minute, hydrogels exposed to shear strains yielded almost 50% of their mechanical strength, and then slowly lost the rest of their mechanical strength over the course of tens of minutes. Once the shear strain was removed almost 50% of the hydrogel strength was recovered in several seconds, and the rest of the strength was recovered over the course of tens of minutes. Strikingly, this process was repeatable for over 12 cycles.57 The fast recovery time of these hydrogels was attributed to rapid diffusion and self-repulsion of the small decapeptides in solution. Significantly, the sheer force sensitivity of these β-sheet derived hydrogels could allow them to undergo a gel-sol transition when passed through a syringe needle and then rapidly recover to form a hydrogel in situ.
Researchers have harnessed specific enzymatic reactions to gain greater control over β-sheet derived hydrogel dynamics. For example, Xu and colleagues have used specific phosphatase and kinase enzymes to control the rate of naphthalene-FFGEY hydrogel assembly and dissociation. Initially, when naphthalene-FFGEY hydrogels were exposed to 3 U of a tyrosine kinase, the enzyme phosphorylated the peptide and dissolved the hydrogels over the course of 24 h. After 24 h, 46% of the peptides were phosphorylated which suggested that a high degree of phosphorylation was not necessary to dissociate the β-sheet crosslinked hydrogel network. Then, 200 U of alkali phosphatase induced peptide dephosphorylation, β-sheet formation, and hydrogel crosslinking in 1 h. During this time 99.1% of the naphthalene-FFGEY peptide was dephosphorylated. The faster recovery time was attributed to the greater activity of alkali phosphatase compared to tyrosine phosphatase.66 These results suggest that balancing the activity of different signal transduction proteins could be used to control the kinetics of β-sheet derived hydrogel formation and dissociation.
3.3 Dynamics of ligand-induced protein conformational changes in hydrogel networks
Dynamic, protein based hydrogels have been designed to respond to specific biochemical ligands. For example, we have developed hydrogels whose dynamic properties have been derived from the pronounced hinge motion of the protein CaM. When CaM was included as an integral component of a hydrogel network, the distance between crosslinks decreased as the protein changed from its “extended dumbbell” conformation to its “collapsed” conformation. Specifically, when mutant CaM(T34C T110C) was in its “extended dumbbell” conformation the two cysteine residues in the protein were 50Å apart, and this Cys-Cys distance decreased to 15Å when CaM bound to its biochemical ligand. Significantly, CaM's nanometer-scale conformational change translated to macroscopic changes in hydrogel volume, optical properties, and transport properties (Fig. 3C).94Previous studies have suggested a mechanism in which CaM's nanometer-scale conformational change scaled to a macroscopic shift in hydrogel properties. Hydrogels formed using a different mutant CaM (CaM(Q41C, K75C)) in which there was little change in the distance between the cysteines in the extended(20Å) and collapsed (18Å) conformation did not undergo ligand-induced volume changes.94 Also, the stoichiometry of ligand-protein binding in these hydrogels (5.494 or 3.125 trifluoperazine (TFP) molecules per CaM molecule) was consistent with the range of CaM to TFP binding ratios observed in solution studies.93,110,111 Additionally, CaM-based hydrogels underwent ligand-induced volume changes when exposed to a CaM-specific peptide ligand, m13 peptide, but did not undergo volume changes when exposed to a scrambled version of the peptide ligand. Furthermore, these hydrogels did not undergo volume changes in response to blood serum or to arginine, which had the same charge as TFP but did not bind CaM.95 Finally, chlorpromazine (CPZ), which had a higher affinity for CaM than TFP, induced larger magnitude hydrogel volume changes than TFP.112 Collectively, these results suggest that CaM retains its ability to bind specific ligands when incorporated in hydrogel networks and CaM's nanometer scale conformational change translated to macroscopic hydrogel volume changes.
The magnitude of CaM-based hydrogels' ligand-induced volume changes has been tuned by varying their initial compositions. Specifically, the magnitude of the ligand-induced volume change has been tuned by varying the concentration of CaM incorporated into the hydrogels, the molecular weight of the PEG chains conjugated to CaM, and the ratio of PEG-CaM-PEG/PEGDA in the hydrogels.25,96,97 In general, hydrogels which had greater Qm underwent larger magnitude ligand-induced volume changes compared to hydrogels with smaller Qm. For example, PEG-CaM-PEG hydrogels with a Qm of 35 underwent 80% ligand-induced volume changes. Alternatively, PEG-CaM-PEG hydrogels with a Qm of 20 and underwent 60% ligand-induced volume changes. There are many different approaches to vary the Qm of hydrogels. Therefore, this approach could be broadly adoptable to create dynamic, protein-based hydrogels with tunable dynamic properties.
The rate of a CaM-based hydrogels' ligand-induced volume changes has been varied by controlling the hydrogels' swelling ratio and size. For example, when crosslinked via a Michael-type addition reaction, CaM-based hydrogels had a Qm of 19, and underwent volume changes over the course of 30 min.94 Alternatively, photocrosslinked PEG-CaM-PEG hydrogels with a Qm of 10 underwent ligand-induced volume decreases over the course of 2 h.25 The smaller Qm of photocrosslinked hydrogels, and thus lower mesh size, could have decreased TFP De and slowed the rate of hydrogel volume changes. TFP diffusion has also been tuned by the hydrogel size. For example, PEG-CaM-PEG microspheres with an average diameter of 38μm underwent their maximum ligand-induced volume decreases in 2 min, whereas volume changes of 5mm diameter macroscopic hydrogel cylinders occurred over the course of tens of minutes to hours. These results were attributed to the decreased time it took for TFP to diffuse throughout the smaller microspheres when compared to large hydrogel cylinders. Together, these results suggest that ligand diffusion through protein-based hydrogels is a key regulator of their dynamic properties.
