Nanoparticles of anionic starch and cationic cyclodextrin derivatives for the targeted delivery of drugs
Carolin
Thiele
a,
Dagmar
Auerbach
b,
Gregor
Jung
b,
Lian
Qiong
c,
Marc
Schneider
c and
Gerhard
Wenz
*a
aOrganic Macromolecular Chemistry, Saarland University, C4.2 Campus Saarbrücken. D-66123, Saarbrücken, Germany. E-mail: g.wenz@mx.uni-saarland.de; Fax: +49 681 302 3909; Tel: +49 681 302 3449
bDepartment of Biophysical Chemistry, Saarland University, B2.2 Campus SaarbrückenD-66123, Saarbrücken, Germany
cDepartment of Biopharmaceutics and Pharmaceutical Technology, Saarland University, A4.1 Campus SaarbrückenD-66123, Saarbrücken, Germany
First published on 23rd October 2010
Abstract
Starch was oxidized with TEMPO for the synthesis of water-soluble copolymers of glucuronic acid and glucose. The carboxylate groups of these copolymers were conjugated with pteroic acid as cell-specific ligand for targeting to cancer cells. Stable spherical nanoparticles (NPs) were formulated mixing aqueous solutions of the anionic copolymers and of a cationic thioether of β-cyclodextrin (β-CD). Particle size distributions of NPs were investigated with DLS as the function of the charge ratio of the constituents. The smallest and most uniform particles with a diameter of about 130 nm were generated at a charge ratio of anion/cation close to 1, preferably 1.2. Stabilities and particle size distributions of these starch NPs were very satisfactory. The starch/β-CD NPs could be loaded with hydrophobic guest molecules like 1,4-dihydroxyanthraquinone (DHA), which served as a model for the important class of anthracycline antibiotics used in cancer therapy.
Introduction
Cyclodextrins (CDs), cyclic oligomers of glucose, are well-known to complex various amphiphilic guest molecules in aqueous solution.1,2 Complexation is mainly driven by hydrophobic interactions and can be supported with both hydrogen bonds and Coulomb interactions exerted by appropriate substituents linked to the cyclodextrin scaffold. Binding constants K exceeding 106 M−1 could be reached accordingly with charged CDs.3–5 The complexation of hydrophobic anticancer drugs in CD hosts is of special interest, since solubility in water can be greatly enhanced leading to better bioavailabilities.6,7 Furthermore, cell-specific carriers were synthesized by conjugation of cell-specific ligands, such as folate,8glucose,9mannose,10 and lactose,11 to a CD. A drug could be targeted with CD–peptide conjugates to the desired location of a test animal.12,13Still, the design of cell-selective molecular carriers is generally hampered by several drawbacks, stated in the following:
• The binding constant of the CD drug complex K has to be high enough, to avoid dissociation of the complex in the body fluid on its way to the target.
• Blood constituents, e.g. albumins, might bind competitively to the drug.
• Most CD derivates cannot overcome the barrier of cell walls and therefore cannot deliver the drug into a cell.
• Syntheses of CD derivatives with high binding potential and conjugated cell specific ligands are generally cumbersome.