The rate of volume recovery in dynamic, protein based hydrogels has been controlled by both ligand diffusion and protein-polymer interactions. For example, when collapsed PEG-CaM-PEG microspheres were moved to an environment devoid of the TFP ligand, volume recovery proceeded slowly over the course of one hour. The differences between the rate of hydrogel swelling (one hour) and collapse (2 min) could be attributed to the rates of ligand binding and release from CaM. Junkers et al. measured CaM's ligand-binding rate to be ∼12.2 s−1 with a lifetime of 102 to 103 s.113 Therefore, collapsed CaM hydrogels may have released their sequestered TFP and underwent swelling over a longer timeframe than it took for TFP to diffuse into hydrogels and induce volume collapse. Also, the slow swelling rate could have been influenced by local protein-polymer interactions in the collapsed hydrogel network, which could have slowed the collective polymer diffusion rate during swelling. This phenomenon has been extensively characterized in a different class of dynamic hydrogels: poly(N-isopropyl acrylamide) (poly(NIPAAM)) hydrogels.114 Several studies have increased the swelling rate of poly(NIPAAM) hydrogels after their temperature-induced collapse by grafting hydrophilic polymer chains to the hydrogel network or including structural inhomogeneities, and these approaches could similarly be adopted in dynamic, protein-based hydrogels.7 It will be interesting to observe whether mechanisms that control the kinetics of CaM-based hydrogel volume changes extend to other dynamic, protein based hydrogels.
Other protein conformational changes have more recently provided additional insights into the mechanisms that control protein-based hydrogel dynamics. For example, Kopeček and colleagues have formed dynamic hydrogels that harnessed AKe's hinge-like conformational change. During AKe's conformational change the separation of the mutant cysteine residues in the protein decreased from 29.5Å to 12.4Å apart. AKe's conformational change translated to an 18% decrease in macroscopic hydrogel volume in the presence of its ligand, ATP. The magnitude of these ATP-induced volume changes was tuned by varying the ATP concentration of in the hydrogel environment.98 ATP is an attractive ligand because it is naturally occurring in the body, but these hydrogels may require greater magnitude ligand-induced volume changes to be applicable in some biomedical applications. AKe hydrogel disks underwent their maximum ATP-induced volume changes over the course of 5 min when the hydrogels were placed in a 4mM ATP solution. During subsequent cycles these hydrogels underwent slower and smaller magnitude ligand-induced volume changes. Similar irreversible volume changes occurred in a limited subset of the aforementioned PEG-CaM-PEG hydrogels.97 These results could be attributed to the proteins becoming insoluble or forming irreversible intermolecular interactions with polymer chains in the hydrogel network. This potential protein precipitation could potentially be addressed in future studies using established techniques to refold precipitated proteins.115
Francis and colleagues have formed dynamic, protein-based hydrogels whose dynamic properties were derived from metallothionein's heavy metal ion-induced hinge-like conformational change. Metallothionein-based hydrogels underwent greater than 80% volume changes in the presence of heavy metal ions like Cd2+, whereas lower molecular weight ions like Ca2+, Mg2+, and Mn2+ did not induce hydrogel volume changes. The magnitude of ligand induced volume change was tuned by the metallothionein concentration in the pre-polymer solution, the ion concentration in solution surrounding the hydrogels, and the metallothionein-ion affinity. These results highlight the high selectivity of protein-based hydrogels' ligand-induced volume changes, which will be crucial for dynamic hydrogels to function in complex environments.102
Alternatively, Daunert and colleagues have formed dynamic, protein-based hydrogels whose dynamic properties were derived from GBP's insulin-induced conformational change. GBP underwent a hinge motion conformation change when it bound to glucose. GBP-based hydrogels underwent reversible 40% volume changes in the presence of glucose and the magnitude of their volume changes was controlled by the glucose concentration in their environment.99 Taken together, studies to date suggest that protein conformational changes will serve as a broadly adaptable mechanism to form dynamic hydrogels that respond to specific biochemical ligands (see summary, Table 1).
| Dynamic Protein | Ligand(s) | Volume Change |
|---|---|---|
| Calmodulin 25,94–97 | Phenothiazine Drugs, Peptide Ligands | 0–85% |
| Adenylate Kinase 98 | ATP | |
| Metallothionein 102 | Heavy Metal Ions | 0–80% |
| Glucose/Galactose Binding Protein99 | Glucose | 0–40% |
4. Influence of conformational change dynamics on protein release
Nanometer scale conformational changes have been extensively used to form hydrogels and tune their dynamic properties. Recently, investigators have begun to harness dynamic hydrogels' unique properties for several different applications, including biosensing116 and bioactuation.117 Here we focus on the use of these materials to modulate the release of therapeutic proteins in biomedical applications. In this section we primarily emphasize studies that have delivered proteins using the dynamic materials introduced in sections 2 and 3 above.4.1 Effect of helix assembly and dissociation dynamics on protein release
Protein release from helix-crosslinked hydrogels has been tuned by controlling the encapsulated proteins' De and hydrogels' erosion rates. In general, small proteins, with molecular weights less than 100kDa, have rapidly diffused out of helix-crosslinked hydrogels.18,118 For example, Vert and colleagues measured that 35–45% of an encapsulated model protein, bovine serum albumin (BSA), was released from 31 helix-crosslinked hydrogels over the course of 10 h. This initial “burst release” of BSA was consistent with protein release profiles observed in other biomaterial systems, and has been attributed to the rapid release of surface-bound proteins.119 After the burst release phase, BSA diffused out of 31 helix-crosslinked hydrogel networks over the course of 170 h. Significantly, increasing the concentration of BSA encapsulated in these hydrogels increased the BSA release rate.18 This observation suggests that proteins smaller than BSA, will release from 31 helix-crosslinked hydrogels primarily via a diffusion mediated mechanism (Fig. 4A Left).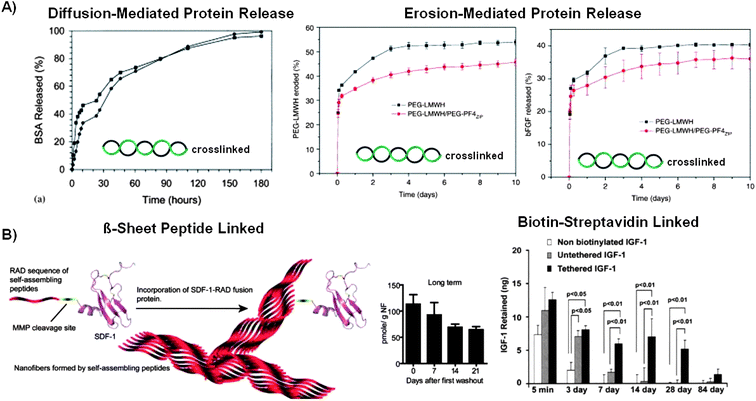 | ||
| Fig. 4 Influence of polymer conformational change dynamics on protein release from helix-crosslinked and β-sheet derived hydrogels. A) In the absence of specific interactions, proteins released primarily via a diffusion-mediated mechanism from 31 helix-crosslinked PLA-PEO-PLA hydrogels (Left),18 and primarily via an erosion-mediated mechanism when bFGF interacted specifically with heparin in a coiled-coil-crosslinked hydrogel (Right).44 B) The protein release rate from β-sheet derived hydrogels was slowed by linking the protein to the hydrogel network. Linker domains were conjugated to therapeutic proteins by genetically engineering the proteins with a β-sheet forming peptide linker (Left) or biotin-streptavidin interactions (Right).126,127 | ||
Alternatively, large proteins, like 400kDa fibrinogen, have had linear “zero order” release kinetics from PLA-PEO-PLA hydrogels over the course of 200–400 h. The zero order release kinetics were attributed to the PEG-induced phase separation of fibrinogen in the hydrogel network. They hypothesized that a fraction of the precipitated fibrinogen could have become soluble and diffused out of the hydrogel network.18 This mechanism is analogous to encapsulating a sustained release device in a hydrogel to enable long-term protein release, which has been achieved in other recent studies.120 The mechanisms that control PEG-induced protein phase separation have been extensively characterized,121 and therefore PEG-induced protein phase separation could be a broadly adaptable technique to create sustained release depots during hydrogel formation without having to encapsulate the protein in another material.
Investigators have developed several techniques to decrease the protein release rate from 31 helix-crosslinked hydrogels. For example, Vert and colleagues have formed PLA-PEG-PLA block copolymers with terminal isocyanate groups that could covalently crosslink proteins into a hydrogel network.122 However, protein bioactivity has often decreased after covalent modification which may limit the utility of this approach.123 In a separate approach, Cheng and colleagues formed PLA-drug conjugates that non-covalently linked the drug into the hydrogel network.124 This approach has yet to be applied to proteins, but could in principle facilitate sustained protein release from 31 helix-crosslinked hydrogels.
Coiled-coil-crosslinks have facilitated nanometer-scale control over hydrogel network structure, and have been used modulate protein release from dynamic hydrogels. For example, Murota and colleagues have used a heterodimeric pair of coiled-coil forming peptides, the 1αE and 1αK peptides, to immobilize green fluorescent protein (GFP) into hydrogels.125 In this approach the 1αK peptide was immobilized to a synthetic hydrogel network and the 1αE peptide was expressed at both the N- and C- termini of GFP. 1αK peptide-modified hydrogels sequestered 30% of the mutant 1αE-GFP-1αE from solution, whereas no unmodified GFP was sequestered into the hydrogels. This result was surprising, as one would expect that a fraction of unmodified GFP would diffuse into the hydrogel network even without coiled-coil interactions.
Mutant 1αE-GFP-1αE released from 1αK peptide-modified hydrogels was triggered by adding either soluble 1αE or 1αK peptides into solution. Significantly, a scrambled version of the 1αK peptide triggered less 1αE-GFP-1αE release than the unmodified 1αK peptide, which indicated the specificity of this triggered release approach. This sequestration approach could be broadly adaptable, as coiled-coil domains could be conjugated to proteins using standard recombinant DNA techniques. Furthermore, harnessing competitive binding events to trigger protein release from hydrogels could be useful in applications where the timing of protein release is important for therapeutic success. However, it may be difficult to use peptide ligands as triggers in the body due to their short in vivo half lives.128
In a separate approach, Kiick and colleagues formed helix-derived hydrogels based on the coiled-coil interactions between heparin and heparin binding peptides, including PF4ZIP. The release of basic fibroblast growth factor (bFGF) from these hydrogels closely matched their erosion profile. This observation could be attributed to heparin's ability to sequester bFGF from solution. The bFGF release rate and hydrogel erosion rate were tuned by varying the affinity of the peptide-heparin interaction (Fig. 4A Right).44 This affinity-modulated approach to tuning hydrogel erosion and protein release could be broadly applicable, as there have been many well characterized peptide-polysaccharide interactions.129 Furthermore, heparin-based materials could be especially useful for controlled growth factor delivery applications, as growth factor binding to heparin has increased the growth factor's stability in solution.130 However, it has been demonstrated that heparin interacts with many growth factors, which may cause specificity problems in the complex in vivo environment.131
4.2 Effect of β-sheet or β-hairpin assembly dynamics on protein release
The mechanisms used to initiate β-sheet assembly in dynamic hydrogels have played a crucial role in controlling the bioactivity of encapsulated proteins. For example, a model protein, ubiquitin, retained its conformation when it was encapsulated in β-sheet derived hydrogels that assembled in response to physical mixing.57 Alternatively, ubiquitin unfolded when it was encapsulated in β-sheet derived hydrogels that assembled in response to changes in pH.59 Many factors have affected protein stability in solution, and often the mechanisms that controlled protein stability have varied for different proteins.132 Together, these findings suggest the mechanisms that mediate hydrogel assembly could be important regulators of protein bioactivity and will be important to characterize in future β-sheet derived hydrogels.Alternatively, proteins have been encapsulated into β-sheet derived hydrogels using an “uptake and release” approach. For example, Semino and colleagues have used different β-sheet derived hydrogels that uptake therapeutic proteins for regenerative medicine purposes.133–138 In one pertinent study, they measured the mass of protein adsorbed into β-sheet derived hydrogels from solutions containing either fibronectin or fetal bovine serum. KFE-8 (FKFEFKFE) and KLD-12 (KLDLKLDLKLDLKLDL) peptide hydrogels absorbed more total protein from 10% fetal bovine serum solutions than RAD16 peptide hydrogels. Alternatively, all of the β-sheet derived hydrogels absorbed statistically equivalent masses of fibronectin from 300μg/ml fibronectin solutions.135 Future characterization of the mechanisms that control protein adsorption into β-sheet derived hydrogels will be necessary to facilitate the formation of hydrogels that selectively adsorb therapeutic proteins from their environment for subsequent therapeutic release.