The avoidance of most of theses drawbacks was accomplished via the incorporation of CD inclusion compounds of drugs within nanoparticles (NPs). Polymers and especially polymeric NPs are the most appropriate vehicles to overcome cell walls since they can enter cells through endocytosis.14 Especially cancer cells are known to take up nanoparticles of sizes between 50 and 150 nm with higher rates than healthy cells. This effect, named enhanced permeability and retention (EPR) effect15–18 is used in cancer therapy to target drugs passively preferentially in cancer cells. Attachment of cell-specific ligands, such as folate,19 and sugars,20 at the outer sphere of nanoparticles further increases the specificity of drug delivery.21 Spheric nanoparticles of well-defined size are obtained by various methods, such as polymerisation in mini-emulsions,22 or dispersion of hydrophobic polymers, e.g. PLGA,23PLA,24 chitosan,25 and starch26 or CD derivates27,28 by emulsion techniques and by interaction of polyelectrolytes, or by combination of oppositely charged polyelectrolytes leading to so-called polyplexes or nanoplexes.29
Nanoplexes from DNA and cationic polymers, such as PEI,30 had been extensively investigated for gene delivery. Nanoparticles from synthetic polymers, e.g. polyamidoamine dendrimers and dicarboxylic acids,31 di- and tri-sulfonic acids32 and of low molecular weight chitosan and polyglutamic acid,33 are also known. Precautions have to be taken to avoid infinite growth, known as Ostwald ripening, which results in the precipitation of the particles. A solution to this problem is the conjugation of neutral water-soluble polymer blocks (e.g. polyethylene glycol) to the polyelectrolyte which shield aggregation sterically.34 In addition, the nanoplexes were chemically crosslinked for reasons of stabilization.35,36 Another solution for this problem is the stepwise layer by layer assembly of polyelectrolytes starting from a template particle, e.g. a drug, leading to microscopic capsules.37
The assembly of nanoplexes from anionic starch derivates and cationic CD thioethers, which can be loaded by hydrophobic drug molecules, will be described in the following. Furthermore, cell specific ligands could be conjugated to the starch derivative to allow cell-specific targeting. Dihydroxyanthraquinone (DHA) was chosen as a model compound for anthraquinone antibiotic drugs, since it is stable, readily available, nontoxic, and very sensitively detectable through its UV-vis absorbance and fluorescence. Furthermore, DHA forms very stable and water soluble inclusion compounds (K = 453![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1) with the cationic CD thioether 1, as described previously (Fig. 1).7
000 M−1) with the cationic CD thioether 1, as described previously (Fig. 1).7
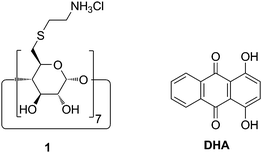 | ||
| Fig. 1 Structure of the cationic CD derivative 1 and dihydroxyanthraquinone (DHA). | ||
Experimental
Materials
Partially hydrolyzed potato starch from Avebe was used for further modifications. The molecular weight of starch was approximately 1300000 g mol−1, and the amylose content 33% as determined from the UV-vis absorption of the iodine inclusion complex.38Pyrofolic acid was synthesized from folic acid according to literature.39 All fine chemicals were purchased from Sigma-Aldrich and used without further purification.
Polymer synthesis
:1 v/v for 16 h at 60 °C measured in D2O/D2SO4 1:1 v/v; standard DSS) 3.17–4.05 (m, glucose), 4.61 (s, H2′ glucuronic acid), 4.79 (m, H1β), 4.90 (d, H4′), 5.19 (m, H5′), 5.22 (m, H3 glucuronic acid), 5.43 (d, H1α), 5.69 (s, H1′). Molar mass weight average Mw = 64000 g mol−1 for a molar ratio of NaOCl per glucose unit 1.8. Other starch polymers were synthesized accordingly.
400 M−1 cm−1).
Nanoparticle preparation
The aqueous CD or CD complex solution (1 mg mL−1) was added dropwise to an aqueous solution of the polymer (concentration 1 mg mL−1) at a stirring speed of 15000 rpm using an ultra turrax from Ika®. After addition was complete, stirring was continued further for 30 s. The volumes of the solutions of the two components were calculated from the molecular mass per charge of each component for the desired charge ratio. The total volume was 20–30 mL.
Nanoparticle characterisation
Particle size d, polydispersity index PDI and ζ potential were determined by dynamic light scattering with a ZetaSizer Nano ZS from Malvern Instruments Ltd. of aqueous dispersions of the nanoparticles, concentration 1 mg mL−1.Nanoparticles were examined by scanning force microscopy (AFM) with a Bioscope equipped with a Nanoscope IV controller from Digital Instruments, Veeco, Santa Barbara, CA, USA. Samples were dried under a stream of compressed air after deposition on muscovite mica (Plano GmbH, Wetzlar, Germany) and investigated under ambient conditions in tapping mode using a scanning probe (Anfatec, Oelsnitz, Germany) with a force constant of 40 N m−1 at a resonant frequency of 170 kHz.