β-sheet derived hydrogels have possessed remarkable stability in vivo, and therefore they have been extensively studied for controlled protein delivery applications. For example, RAD16 peptide hydrogels have been used to deliver therapeutically important proteins including insulin like growth factor-1 (IGF-1),126 vascular endothelial growth factor (VEGF), bFGF, angiopoietin-1 (Ang-1), serum albumin, platelet derived growth factor-BB (PDGF-BB),139 stromal cell derived factor-1 (SCF-1),127 and epidermal growth factor (EGF).56 The fundamental mechanisms that controlled protein release from RAD16 peptide hydrogels may direct the development of future β-sheet derived hydrogels for protein delivery applications.
Typically, proteins have rapidly released from β-sheet derived hydrogels. 99% of encapsulated PDGF-BB was released from RAD16 peptide hydrogels over the course of 3 h into PBS.139 Similarly, greater than 75% of encapsulated VEGF, bFGF, and Ang-1 have been released from RAD16 peptide hydrogels during the same time period. Alternatively, only 50% of encapsulated BSA was released from RAD16 peptide hydrogels in the 3 h release period. BSA has a larger molecular weight (66.4kDa) than the growth factors (7.7–57KDa), which suggested that protein release from RAD16 peptide hydrogels was diffusion mediated. Also, the rapid release profiles from RAD16 peptide hydrogels suggests that these β-sheet derived hydrogels did not extensively interact with encapsulated therapeutic proteins, which is consistent with their ability to resist enzymatic degradation.
The rate of protein release from RAD16 peptide hydrogels has been environment dependent. For example, 91.8% of PDGF-BB encapsulated in RAD16 peptide hydrogels has been retained at the injection site after 10 min and 16.1% of the PDGF-BB remained after 14 days. Therefore, the rapid PDGF-BB release rate from RAD16 peptide hydrogels observed in PBS may become slower if characterized in vivo.139 In a separate study, no EGF was released from RAD16 peptide hydrogels in PBS, but 65.5% of the encapsulated EGF was released over 24 h from RAD16 peptide hydrogels in a protease containing in vitro wound model.56 The increased EGF release rate in the wound site was unexpected, as β-sheet derived hydrogels have been resistant to protease activity, and therefore they were unlikely to erode or dissociate to form hydrogels with larger mesh sizes. Future research will be necessary to characterize the mechanisms that control protein release from RAD16 peptide hydrogels in different environments.
Proteins have also rapidly released from β-hairpin derived hydrogels. For example, greater than 70% of a negatively charged protein, lactoferrin, was released from MAX1 hydrogels over the course of 2 days. Alternatively, less than 40% of a similarly sized, but oppositely charged, dextran was released over the course of 2 days. This result suggested that electrostatic interactions could have a significant role in tuning the protein release rate from β-hairpin derived hydrogels.140 The results of this study and others motivate the need to form β-sheet and β-hairpin derived hydrogels that possess specific intermolecular interactions to slow the release of encapsulated proteins for sustained protein release applications. Applications such as tissue engineering are particularly sensitive to the need for sustained delivery, since new tissue development typically occurs over weeks to months.
To address this challenge investigators have recently begun to form hydrogels that harness specific intermolecular interactions between encapsulated proteins and the hydrogel network to slow the protein release rate. For example, Segers et al. formed a SCF-1 mutant with an N-terminal linker domain to incorporate SCF-1 into β-sheet derived hydrogels. The N-terminal linker domain was composed of a RAD16 peptide conjugated to a matrix metalloprotease (MMP) cleavable peptide. RAD16 peptide hydrogels formed in the presence of RAD16-SCF-1 mutant proteins or unmodified SCF-1, and were repeatedly washed with PBS to quantify the mass of SCF-1 incorporated and released. After the first wash, 2% of the original RAD16-SCF-1 mutant protein was encapsulated into the hydrogels, whereas only 0.25% of the unmodified SCF-1 was incorporated into the hydrogels. After 32 washes, the original mass of RAD16-SCF-1 was retained in the RAD16 peptide hydrogels, whereas no SCF-1 was retained without the RAD16 linker. This result was consistent with previous studies in which proteins rapidly released from β-sheet derived hydrogels unless they were linked to the hydrogel network.139
Increasing the RAD16 peptide concentration in the pre-polymer solution enhanced the RAD16-SCF-1 encapsulation efficiency compared to hydrogels formed with lower RAD16 peptide concentrations. This capability to tune the protein encapsulation efficiency in β-sheet derived hydrogels could be a valuable tool in dose dependant applications. When linked to the hydrogel network, 30% of the encapsulated RAD16-SCF-1 was released over the course of 21 days in the absence of MMPs (Fig. 4B Left). Interestingly, the addition of MMPs induced the release of just 22% of the encapsulated protein during the same time period, and the magnitude of MMP triggered protein release was independent of the MMP concentration.127 These results suggest that RAD16-peptide based materials could be useful in applications where the material needs to resist MMP degradation for long term protein release, such as implantation at wound sites.
In an alternative approach, Davis et al. decreased the protein release rate from RAD16 peptide hydrogels using biotin-streptavidin interactions. In this approach, biotinylated IGF-1 was linked with tetravalent streptavidin, and then linked to biotinylated RAD16 peptides to form RAD16-IGF-1 conjugates. RAD16 hydrogels that contained 25ng of either IGF-1 or Streptavidin-IGF-1 were injected into the rat myocardium and the concentration of IGF-1 was quantified. After 3 days, 1.97 ± 1.18 ng of unmodified IGF-1 remained in the rat myocardium, whereas 8.08 ± 0.60 ng of RAD16-IGF-1 was detected around the injection site. After 28 days there was still more RAD16-IGF-1 than unmodified IGF-1 in the rat myocardium.126 These studies indicated that proteins can readily be linked into β-sheet derived hydrogels using modular, non-covalent interactions which could be adapted for a broad array of clinically relevant proteins (Fig. 4B Right).
4.3 Effect of protein conformational change dynamics on protein release
Protein conformational changes have emerged as a potentially useful tool to vary the timing of protein release from dynamic hydrogels. We have characterized the mechanisms by which CaM's protein conformational change modulated the release of an important therapeutic protein, VEGF.95,96 Significantly, CaM's ligand-induced conformational change translated to macroscopic hydrogel volume changes that induced the release of encapsulated VEGF (Fig. 5). This protein release mechanism may be analogous to an approach described by Mooney and colleagues, in which a VEGF-laden alginate hydrogel was mechanically loaded, which induced fluid exclusion and VEGF release.141 However, controlling this mechanical stimulus may be difficult in the body due to the complex in vivo environment. Interestingly, the mechanism that controlled protein release from dynamic, protein-based hydrogels is quite distinct from the mechanisms that have controlled protein release from hydrogels that underwent phase transition-induced volume changes. In hydrogels that underwent temperature-induced phase transitions, protein release was restricted when the hydrogels collapsed due to the hydrogels' decreased mesh size and the encapsulated protein's interaction with hydrophobic polymers.142 Therefore, dynamic protein-based hydrogels could enable therapeutic strategies in which protein release is triggered by a specific biochemical stimulus. | ||
| Fig. 5 The VEGF release rate from dynamic, protein-based hydrogels was triggered by their ligand-induced conformational change. A) TFP induced an 8-fold increase in the release rate of VEGF from CaM-based hydrogels. B) The timing of VEGF release from CaM-based hydrogels was modulated by varying the timing of the TFP “trigger”. C) Schematic representation of protein release from dynamic hydrogels in which a protein conformational change induced hydrogel volume decreases, fluid exclusion, and protein release.96 | ||
The absorption and release of VEGF from CaM-based hydrogels has been modulating by varying the hydrogel material properties. First, hydrogels with increasing concentrations of CaM absorbed greater masses of VEGF than hydrogels with lower CaM concentrations.96 This trend was attributed to electrostatic attraction between CaM and VEGF, as they are oppositely charged at physiological pH. Also, the mass of VEGF released from CaM-based hydrogels was tuned by both the concentration of CaM in the hydrogel network and the magnitude of their ligand-induced volume change. For example, while CaM-based hydrogel disks formed from pre-polymer solutions containing 10% or 12.5% (w/v) PEG-CaM-PEG each released 100% of their encapsulated VEGF, the 12.5% (w/v) hydrogels released a greater mass of VEGF because they initially absorbed a greater total mass of VEGF. Alternatively, 15% (w/v) CaM-based hydrogels absorbed the greatest mass of VEGF, but released the smallest fraction of their encapsulated VEGF because of their small magnitude ligand-induced volume changes.96 Together, these results suggest that the network properties of dynamic, protein-based hydrogels can be tuned to control the mass of therapeutic protein released in response to a specific biochemical ligand.
The VEGF release rate from CaM-based hydrogels has been tuned by varying the rate of their volume changes. For example, TFP induced the collapse of CaM-based hydrogel disks over the course of 2 h,25 and increased the VEGF release rate over the course of 12 h.96 Alternatively, TFP induced CaM-based microspheres to collapse in 1 min, and increased the VEGF release rate over the course of 30 min.95 These results indicated that therapeutic protein release from dynamic hydrogels was accelerated from hydrogels that underwent faster ligand-induced volume changes. There are many techniques to control the volume and geometry of hydrogels,143 therefore this approach to varying the protein release rate from dynamic hydrogels may be broadly adaptable. Importantly, ligand-induced protein conformational changes provide stimulus specificity, temporal control, and tunable release kinetics, and these collective capabilities are difficult to achieve using other mechanisms.
5. Conclusions and perspective
Nanometer scale polymer conformational changes have supplied investigators with new mechanisms to control protein release from dynamic hydrogels. First, the network structure of dynamic hydrogels has been tuned by varying the fundamental principles that control helix and β-sheet assembly and disassociation. Protein release from helix-crosslinked or β-sheet derived hydrogels has typically proceeded rapidly via diffusion mediated mechanisms unless the protein was linked somehow to the hydrogel network. Alternatively, specific interpolymer interactions were used to tether proteins into hydrogels networks and slow the protein release rate from dynamic hydrogels. Significantly, nanometer scale protein conformational changes have induced macroscopic changes in hydrogel properties and varied the protein release rate. Protein release from dynamic, protein-based hydrogels was tuned by the rate and magnitude of their volume changes. Taken together, results to date demonstrate that polymer conformational changes have already provided investigators with unique mechanisms to control protein release from dynamic hydrogels.There are still challenges to overcome in order to obtain controlled protein release from dynamic hydrogels. First, many of the chemistries used to form hydrogel networks have been shown to decrease the activity of encapsulated proteins.144 Also, many of the self-assembling peptides discussed in this review are sensitive to slight changes in pH, salt concentration, and temperature, which could affect their in vivo stability.35,66 Furthermore, many of the protein- or peptide-based hydrogels discussed in this manuscript will need to be characterized to observe if they induce an immune response or if they have an unexpected biological effect. Together, future hydrogels will need to be developed that promote therapeutic protein activity and maintain their in vivo stability.
Characterizing how these materials change their drug release rates in response to changes in their in vivo environment will also be critical for their clinical success. Slowing protein release from helix-crosslinked or β-sheet derived hydrogels has required the use of mutant proteins with linker domains. These linker domains could alter the protein's bioactivity. Therefore, new mechanisms to control release of unmodified therapeutic proteins from hydrogels could be important for clinical applications. Also, the relationship between protein release into PBS, the most commonly used buffer for in vitro protein release experiments, and protein release in vivo has not been mechanistically established. Although technically challenging, characterizing the differences in protein release in vitro and in vivo would be of great benefit to the drug delivery community.