Fluorescence correlation spectroscopy (FCS) was performed on a home-built setup.40 The nanoparticles were excited by a frequency-doubled diode laser operating at λ = 488 nm (Picarro, Soliton GmbH). Focussing as well as fluorescence light collection was achieved by an objective lens (63× WI, NA 1.2, Zeiss). After passing a confocal pinhole with a diameter of 50 µm and a fluorescence filter (HQ 590/70, AHF Analysentechnik), the fluorescence photons were detected by a pair of avalanche photodiodes (SPCM-14-AQR, Perkin-Elmer Optoelectronics) and cross-correlated by a hardware correlator (Flex-02-D, http://correlator.com/). The obtained autocorrelation traces were analyzed according to the two-dimensional diffusion model g(τ) = 1 + N−1(1 + τ/τdiff)−1.41 The hydrodynamic diameter was calculated using green fluorescent protein as the reference with a hydrodynamic radius of 2.8 nm.42 Details are reported elsewhere.40
Nanoparticles were centrifuged to bottom at 13000 rpm for 20 h using a Biofuge A from Heraeus Sepatech to separate the nanoparticles from the supernatant.
Results and discussion
Polymer synthesis and characterisation
The anionic copolymers of glucuronic acid and glucose 2 (Fig. 2) were synthesized by oxidation of partially hydrolyzed potato starch with NaOCl and catalytic amounts of TEMPO in 60–75% yield. The oxidation occurs selectively at the primary carbon atoms of starch leading to carbaldehyde and carboxylate groups.43,44 The carbaldehyde groups were reduced back to carbinol groups by NaBH4. The final degree of substitution (DS) of carboxylate groups, determined according to the Blumenkrantz assay,45 could be controlled between 0.06 and 0.75 by the molar ratio of NaOCl per glucose unit as shown in Fig. 3. The molecular weights, determined by GPC, of copolymers 1, ranging between 33000 g mol−1 and 63000 g mol−1, were significantly lower than the one of the original starch (1300000 g mol−1). This decrease was attributed to some degradation occurring during the oxidation process. These oxidised starches rapidly dissolve in water leading to solutions without turbidity.
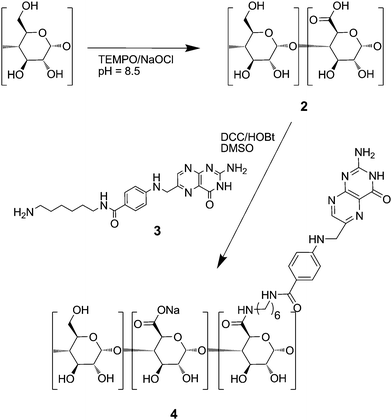 | ||
| Fig. 2 Scheme of the syntheses of anionic starch derivatives 2 and 4. | ||
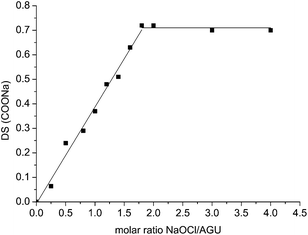 | ||
| Fig. 3 Influence of the molar ratio of the oxidative agent NaOCl per glucose unit on the DS of COOH groups in resulting polymer 2. | ||
The anionic polymers 2 were partially conjugated at the carboxylate groups with N-(6-aminohexyl)-pteroylamide 3. The latter was synthesized by reaction of pyrofolic acid39 and 1,6-diaminohexane. The pteroyl group is known to be a specific ligand for cancer cells similar to folate groups, especially if it is conjugated via a spacer group.46 The pteroyl derivative was chosen since its conjugation chemistry was easier to perform than the one of folate. The conjugation was performed in DMSO solution at 50 °C with N,N′-dicyclohexylcarbodiimide DCC and 1-hydroxybenzotriazole HOBt as the coupling agents.