Future dynamic hydrogels could enable even greater control over therapeutic protein delivery. There are hundreds of polymers that could be used either independently, or as block copolymers, to form tailored, multifunctional materials for protein delivery applications. Recently discovered protein-peptide interactions may supply investigators with new stimuli to trigger or temporally control protein release from dynamic hydrogels. The publication rate of new protein-peptide interactions in the Protein Data Bank has increased over 250% from 2000 to 2008.145 Furthermore, there are over 200 proteins that undergo well characterized conformational changes that could be used to form dynamic hydrogels,146 and a large percentage of these changes are “hinge motions” similar to those used in recent studies described in this review.25,94–99,102 Therefore, recombinant DNA techniques could be used to synthesize peptide block copolymers that contain: 1) coiled-coil domains for precise control over crosslink structures, 2) β-sheet domains that impart stability, and/or 3) proteins that undergo ligand-induced conformational changes to facilitate selectivity and reversible hydrogel volume changes. These diverse building blocks may facilitate the combinatorial synthesis of dynamic hydrogels to screen for materials with unique protein delivery capabilities.147 Alternatively, the intelligent selection of these polymer components could enable the formation of hydrogel networks that tune their protein release rate in response to physiologically relevant stimuli. Together, polymer conformational changes could provide polymer scientists with a broadly adaptable mechanism to control protein release from dynamic hydrogels.
Acknowledgements
The authors are grateful for support from the National Science Foundation (Materials Research Science and Engineering Center (MRSEC) seed grant to W.L.M., Graduate Research Fellowship to W.J.K, and CAREER award #0745563 to W.L.M.)Notes and references
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS.
- R. L. Lalonde and P. Honig, Clin. Pharmacol. Ther., 2008, 84, 533–536 CrossRef CAS.
- J. Stone, A. Itin, T. Alon, J. Peer, H. Gnessin, T. Chanling and E. Keshet, J. Neurosci., 1995, 15, 4738–4747 CAS.
- E. Aiba-Kojima, N. H. Tsuno, K. Inoue, D. Matsumoto, T. Shigeura, T. Sato, H. Suga, H. Kato, T. Nagase, K. Gonda, I. Koshima, K. Takahashi and K. Yoshimura, Wound Repair Regener., 2007, 15, 511–520 Search PubMed.
- J. L. Shifren, J. F. Tseng, C. J. Zaloudek, I. P. Ryan, Y. G. Meng, N. Ferrara, R. B. Jaffe and R. N. Taylor, J. Clin. Endocrinol. Metab., 1996, 81, 3112–3118 CrossRef CAS.
- C. E. Murry and G. Keller, Cell, 2008, 132, 661–680 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- J. Shaikh Mohammed and W. L. Murphy, Adv. Mater., 2009, 21, 2361–2374 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS.
- A. A. Adzhubei and M. J. E. Sternberg, J. Mol. Biol., 1993, 229, 472–493 CrossRef CAS.
- F. Li, S. M. Li and M. Vert, Macromol. Biosci., 2005, 5, 1125–1131 CrossRef CAS.
- T. Okihara, M. Tsuji, A. Kawaguchi, K. Katayama, H. Tsuji, S. H. Hyon and Y. Ikada, J. Macromol. Sci., Part B: Phys., 1991, 30, 119–140 Search PubMed.
- D. Brizzolara, H. J. Cantow, K. Diederichs, E. Keller and A. J. Domb, Macromolecules, 1996, 29, 191–197 CrossRef CAS.
- W. Hoogsteen, A. R. Postema, A. J. Pennings, G. Tenbrinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642 CrossRef CAS.
- S. M. Li, S. Anjard, I. Rashkov and M. Vert, Polymer, 1998, 39, 5421–5430 CrossRef CAS.
- S. M. Li, I. Rashkov, J. L. Espartero, N. Manolova and M. Vert, Macromolecules, 1996, 29, 57–62 CrossRef CAS.
- I. Rashkov, N. Manolova, S. M. Li, J. L. Espartero and M. Vert, Macromolecules, 1996, 29, 50–56 CrossRef CAS.
- I. Molina, S. M. Li, M. B. Martinez and M. Vert, Biomaterials, 2001, 22, 363–369 CrossRef CAS.
- C. Hiemstra, Z. Y. Zhong, P. Dijkstra and J. Feijen, Macromol. Symp., 2005, 224, 119–131 CrossRef CAS.
- S. R. Van Tomme and W. E. Hennink, Expert Rev. Med. Devices, 2007, 4, 147–164 Search PubMed.
- J. Li, X. Li, X. P. Ni, X. Wang, H. Z. Li and K. W. Leong, Biomaterials, 2006, 27, 4132–4140 CrossRef CAS.
- C. Fraschini and R. E. Prud'homme, Eur. Polym. J., 2008, 44, 653–655 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- S. G. Zhang, T. C. Holmes, C. M. Dipersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv. Mater., 2007, 19, 3377–3380 CrossRef CAS.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- C. Cohen and D. A. D. Parry, Proteins: Struct., Funct., Genet., 1990, 7, 1–15 CrossRef CAS.
- C. Wang, R. J. Stewart and J. Kopecek, Nature, 1999, 397, 417–420 CrossRef CAS.
- J. Stetefeld, M. Jenny, T. Schulthess, R. Landwehr, J. Engel and R. A. Kammerer, Nat. Struct. Biol., 2000, 7, 772–776 CrossRef CAS.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS.
- E. K. Oshea, R. Rutkowski and P. S. Kim, Science, 1989, 243, 538–542 CAS.
- W. Shen, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2007, 40, 689–692 CrossRef CAS.
- J. C. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897–1904 RSC.
- W. Shen, J. A. Kornfield and D. A. Tirrell, Soft Matter, 2007, 3, 99–107 RSC.
- W. Shen, K. C. Zhang, J. A. Kornfield and D. A. Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS.
- K. J. Lumb and P. S. Kim, Science, 1995, 268, 436–439 CrossRef CAS.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CAS.
- S. Y. M. Lau, A. K. Taneja and R. S. Hodges, J. Biol. Chem., 1984, 259, 3253–3261.
- K. J. Lumb and P. S. Kim, Biochemistry, 1995, 34, 8642–8648 CrossRef CAS.