The DS of pteroate groups conjugated to the copolymer backbone was determined through its absorption band at λ = 280 nm (ε = 23400 M−1 cm−1) in aqueous solution. DS values per repeat unit (Table 1) ranged from 0.008 to 0.25 in copolymer 4 depending on the stoichiometric ratio of pteroyl derivative 3 over the carboxylate groups of copolymer 2, ranging from 0.1 to 0.5. Solubilities of the products in water decreased with increasing DS of pteroyl groups. Polymer 4d with DSpteroate = 0.25 was only sparingly water soluble, while polymers with DSpteroate > 0.25 were insoluble in water. The pteroyl moiety resembles the guanidine group which forms strong intermolecular hydrogen bonds, so-called G-quartets.47 Therefore polymer 4b with a low DSpteroate = 0.023 was chosen for the following experiments to avoid aggregation in water.
| Molar ratio ligand/COOH | DS (pteroate) | |
|---|---|---|
| 4a | 0.1 | 0.008 |
| 4b | 0.2 | 0.023 |
| 4c | 0.3 | 0.077 |
| 4d | 0.5 | 0.250 |
| 4e | 1.0 | Insoluble |
Synthesis of the cationic CD derivative 1 and its loading with DHA
Heptakis(6-deoxy-6-aminoethylsulfanyl)-β-CD 1 was synthesized from heptakis(6-deoxy-6-iodo)-β-CD according to literature and saturated with DHA in aqueous solution at 25 °C. Almost 20% of the cavities of 1 were occupied by DHA, as determined by UV-vis spectroscopy from the absorption peak of DHA at λ = 480 nm (ε = 8240 M−1 cm−1).Assembly and characterization of nanoparticles
Nanoparticles were obtained by mixing 0.1 wt% aqueous solutions of the anionic polymers 2 and 4 each with the 0.1 wt% aqueous solution of the cationic CD derivate 1 under high speed agitation with an ultra turrax stirrer. The speed of stirring had no influence on the final particle size. Furthermore, the particles were stable for one week, leading to the conclusion that the formation of the particles is thermodynamically controlled. For various molar ratios of the components slightly opaque stable dispersions were obtained. The particle size distributions were measured by dynamic light scattering DLS.Most particle size distributions were monomodal with polydispersity indices 0.05 < PDI < 0.3. Broad bimodal size distributions were found when the charge ratio a/c was higher than 10. For both polymers 2 and 4 the obtained main particle diameter d depended on the stoichiometric ratio a/c of anionic groups over cationic groups of the constituents as shown in Fig. 4. The smallest particle sizes d and the lowest PDIs were found around a/c = 1, see Fig. 4. Also the ζ potentials of the nanoparticles, shown in Fig. 4, varied with increasing charge ratio a/c from positive to negative. The negative value of the ζ potential at a/c = 1 might be due to incomplete uptake of 1 in the nanoparticles. The charge ratio was fixed at a/c = 1.2 in the following investigations to make sure that the resulting particles were anionic which was later important in order to avoid unspecific interactions with proteins and cells in living systems. Scanning force microscopy (AFM) investigations revealed that the particles were nearly spherically shaped (Fig. 5).
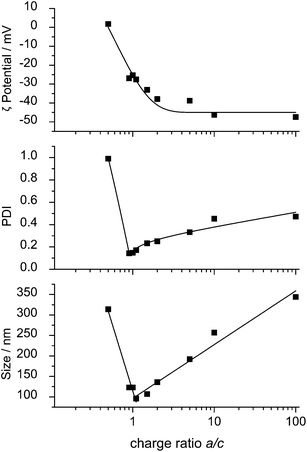 | ||
| Fig. 4 ζ Potential, PDI and particle size d of nanoparticles from 1 and 4 as a function of the charge ratio a/c. | ||
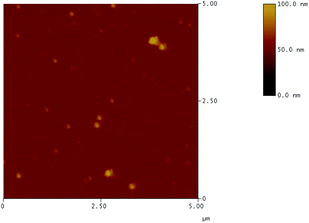 | ||
| Fig. 5 AFM image of nanoparticles 1·4b. | ||
Long term stability studies performed with those nanoparticle dispersions (a/c = 1.2) did not show any precipitation nor any significant (±10%) changes in both d and PDI during a week of observation at rt. A possible reason for the observed stability of the nanoparticle dispersions could be the structural similarity of the anionic starch polymers 2 or 4 and the cationic CD derivative 1 both consisting of α(1 → 4) linked glucose units. The structural similarity could allow the polymer chain wrapping around the CD derivative as depicted in Fig. 6. Futhermore, it should be taken into account that these charged molecules are rather stiff like the so-called “structural counterions” already described by Gröhn et al.32 Interaction of rigid counterions of finite sizes can cause geometrical constraints and thereby incomplete charge neutralization which results in finite-diameter bundles as Pincus already suggested in a theoretical consideration.48
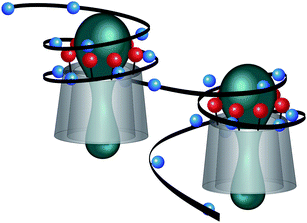 | ||
| Fig. 6 Fragment of the possible structure of the nanoplexes from 1 and 4, positive charges are shown in red, negative charges in blue. | ||
Consequently, the stability and size of the nanoplexes should depend on the number of possible Coulomb interactions per polymer molecule, which is controlled by the DS of the anionic carboxylate groups attached to the polymer. Therefore we determined the particle sizes d for various DS(COOH) values of polymer 2 for a constant a/c = 1.2, shown in Fig. 7, and found that the particle size d indeed increases with decreasing DS(COOH). Precipitation occurred for DS < 0.29. Furthermore we investigated the interaction of the mono-cationic mono-6-deoxy-6-aminoethylsulfanyl-β-CD, with oxidized starch 2 (DS = 0.65) and could not detect any formation of defined nanoparticles by both turbidity and DLS. Therefore hepta-cationic CD derivative 1 and starch derivatives 2 and 4 with high DS > 0.6 were chosen for the following experiments to achieve particles as small as possible, suitable for entering cells.
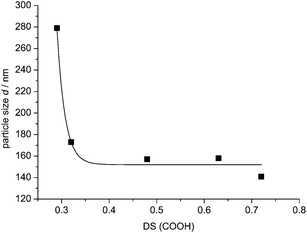 | ||
| Fig. 7 Particle size d of nanoparticles 1·2 for various DS(COOH) values at constant a/c = 1.2. | ||
Loading the nanoplexes with the model drug DHA
After loading CD derivate 1 with the guest DHA, nanoparticles were formed with polymers 2 and 4, respectively. Inclusion of the guest DHA had no effect on both the particle size (shown in Table 2) and stability. The nanoparticle dispersions were slightly opaque with orange color due to the presence of DHA. No precipitation was visible within one week at 25 °C. The dispersions were characterized by UV-vis spectroscopy (Fig. 8). The nanoplexes of DHA complexed in 1 with polyanion 2 and 4, 1(DHA)·2 and 1(DHA)·4, still showed the absorption peak at λ = 480 nm superimposed with the typical turbidity curve of the nanoplexes. In general, turbidity due to particle scattering strongly increases with decreasing wavelength according to Rayleigh's law. Turbidity also increases with both particle size and concentration.49,50 Aqueous solutions of the constituents 1, DHA, 2 and 4 by themselves did not show any turbidity.| Size d/nm | PDI | |
|---|---|---|
| 1·2 | 113 | 0.116 |
| 1(DHA)·2 | 122 | 0.139 |
| 1·4 | 151 | 0.122 |
| 1(DHA)·4 | 154 | 0.101 |
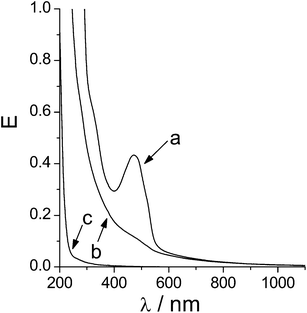 | ||
| Fig. 8 UV-vis spectra of (a) nanoparticles 1(DHA)·2, (b) nanoparticles 1·2 and (c) 2. | ||
After subtraction of the turbidity spectrum of the empty nanoparticles 1·2 from the spectrum of 1(DHA)·2 the spectrum of DHA remained. This way the concentration of DHA in the nanoplex dispersion could be calculated assuming the same extinction coefficient (ε = 8240 M−1 cm−1) like in solution. Indeed the employed initial concentration of DHA was found again (see Table 3) showing that the assumption was right. The nanoplexes 1(DHA)·2 and 1(DHA)·4 could be separated from the solution by extensive centrifugation. The nanoplexes settled down as a solid red spot, insoluble in both water and 1-octanol, solvents for 1, 2, 4, and DHA, respectively. The supernatant after centrifugation did not show any turbidity but only a slight absorption of DHA (see Fig. 9), showing that nearly all DHA could be removed from the aqueous phase. Results from the UV-vis investigations are compiled in Table 3. It was found, that more DHA (88%) was bound in the nanoparticles 1(DHA)·4 compared with 1(DHA)·2 (56%). The slightly larger particle size of 1(DHA)·4 was made responsible for its higher loading efficiency.
| c DHA total/µM | c DHA in supernatant/µM | Loading efficiency (%) | |
|---|---|---|---|
| 1(DHA)·2 | 37.8 | 16.8 | 56 |
| 1(DHA)·4 | 33.6 | 4.0 | 88 |
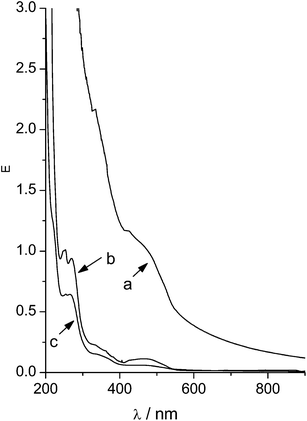 | ||
| Fig. 9 UV-vis spectra of (a) nanoparticles 1(DHA)·4, (b) supernatant of nanoparticles 1(DHA)·4 after 30 min centrifugation and (c) supernatant of nanoparticles 1(DHA)·4 after 20 h centrifugation. | ||
Fluorescence correlation spectroscopy of DHA nanoplexes
FCS measurements were performed to unambiguously prove that the DHA molecules are trapped in the nanoplex dispersions. Autocorrelation traces were recorded from the nanoparticles. They could be described by solely one diffusing species (see Fig. 10). From comparison with the diffusion of green fluorescent protein, a hydrodynamic diameter of 140 ± 30 nm was determined for nanoparticles 1(DHA)·2 in good agreement with the outcome of the DLS experiments.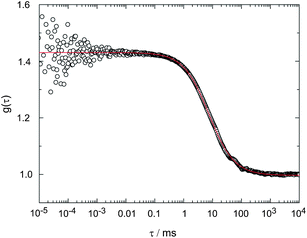 | ||
| Fig. 10 FCS-trace of the nanoparticles 1(DHA)·4 (open symbols) with the fit curve (line) according to the two-dimensional diffusion model. The extracted diffusion time τdiff is 7.4 ms. | ||
Conclusions
We found a new modular concept for the formation of nanoparticles from poorly soluble drugs using readily available building blocks. Organic solvents, emulsifiers and other reagents harmful for biological systems could be avoided completely for the use in nanoparticle formulation. Since there are many choices of cationic CD derivatives and anionic polymers there are plenty of combinations possible to adapt those nanoplexes to a desired task. Release of a complexed drug might be triggered by pH like already shown for other polyelectrolyte complexes.51 Biological studies have to be performed now to show whether the nanoplexes loaded with a drug can enter cancer cells selectively sparing healthy cells.Acknowledgements
This work was supported by the Federal Ministry of Education and Research BMBF, Germany, project number 13N9133. The authors thank A. Engelke and T. Stauner for assistance.References
- G. Wenz, Adv. Polym. Sci., 2009, 222, 1–54 CAS.
- G. Wenz, Angew. Chem., Int. Ed. Engl., 1994, 33, 803–822 CrossRef.
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J.v. Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M.-Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265–270 CrossRef CAS.
- V. Zia, R. A. Rajewski and V. J. Stella, Pharm. Res., 2001, 18, 667–673 CrossRef CAS.
- G. Wenz, C. Strassnig, C. Thiele, A. Engelke, B. Morgenstern and K. Hegetschweiler, Chem.–Eur. J., 2008, 14, 7202–7211 CrossRef CAS.
- J. M. G. Fernandez, C. O. Mellet, S. Maciejewski and J. Defaye, Chem. Commun., 1996, 2741–2743 RSC.
- C. Thiele, D. Auerbach, G. Jung and G. Wenz, J. Inclusion Phenom. Mol. Recognit. Chem., 2010 DOI:10.1007/s10847-010-9741-4.
- A. Clementi, M. C. Aversa, C. Corsaro, J. Spooren, R. Stancanelli, C. O'Connor, M. McNamara and A. Mazzaglia, J. Inclusion Phenom. Macrocyclic Chem., 2010 DOI:10.1007/s10847-010-9738-z.
- Y. Oda, N. Kobayashi, T. Yamanoi, K. Katsuraya, K. Takahashi and K. Hattori, Med. Chem., 2008, 4, 244–255 CrossRef CAS.
- J. M. Benito, M. Gomez-Garcia, C. Ortiz Mellet, I. Baussanne, J. Defaye and J. M. G. Fernandez, J. Am. Chem. Soc., 2004, 126, 10355–10363 CrossRef CAS.
- M. Ortega-Munoz, J. Morales-Sanfrutos, F. Perez-Balderas, F. Hernandez-Mateo, M. D. Giron-Gonzalez, N. Sevillano-Tripero, R. Salto-Gonzalez and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2007, 5, 2291–2301 RSC.
- C. Pean, A. Wijkhuisen, F. Djedaini-Pilard, J. Fischer, S. Doly, M. Conrath, J.-Y. Couraud, J. Grassi, B. Perly and C. Creminon, Biochim. Biophys. Acta, Mol. Cell Res., 2001, 1541, 150–160 CrossRef CAS.
- C. Pean, C. Creminon, A. Wijkhuisen, J. Grassi, P. Guenot, P. Jehan, J.-P. Dalbiez, B. Perly and F. Djedaini-Pilard, J. Chem. Soc., Perkin Trans. 2, 2000, 853–863 RSC.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS.
- H. Maeda, K. Greish and J. Fang, Adv. Polym. Sci., 2006, 193, 103–121 CAS.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, Adv. Polym. Sci., 2006, 193, 1–65 CAS.
- M. J. Vicent and R. Duncan, Trends Biotechnol., 2006, 24, 39–47 CrossRef CAS.
- S. H. Kim, J. H. Jeong, K. W. Chun and T. G. Park, Langmuir, 2005, 21, 8852–8857 CrossRef CAS.
- N. Nafee, M. Schneider and C. M. Lehr, in Multifunctional Pharmaceutical Nanocarriers, 2008, pp. 337–362 Search PubMed.
- R. Solaro, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1–11 CrossRef CAS.
- V. Mailander and K. Landfester, Biomacromolecules, 2009, 10, 2379–2400 CrossRef.
- C. E. Astete and C. M. Sabliov, J. Biomater. Sci., Polym. Ed., 2006, 17, 247–289 CrossRef CAS.
- R. Tong and J. Cheng, Bioconjugate Chem., 2010, 21, 111–121 CrossRef CAS.
- R. Jayakumar, K. P. Chennazhi, R. A. A. Muzzarelli, H. Tamura, S. V. Nair and N. Selvamurugan, Carbohydr. Polym., 2010, 79, 1–8 CrossRef CAS.
- M. J. Santander-Ortega, T. Stauner, B. Loretz, J. L. Ortega-Vinuesa, D. Bastos-González, G. Wenz, U. F. Schaefer and C. M. Lehr, J. Controlled Release, 2010, 141, 85–92 CrossRef CAS.
- E. Memisoglu, A. Bochot, M. Sen, D. Duchene and A. A. Hincal, Int. J. Pharm., 2003, 251, 143–153 CrossRef CAS.
- D. Duchene, G. Ponchel and D. Wouessidjewe, Adv. Drug Delivery Rev., 1999, 36, 29–40 CrossRef CAS.
- A. Ranjan, N. Pothayee, M. Seleem, N. Jain, N. Sriranganathan, J. S. Riffle and R. Kasimanickam, J. Nanopart. Res., 2010, 12, 905–914 CrossRef CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CAS.
- F. Grohn, K. Klein and S. Brand, Chem.–Eur. J., 2008, 14, 6866–6869 CrossRef.
- I. Willerich, H. Ritter and F. Gröhn, J. Phys. Chem. B, 2009, 113, 3339–3354 CrossRef CAS.
- Y. Lin, F. Mi, C. Chen, W. Chang, S. Peng, H. Liang and H. Sung, Biomacromolecules, 2007, 8, 146–152 CrossRef CAS.
- Y. Li, T. K. Bronich, P. S. Chelushkin and A. V. Kabanov, Macromolecules, 2008, 41, 5863–5868 CrossRef CAS.
- J. O. Kim, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2009, 138, 197–204 CrossRef CAS.
- J. O. Kim, N. V. Nukolova, H. S. Oberoi, A. V. Kabanov and T. K. Bronich, Polym. Sci., Ser. A, 2009, 51, 708–718 CrossRef.
- X. P. Qiu, S. Leporatti, E. Donath and H. Mohwald, Langmuir, 2001, 17, 5375–5380 CrossRef CAS.
- N. L. Hoang, A. Landolfi, A. Kravchuk, E. Girard, J. Peate, J. M. Hernandez, M. Gaborieau, O. Kravchuk, R. G. Gilbert, Y. Guillaneuf and P. Castignolles, J. Chromatogr., A, 2008, 1205, 60–70 CrossRef CAS.
- J. Luo, M. D. Smith, D. A. Lantrip, S. Wang and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 10004–10013 CrossRef CAS.
- B. Hinkeldey, A. Schmitt and G. Jung, ChemPhysChem, 2008, 9, 2019–2027 CrossRef CAS.
- R. Rigler, Ü. Mets, J. Widengren and P. Kask, Eur. Biophys. J., 1993, 22, 169–175 CrossRef CAS.
- B. Terry, E. Matthews and J. Haseloff, Biochem. Biophys. Res. Commun., 1995, 217, 21–27 CrossRef CAS.
- P. L. Bragd, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 2000, 328, 355–363 CrossRef CAS.
- P. L. Bragd, H. van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49–66 CrossRef CAS.
- N. Blumenkrantz and G. Asboe-Hansen, Anal. Biochem., 1973, 54, 484–489 CrossRef CAS.
- C. P. Leamon and J. A. Reddy, Adv. Drug Delivery Rev., 2004, 56, 1127–1141 CrossRef CAS.
- N. Sreenivasachary and J. M. Lehn, Chem.–Asian J., 2008, 3, 134–139 CrossRef CAS.
- M. Henle and P. Pincus, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 60801 CrossRef.
- U. Apfel, R. Grunder and M. Ballauff, Colloid Polym. Sci., 1994, 272, 820–829 CrossRef CAS.
- U. Apfel, K. D. Horner and M. Ballauff, Langmuir, 1995, 11, 3401–3407 CrossRef CAS.
- M. Soliman, S. Allen, M. C. Davies and C. Alexander, Chem. Commun., 2010, 46, 5421–5433 RSC.
| This journal is © The Royal Society of Chemistry 2011 |