- N. Yamaguchi, B. S. Chae, L. Zhang, K. L. Kiick and E. M. Furst, Biomacromolecules, 2005, 6, 1931–1940 CrossRef CAS.
- L. Zhang, E. M. Furst and K. L. Kiick, J. Controlled Release, 2006, 114, 130–142 CrossRef CAS.
- S. H. Kim and K. L. Kiick, Peptides, 2007, 28, 2125–2136 CrossRef CAS.
- D. J. Butcher, M. A. Kowalska, S. Li, Z. Luo, S. Shan, Z. Lu, S. Niewiarowski and Z. Huang, FEBS Lett., 1997, 409, 183–187 CrossRef CAS.
- J. R. Fromm, R. E. Hileman, E. E. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1997, 343, 92–100 CrossRef CAS.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390–412 CrossRef CAS.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- S. Zhang, F. Gelain and X. Zhao, Semin. Cancer Biol., 2005, 15, 413–420 CrossRef.
- N. J. Trengove, M. C. Stacey, S. Macauley, N. Bennett, J. Gibson, F. Burslem, G. Murphy and G. Schultz, Wound Repair Regener., 1999, 7, 442–452 Search PubMed.
- T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- E. Fournier, C. Passirani, C. N. Montero-Menei and J. P. Benoit, Biomaterials, 2003, 24, 3311–3331 CrossRef CAS.
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CAS.
- S. G. Zhang, C. Lockshin, R. Cook and A. Rich, Biopolymers, 1994, 34, 663–672 CrossRef CAS.
- A. Schneider, J. A. Garlick and C. Egles, PLoS One, 2008, 3, e1410 CrossRef.
- S. Ramachandran, Y. Tseng and Y. B. Yu, Biomacromolecules, 2005, 6, 1316–1321 CrossRef CAS.
- S. Ramachandran, J. Trewhella, Y. Tseng and Y. B. Yu, Chem. Mater., 2006, 18, 6157–6162 CrossRef CAS.
- S. Rarnachandran, P. Flynn, Y. Tseng and Y. B. Yu, Chem. Mater., 2005, 17, 6583–6588 CrossRef CAS.
- X. D. Xu, C. S. Chen, B. Lu, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, J. Phys. Chem. B, 2010, 114, 2365–2372 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS.
- B. L. Seal, T. C. Otero and A. Panitch, Mater. Sci. Eng., R, 2001, 34, 147–230 CrossRef.
- Z. M. Yang, G. L. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
- J. C. Garcia-Pagan, R. Morillas, R. Banares, A. Albillos, C. Villanueva, C. Vila, J. Genesca, M. Jimenez, M. Rodriguez, J. L. Calleja, J. Balanzo, F. Garcia-Duran, R. Planas and J. Bosch, Hepatology, 2003, 37, 1260–1266 CrossRef CAS.
- J. Miller and E. H. Wolf, Ann. Allergy, 1975, 35, 81–86 Search PubMed.
- B. Girolami, E. Bernardi, M. H. Prins, J. W. ten Cate, R. Hettiarachchi, P. Prandoni, A. Girolami and H. R. Buller, Arch. Intern. Med., 1999, 159, 337–345 CrossRef CAS.
- X. M. Li, Y. A. Gao, Y. Kuang and B. Xu, Chem. Commun., 2010, 46, 5364–5366 RSC.
- Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS.
- G. L. Liang, Z. M. Yang, R. J. Zhang, L. H. Li, Y. J. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS.
- Q. G. Wang, Z. M. Yang, Y. Gao, W. W. Ge, L. Wang and B. Xu, Soft Matter, 2008, 4, 550–553 RSC.
- P. J. Kennelly and E. G. Krebs, J. Biol. Chem., 1991, 266, 15555–15558 CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS.
- K. L. Niece, J. D. Hartgerink, J. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS.
- H. A. Behanna, J. Donners, A. C. Gordon and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef CAS.
- S. Takada, Z. Luthey-Schulten and P. G. Wolynes, J. Chem. Phys., 1999, 110, 11616–11629 CrossRef CAS.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS.
- A. Roth and R. R. Breaker, Annu. Rev. Biochem., 2009, 78, 305–334 CrossRef CAS.
- D. Chin and A. R. Means, Trends Cell Biol., 2000, 10, 322–328 CrossRef CAS.
- M. Gerstein and W. Krebs, Nucleic Acids Res., 1998, 26, 4280–4290 CrossRef CAS.
- C. S. Goh, D. Milburn and M. Gerstein, Curr. Opin. Struct. Biol., 2004, 14, 104–109 CrossRef CAS.
- S. Hayward, Proteins: Struct., Funct., Genet., 1999, 36, 425–435 CrossRef CAS.
- M. Gerstein, A. M. Lesk and C. Chothia, Biochemistry, 1994, 33, 6739–6749 CrossRef CAS.
- S. Lindquist and E. A. Craig, Annu. Rev. Genet., 1988, 22, 631–677 CrossRef CAS.
- E. P. Widmaier, H. Raff, K. T. Strang and A. J. Vander, Vander's human physiology: the mechanisms of body function, McGraw-Hill, New York, 2011 Search PubMed.
- M. Vandonselaar, R. A. Hickie, J. W. Quail and L. T. J. Delbaere, Nat. Struct. Biol., 1994, 1, 795–801 CrossRef CAS.
- W. L. Murphy, W. S. Dillmore, J. Modica and M. Mrksich, Angew. Chem., Int. Ed., 2007, 46, 3066–3069 CrossRef CAS.
- W. J. King, J. N. Pytel, K. Ng and W. L. Murphy, Macromol. Biosci., 2010, 10, 580–584 CrossRef CAS.
- W. King, J. Mohammed and W. Murphy, Soft Matter, 2009, 5, 2399–2406 RSC.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv. Funct. Mater., 2008, 18, 1824–1831 CrossRef CAS.
- W. W. Yuan, J. Y. Yang, P. Kopeckova and J. Kopecek, J. Am. Chem. Soc., 2008, 130, 15760–15761 CrossRef CAS.
- J. D. Ehrick, M. R. Luckett, S. Khatwani, Y. Wei, S. K. Deo, L. G. Bachas and S. Daunert, Macromol. Biosci., 2009, 9, 864–868 CrossRef CAS.
- J. Jose and S. Handel, ChemBioChem, 2003, 4, 396–405 CrossRef CAS.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- A. P. Esser-Kahn, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 15820–15822 CrossRef CAS.
- S. B. Chen, R. Pieper, D. C. Webster and J. Singh, Int. J. Pharm., 2005, 288, 207–218 CrossRef CAS.
- N. Sanabria-DeLong, S. K. Agrawal, S. R. Bhatia and G. N. Tew, Macromolecules, 2007, 40, 7864–7873 CrossRef CAS.
- J. D. Clapper, J. M. Skeie, R. F. Mullins and C. A. Guymon, Polymer, 2007, 48, 6554–6564 CrossRef CAS.
- H. Yokoi, T. Kinoshita and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS.
- J. Y. Yang, C. Y. Xu, C. Wang and J. Kopecek, Biomacromolecules, 2006, 7, 1187–1195 CrossRef CAS.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- C. M. C. Tempany, E. A. Stewart, N. McDannold, B. J. Quade, F. A. Jolesz and K. Hynynen, Radiology, 2003, 226, 897–905 Search PubMed.
- N. Matsushima, N. Hayashi, Y. Jinbo and Y. Izumi, Biochem. J., 2000, 347, 211–215 CrossRef CAS.
- A. E. Jackson and D. Puett, Biochem. Pharmacol., 1986, 35, 4395–4400 CrossRef CAS.
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Nat. Mater., 2005, 4, 298–302 CrossRef CAS.
- J. P. Junker, F. Ziegler and M. Rief, Science, 2009, 323, 633–637 CrossRef CAS.
- E. S. Matsuo and T. Tanaka, J. Chem. Phys., 1988, 89, 1695–1703 CrossRef CAS.
- S. M. Singh and A. K. Panda, J. Biosci. Bioeng., 2005, 99, 303–310 CrossRef CAS.
- G. R. Hendrickson and L. A. Lyon, Soft Matter, 2009, 5, 29–35 RSC.
- J. M. G. Swann and A. J. Ryan, Polym. Int., 2009, 58, 285–289 CrossRef CAS.
- M. Rafat, C. A. Cleroux, W. G. Fong, A. N. Baker, B. C. Leonard, M. D. O'Connor and C. Tsilfidis, Biomaterials, 2010, 31, 3414–3421 CrossRef CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS.
- B. J. Peret and W. L. Murphy, Adv. Funct. Mater., 2008, 18, 3410–3417 CrossRef CAS.
- A. M. Kulkarni, A. P. Chatterjee, K. S. Schweizer and C. F. Zukoski, J. Chem. Phys., 2000, 113, 9863–9873 CrossRef CAS.
- H. Van den Berghe, J. Coudane and M. Vert, J. Bioact. Compat. Polym., 2007, 22, 637–650 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- R. Tong, L. D. Yala, T. M. Fan and J. J. Cheng, Biomaterials, 2010, 31, 3043–3053 CrossRef CAS.
- K. Murota, S. Sakamoto and K. Kudo, Chem. Lett., 2007, 36, 1320–1321 CrossRef CAS.
- M. E. Davis, P. C. H. Hsieh, T. Takahashi, Q. Song, S. G. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS.
- V. F. M. Segers, T. Tokunou, L. J. Higgins, C. MacGillivray, J. Gannon and R. T. Lee, Circulation, 2007, 116, 1683–1692 CrossRef CAS.
- V. P. Torchilin and A. N. Lukyanov, Drug Discovery Today, 2003, 8, 259–266 CrossRef CAS.
- A. Rullo and M. Nitz, Biopolymers, 2010, 93, 290–298 CrossRef CAS.
- T. A. McCaffrey, D. J. Falcone, D. Vicente, B. H. Du, S. Consigli and W. Borth, J. Cell. Physiol., 1994, 159, 51–59 CrossRef CAS.
- D. P. Witt and A. D. Lander, Curr. Biol., 1994, 4, 394–400 CrossRef CAS.
- J. L. Cleland, M. F. Powell and S. J. Shire, Crit. Rev. Ther. Drug Carrier Syst., 1993, 10, 307–377 CAS.
- E. Genove, C. Shen, S. G. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341–3351 CrossRef CAS.
- C. E. Semino, J. Kasahara, Y. Hayashi and S. G. Zhang, Tissue Eng., 2004, 10, 643–655 CrossRef CAS.
- A. L. Sieminski, C. E. Semino, H. Gong and R. D. Kamm, J. Biomed. Mater. Res., Part A, 2008, 87a, 494–504 CrossRef CAS.
- E. Genove, S. Schmitmeier, A. Sala, S. Borros, A. Bader, L. G. Griffith and C. E. Semino, J. Cell. Mol. Med., 2009, 13, 3387–3397 CrossRef.
- E. Garreta, E. Genove, S. Borros and C. E. Semino, Tissue Eng., 2006, 12, 2215–2227 CrossRef CAS.
- L. Quinatana, T. F. Muinos, E. Genove, M. D. Olmos, S. Borros and C. E. Semino, Tissue Eng. A, 2009, 15, 45–54 Search PubMed.
- P. C. H. Hsieh, M. E. Davis, J. Gannon, C. MacGillivray and R. T. Lee, J. Clin. Invest., 2006, 116, 237–248 CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS.
- K. Y. Lee, M. C. Peters, K. W. Anderson and D. J. Mooney, Nature, 2000, 408, 998–1000 CrossRef CAS.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- C. C. Lin, S. M. Sawicki and A. T. Metters, Biomacromolecules, 2008, 9, 75–83 CrossRef CAS.
- H. Berman, K. Henrick and H. Nakamura, Nat. Struct. Biol., 2003, 10, 980–980 CrossRef CAS.
- V. Alexandrov, U. Lehnert, N. Echols, D. Milburn, D. Engelman and M. Gerstein, Protein Sci., 2005, 14, 633–643 CrossRef CAS.
- A. L. Hook, D. G. Anderson, R. Langer, P. Williams, M. C. Davies and M. R. Alexander, Biomaterials, 2010, 31, 187–198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |