Protein conjugation of thermoresponsive amine-reactive polymers prepared by RAFT†
Hongmei
Li
,
Abhijeet P.
Bapat
,
Ming
Li
and
Brent S.
Sumerlin
*
Department of Chemistry, Southern Methodist University, Dallas, Texas, USA. E-mail: bsumerlin@smu.edu; Fax: +1-214-768-4089; Tel: +1-214-768-8802
First published on 31st August 2010
Abstract
Reversible addition–fragmentation chain transfer (RAFT) of N-isopropylacrylamide with a chain transfer agent containing an activated-ester moiety was employed to prepare well-defined temperature-responsive polymers for conjugation to amine-containing proteins. Bioconjugation efficiency of the polymers with lysozyme was investigated as a function of stoichiometry, polymer molecular weight, and reaction pH. The resulting polymer–protein conjugates retained the temperature-responsive nature of the original polymers.
Introduction
Covalent conjugation to synthetic polymers is a facile method to modify the stability, activity, biocompatibility, or solubility of many biomacromolecules. Specifically, polymer–protein conjugates have demonstrated promise in a variety of applications, including drug delivery,1,2 biocatalysis,3,4 and biosensing.5,6 Because of its hydrophilicity and biocompatibility, poly(ethylene glycol) (PEG) is the polymer most commonly employed for the modification of proteins for therapeutic applications. Conjugates containing a responsive (smart) synthetic component with properties that can be tuned by applying an appropriate stimulus are also of interest.7,8 For instance, polymers that alter their solubility or propensity for self-assembly when exposed to changes in pH or temperature allow their responsive nature to be conferred to the protein to which they are attached. Controlled radical polymerization9–14 has proven to be especially useful during the construction of such polymer–protein conjugates.15,16 In particular, atom transfer radical polymerization (ATRP)4,17–23 and reversible addition–fragmentation chain transfer (RAFT)24–32 have been successfully employed to prepare conjugates by either a grafting-to or grafting-from approach.The diversity of functional groups present in proteins allows a wide variety of chemistries to be employed during polymer conjugation.33 Free thiols on cysteine residues and amines at the N-terminus or on lysine residues are common targets. Thiols are typically functionalized by Michael addition with α,β-unsaturated carbonyl compounds,10,11,18,34–45 or by thiol-disulfide exchange.17,46–58 However, not all proteins contain free thiols in their native state. On the other hand, amine residues are generally plentiful, and conjugationviaε-lysine residues or N-termini is a more widely applicable method. For example, Pound et al. modified the ω-end of poly(N-vinylpyrrolidone) to contain aldehyde groups, and conjugated the resulting polymers to protein amines.59Polymers functionalized with activated esters60–70 are also capable of reacting with protein amines. N-Hydroxysuccinimidyl (NHS) esters have been most heavily considered, though several other convenient activated esters have received recent attention.60,61,63,64,66–70Polymers prepared from NHS-functionalized ATRP initiators and RAFT chain transfer agents (CTAs) have been functionalized with a variety of small molecule and (bio)macromolecular amines. For instance, D'Agosto, Charreyre, and coworkers first reported that NHS-RAFT agents can be selectively reacted with primary amines at the activated ester site with retention of the thiocarbonylthio moiety.71 Stenzel et al. prepared stars by immobilizing NHS-terminated polymers to lysozyme in dimethyl sulfoxide.72 Kiick and Theato et al. prepared poly(diethylene glycol methyl ether methacrylate) with a pentafluorophenyl activated ester RAFT agent and conjugated the polymer to amine groups on collagen-like peptides.64
Here, we report an NHS-containing trithiocarbonate RAFT agent that can be employed to prepare polymers for selective functionalization of protein amines in water. Poly(N-isopropylacrylamide) (PNIPAM), a thermoresponsive polymer with a lower critical solution temperature (LCST) of approximately 32 °C, was prepared with this CTA, and its conjugation to lysozyme (LYS) in aqueous media led to conjugates that retained the responsive solution behavior of the polymer. Control of solution pH during the conjugation reaction allowed control over the multiplicity of immobilized chains.
Experimental
Materials
2,2′-Azobisisobutyronitrile (AIBN, 98%, Aldrich) was recrystallized from ethanol, and N-isopropylacrylamide (NIPAM, TCI America) was recrystallized from hexanes. Lysozyme (LYS, MP Biochemicals), N-hydroxylsuccinimide (98%, Aldrich), and N,N′-dicyclohexylcarbodiimide (DCC, PIERCE) were used as received. All other chemicals were purchased from VWR or Fisher and used without further purification. Dialysis membranes were purchased from Fisher (MWCO = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 14000 Da) and Spectrum Laboratories (MWCO = 25000 Da).
000 and 14000 Da) and Spectrum Laboratories (MWCO = 25000 Da).
Instrumentation
PNIPAM molecular weight and polydispersity were characterized by size exclusion chromatography (SEC) in N,N-dimethylformamide (DMF) with 50 mM LiBr at 55 °C using a flow rate of 1.0 mL min−1 (Viscotek SEC pump, columns: ViscoGel I-series G3000 and G4000 mixed bed columns: molecular weight range 0–60 × 103 and 0–400 × 103 g mol−1, respectively). Detection consisted of a Viscotek refractive index detector operating at 660 nm, and a Viscotek model 270 series platform with a laser light scattering detector (operating at 3 mW, 670 nm with detection angles of 7° and 90°) and a four-capillary viscometer. Molecular weights were determined by the triple detection method using a dn/dc = 0.077 mL g−1 for PNIPAM. PNIPAM–lysozyme conjugates were characterized by SEC in an aqueous eluent (0.5 w/v% aqueous NaN3) at 25 °C with a flow rate of 0.7 mL min−1 (Viscotek SEC pump, column: Biosep-SEC-S 3000). Detection consisted of a Viscotek refractive index detector operating at 660 nm and a Viscotek UV-Vis detector operating at 280 nm. UV-Vis spectroscopic measurements were conducted with a Beckman Coulter DU Series 800 spectrophotometer equipped with a Peltier temperature controller. 1H-NMR spectroscopy was conducted with a JEOL Delta 500 spectrometer at 500 MHz. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a Bio-Rad electrophoresis apparatus with a resolving gel containing 12% polyacrylamide.Synthesis of S-butyl-S′-(α,α′-dimethyl-α″-acetic acid)-trithiocarbonate CTA (1)
Butanethiol (18.0 g, 0.200 mol), acetone (96.4 g, 1.66 mol), and trioctylmethylammonium chloride (Aliquat 336, 3.23 g, 8.00 mmol) were mixed in a round-bottom flask, and the mixture was cooled below 10 °C under a nitrogen atmosphere. A solution of sodium hydroxide (50 w/v%) (16.8 g, 0.210 mol) was slowly added over 20 min. The reaction was stirred for an additional 20 min. A solution of carbon disulfide (15.2 g, 0.200 mol) in acetone (20.3 g, 0.350 mol) was added over a 20 min period. After 20 min, chloroform (35.8 g, 0.300 mol) was added, followed by dropwise addition of a solution of sodium hydroxide (50 w/v%, 80.0 g, 1.00 mol) over 30 min. The reaction mixture was stirred overnight at room temperature. Acetone was removed viarotary evaporation, and the residue was dissolved in water (250 mL) and cooled below 10 °C. Concentrated HCl was added dropwise during vigorous stirring to reach a pH of 2. The aqueous solution was extracted with hexane (×4), concentrated viarotary evaporation, and washed (×3) with water. The product was isolated by column chromatography (silica gel 240–400 mesh) using hexane:ethyl acetate (95:5 v/v) as an eluent. The product was obtained by recrystallization from hexanes, drying under vacuum, and collection in the form of yellow crystals. The crystals were dried by evaporation of trace solvent under vacuum (3.5 g, 7% yield). The product was characterized by 1H- and 13C-NMR spectroscopy and elemental analysis. 1H-NMR (500 MHz, ppm, CDCl3): 3.27 (t, 2H, –CH2–S), 1.71 (s, 6H, (CH3)2–C–S), 1.65 (q, 2H, –CH2–CH2–CH2–CH3), 1.41 (m, 2H, –CH2–CH2–CH2–CH3), 0.91 (t, 3H, –CH2–CH2–CH2–CH3). 13C-NMR (125.8 MHz, ppm, CDCl3): 220.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 171.8 (–COOH), 55.3 (–C(CH3)2), 37.0 (CH3–CH2–CH2–CH2–S), 29.9 (CH3–CH2–CH2–CH2–S), 25.5 (–C(CH3)2), 22.3 (CH3–CH2–CH2–CH2–S), 13.4 (CH3–CH2–CH2–CH2–S). Elemental analysis: calculated for C9H16O2S3: C = 42.82%, H = 6.39%. Found: C = 42.47%, H = 6.30%.
S), 171.8 (–COOH), 55.3 (–C(CH3)2), 37.0 (CH3–CH2–CH2–CH2–S), 29.9 (CH3–CH2–CH2–CH2–S), 25.5 (–C(CH3)2), 22.3 (CH3–CH2–CH2–CH2–S), 13.4 (CH3–CH2–CH2–CH2–S). Elemental analysis: calculated for C9H16O2S3: C = 42.82%, H = 6.39%. Found: C = 42.47%, H = 6.30%.
Synthesis of the N-hydroxylsuccinimide-containing CTA (2)
The NHS RAFT agent (2) was prepared as follows. CTA 1 (400 mg, 1.59 mmol) and NHS (183 mg, 1.59 mmol) were added to a round-bottom flask and dissolved in dichloromethane (DCM, 18 mL). The mixture was held at 4 °C and purged with N2 for 30 min. A solution of DCC (327 mg, 1.59 mmol) in DCM (2 mL) was injected, and the mixture was purged with N2 for 20 min. The reaction mixture was kept at 4 °C for 2 h and continued at room temperature for 24 h. The resulting solution was filtered (×2) with a 0.45 nm filter, and the solvent was evaporated under vacuum. The crude product was purified by column chromatography on silica gel with a mobile phase of hexanes:ethyl acetate (1/1, v/v). After recrystallization (×2) from a toluene:hexanes (50:50) mixture, the product was dried under vacuum to yield a solid yellow powder (290 mg, 52% yield). 1H-NMR (500 MHz, ppm, CDCl3): 3.31 (t, 2H, –CH2–CH2–CH2–CH3), 2.80 (duplicate t, 4H, CO–CH2–CH2–CO), 1.86 (s, 6H, (CH3)2–C–S), 1.68 (q, 2H, –CH2–CH2–CH2–CH3), 1.42 (m, 2H, –CH3–CH2–CH2–CH2), 0.92 (t, 3H, –CH2–CH2–CH2–CH3). 13C-NMR (125.8 MHz, ppm, CDCl3): 218.7 (CS), 169.2 (–O–CO–), 168.8 (–N–CO–), 54.4 (–C(CH3)2), 36.8 (CH3–CH2–CH2–CH2–S), 29.9 (CH3–CH2–CH2–CH2–S), 25.7 (–C(CH3)2), 22.2 (CH3–CH2–CH2–CH2–S), 13.6 (CH3–CH2–CH2–CH2–S). Elemental analysis: calculated for C13H19NO4S3: C = 44.68%, H = 5.48%, N = 4.01%. Found: C = 43.91%, H = 5.44%, N = 3.84%.
RAFT polymerization of NIPAM with CTA 2
An example RAFT polymerization procedure is as follows. NIPAM (1.50 g, 13.3 mmol), 2 (54.5 mg, 0.156 mmol), 1,3,5-trioxane (20.0 mg, 0.220 mmol, internal standard), AIBN (2.6 mg, 0.016 mmol), and 1,4-dioxane (4.33 mL) were sealed in a 20 mL vial. After purging with N2 for 15 min, the solution was heated at 60 °C for a predetermined time (Table 1) before being quenched by cooling in an ice water bath and exposing the polymerization solution to air. The reaction solutions were diluted with tetrahydrofuran, and the polymeric product was precipitated into ether and dried under vacuum to yield succinimidyl-ester terminated PNIPAM (NHS–PNIPAM).| Entry | [M]/[CTA]/[I]a | Time/min | Conv.b (%) | M n theory/g mol−1 | M n c/g mol−1 | M w/Mnc |
|---|---|---|---|---|---|---|
| a Molar ratio of monomer (M)/chain transfer agent (CTA)/initiator (I). b Monomer conversion determined by 1H-NMR spectroscopy. c Determined by size exclusion chromatography. | ||||||
| A | 85/1/0.10 | 160 | 49.4 | 5100 | 5100 | 1.10 |
| B | 125/1/0.14 | 141 | 71.1 | 10400 |
10700 |
1.10 |
| C | 208/1/0.17 | 129 | 65.7 | 15800 |
15700 |
1.07 |
Conjugation of NHS–PNIPAM to lysozyme
An example PNIPAM–LYS conjugation procedure is as follows. Hen egg white lysozyme (LYS, 4.0 mg, 2.8 × 10−4 mmol) and NHS–PNIPAM (14.9 mg, 1.4 × 10−3 mmol) were dissolved in phosphate buffer (3 mL, 0.1 M, pH 7.5), and the reaction solution was incubated for 20 h at 25 °C. The progress of the conjugation reaction was monitored by SDS-PAGE. Following the reactions, the conjugate solutions were heated to 40 °C for 5 min and centrifuged at 13200 rpm for 3 min at 40 °C. The resulting precipitate was redissolved in deionized water at room temperature and dialyzed (MWCO = 10, 12–14, or 25 kDa) against deionized water overnight. The resulting product was isolated by lyophilization.
Results and discussion
CTA 1 was reacted with NHS by DCC coupling to give the activated ester CTA 2 (Scheme 1). This CTA successfully controlled the polymerization of NIPAM, leading to well-defined NHS-terminated polymers (3) of 5.1, 10.7, and 15.7 kg mol−1, as determined by SEC (Fig. 1 and Table 1). Efficient retention of the NHS groups during the polymerization and purification was confirmed by 1H-NMR spectroscopy by comparing the signals of the succinimidyl methylene protons (–(CO)–CH2CH2–(CO)–) at δ = 2.86 ppm and the methylene adjacent to the trithiocarbonate (C3H7–CH2–S(CS)S–) in the CTA Z-group at δ = 3.34 ppm. LYS served as a convenient model for conjugation to NHS-functional polymers, as it has seven primary amines, including six lysine residues and the N-terminus. While small molecules containing activated esters can quantitatively react with the amino groups, sterics must be considered when conjugating high molecular weight compounds to the protein in its native state. The conjugation reaction was investigated under a variety of conditions. The coupling efficiency was observed to depend on polymer molecular weight, solution pH, and stoichiometry. Following the reactions, the solutions were heated to 40 °C and centrifuged. Because of the LCST behavior, unreacted PNIPAM and the targeted PNIPAM–LYS conjugates precipitated under these conditions, while the unreacted protein remained in the supernatant. Unreacted polymer was removed from the remaining mixture by dialysis to afford the targeted PNIPAM–LYS conjugates.
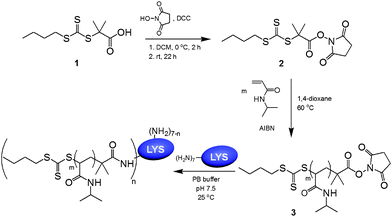 | ||
| Scheme 1 Synthesis of NHS-functionalized chain transfer agent, polymerization of NIPAM, and conjugation of the resulting amine-reactive PNIPAM to lysozyme. | ||
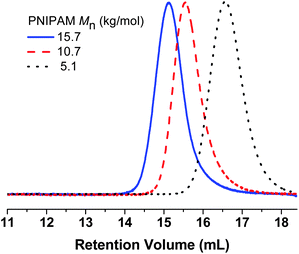 | ||
| Fig. 1 Organic SEC refractive index traces of NHS-terminated PNIPAM prepared from CTA 2. | ||
NHS-terminated PNIPAM of three different molecular weights was reacted with LYS at various stoichiometric ratios. The conjugates were characterized by aqueous SEC (Fig. 2) and SDS-PAGE (Fig. 3). As a control experiment, LYS was also incubated with PNIPAM containing a –COOH end group instead of the activated ester functionality, and under these conditions, no increase in apparent molecular weight was observed. When the reactions with the NHS-terminated polymers were conducted at pH 7.5, the conjugation products (lanes 2–8) demonstrated an increase in molecular weight as compared to native LYS (lane 1, 14.3 kDa), indicating successful conjugate formation (Fig. 3). With NHS-terminated PNIPAM of Mn = 5.1 kg mol−1 and [PNIPAM]:[LYS] = 5:1, the band attributed to PNIPAM–LYS conjugates was relatively broad, which suggested the presence of conjugates with multiple chains attached (Fig. 3, lane 2). On the other hand, using the same stoichiometry for reactions of PNIPAM with Mn = 10.7 and 15.7 kg mol−1 resulted in conjugates with relatively narrow bands and apparent molecular weights consistent with the attachment of single chains, though a small amount of higher order conjugates was also visible (Fig. 3, lanes 3 and 4).
![Aqueous SEC UV traces of the conjugation reaction products of PNIPAM (Mn = 5.1, 10.7, and 15.7 kg mol−1) and LYS at molar ratios of [PNIPAM]/[LYS] = 5/1 and 10/1.](/image/article/2011/PY/c0py00178c/c0py00178c-f2.gif) | ||
| Fig. 2 Aqueous SEC UV traces of the conjugation reaction products of PNIPAM (Mn = 5.1, 10.7, and 15.7 kg mol−1) and LYS at molar ratios of [PNIPAM]/[LYS] = 5/1 and 10/1. | ||
![SDS-PAGE results from the reactions of NHS-terminated PNIPAM with LYS (pH = 7.5, t = 20 h). [Native LYS (lane 1), conjugates from the reactions with NHS–PNIPAM of Mn = 5.1 kg mol−1 (lanes 2, 5, and 8), 10.7 kg mol−1 (lanes 3 and 6), and 15.7 kg mol−1 (lanes 4 and 7).] Negative control reaction of HO2C–PNIPAM (Mn = 10 kg mol−1) with LYS (pH = 7.5, t = 120 h) (lane 9). Reactions were conducted with [PNIPAM] : [LYS] = 5 : 1 (lanes 2–4) and 10 : 1 (lanes 5–7 and 9) and 100 : 1 (lane 8).](/image/article/2011/PY/c0py00178c/c0py00178c-f3.gif) | ||
| Fig. 3
SDS-PAGE results from the reactions of NHS-terminated PNIPAM with LYS (pH = 7.5, t = 20 h). [Native LYS (lane 1), conjugates from the reactions with NHS–PNIPAM of Mn = 5.1 kg mol−1 (lanes 2, 5, and 8), 10.7 kg mol−1 (lanes 3 and 6), and 15.7 kg mol−1 (lanes 4 and 7).] Negative control reaction of HO2C–PNIPAM (Mn = 10 kg mol−1) with LYS (pH = 7.5, t = 120 h) (lane 9). Reactions were conducted with [PNIPAM]:[LYS] = 5:1 (lanes 2–4) and 10:1 (lanes 5–7 and 9) and 100:1 (lane 8). | ||
For PNIPAM of 5.1 kg mol−1, increasing the concentration of polymer during the reaction with LYS seemingly resulted in conjugates with more than one chain attached. For instance, increasing the ratio of [PNIPAM]:[LYS] to 10:1 yielded a band of higher molecular weight than was observed for the previous reaction with [PNIPAM]:[LYS] = 5:1 (compare lane 2 to lane 5 in Fig. 3). In fact, the higher ratio of polymer to protein with this 5.1 kg mol−1polymer demonstrated a remarkably similar shift in the SDS-PAGE to that of the conjugate with the 10.7 kg mol−1polymer. This result may be consistent with two chains of the smaller polymer conjugating with the protein. On the other hand, increasing the ratio of the higher molecular weight polymers seemed to have no significant effect on the products obtained. With PNIPAM of 10.7 and 15.7 kg mol−1, ratios of [PNIPAM]:[LYS] = 10:1 and 5:1 yielded similar results, as seen by comparing lanes 3 and 4 to lanes 6 and 7 in Fig. 3. These results may elucidate the role of sterics during the conjugation reactions. While increasing the ratio of polymer to protein results in enhanced conjugation efficiency for low molecular weight polymers, relatively little advantage is gained by employing higher ratios with larger polymers, presumably because their sterics limit access of additional chains to unreacted amines. Dramatically increasing the ratio of [PNIPAM]:[LYS] to 100:1 did not significantly increase the number of conjugated chains, even with the low molecular weight polymer (Fig. 3, lane 8), though there was an apparent increase in conjugation efficiency.
The effect of pH during the conjugation reaction was also investigated. Because the basicity of the amino group at the N-terminus (pKa = 7.6–8.0)73 is considerably lower than that of the ε-amino group of the lysine residues (pKa = 10.5), control of the reaction pH can favor deprotonation of the terminal amine, potentially allowing site-specific conjugation.74 To investigate the effect of pH, NHS-terminated PNIPAM was mixed with LYS at pH 7.5, 8.0, and 9.0. The conjugate products obtained after 5 days under each condition were investigated by SDS-PAGE (Fig. 4). Samples were taken at various times, and the conjugation reaction was observed to be essentially complete after 24 h. No significant difference was observed in terms of conjugation efficiency by increasing the pH from 7.5 to 8.0. However, reactions conducted at pH 9.0 clearly indicated a mixture of products containing multiple conjugated chains (Fig. 4, lanes 3, 6, and 9). In addition to enhancing conjugation efficiency, increased pH also led to accelerated reaction kinetics. This behavior may indicate preferential functionalization of the N-terminus at low pH and functionalization with multiple chains when the reactions are conducted at high pH conditions at which lysine amino groups are also significantly deprotonated. Interestingly, the attachment of multiple chains at pH 9.0 occurred even with the higher molecular weight polymers. This may indicate a more complex relationship between sterics and conjugation efficiency, with pH also playing a significant role. It should be noted that conducting reactions at pH greater than 9.0 was complicated by rapid hydrolysis of the NHS polymer end groups.
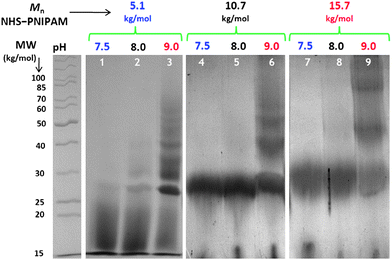 | ||
| Fig. 4 SDS-PAGE results from the reactions of NHS-terminated PNIPAM with LYS in phosphate buffer at pH 7.5 (lanes 1, 4, and 7), 8.0 (lanes 2, 5, and 8), and 9.0 (lanes 3, 6, and 9). Reactions were conducted with NHS–PNIPAM with Mn = 5.1 kg mol−1 (1–3), 10.7 kg mol−1 (4–6), and 15.7 kg mol−1 (7–9). | ||
Conclusions
Using the methods reported here, a wide variety of proteins should be capable of being modified with well-defined polymers prepared by RAFT polymerization. During the conjugation of NHS-terminated PNIPAM with LYS, the efficiency of conjugation was dependent on several factors, including polymer molecular weight, stoichiometry, and pH. Therefore, careful selection of the reaction conditions may allow tailored synthesis of other polymer–protein conjugates. Currently we are investigating regulation of protein bioactivity by exploiting the stimuli-responsive nature of the immobilized polymer.Acknowledgements
This material is based upon work supported by the National Science Foundation (CAREER DMR-0846792) and an Alfred P. Sloan Research Fellowship (BSS).References
- R. Duncan, H. Ringsdorf and R. Satchi-Fainaro, J. Drug Targeting, 2006, 14, 337–341 Search PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- F.-Y. Jeng and S.-C. Lin, Process Biochem., 2006, 41, 1566–1573 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697–4699 RSC.
- A. M. Dattelbaum, G. A. Baker, J. M. Fox, S. Iyer and J. D. Dattelbaum, Bioconjugate Chem., 2009, 20, 2381–2384 CrossRef CAS.
- E. G. R. Fernandes and A. A. A. Queiroz, J. Mater. Sci.: Mater. Med., 2009, 20, 473–479 CrossRef CAS.
- A. S. Hoffman, J. Controlled Release, 2008, 132, 153–163 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148–2150 RSC.
- L. Tao, C. S. Kaddis, R. R. V. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028–8033 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650–7656 CrossRef CAS.
- H. D. Maynard, K. L. Heredia, R. C. Li, D. P. Parra and V. Vazquez-Dorbatt, J. Mater. Chem., 2007, 17, 4015–4017 RSC.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, ACS Nano, 2008, 2, 757–765 CrossRef CAS.
- B. L. Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207–2212 CrossRef CAS.
- Z. P. Tolstyka, J. T. Kopping and H. D. Maynard, Macromolecules, 2008, 41, 599–606 CrossRef CAS.
- R. M. Broyer, G. M. Quaker and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 1041–1047 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916–1922 RSC.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004, 2026–2027 RSC.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- L. Tao, J. Liu, J. Xu and T. P. Davis, Org. Biomol. Chem., 2009, 7, 3481–3485 RSC.
- L. Tao, J. Liu, J. Xu and T. P. Davis, Chem. Commun., 2009, 6560–6562 RSC.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- J. Hwang, R. C. Li and H. D. Maynard, J. Controlled Release, 2007, 122, 279–286 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Mueller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736–2741 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 1996 Search PubMed.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360–2367 CrossRef CAS.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS.
- E. Bays, L. Tao, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777–1781 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- K. L. Christman, E. Schopf, R. M. Broyer, R. C. Li, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2009, 131, 521–527 CrossRef CAS.
- C. Da Pieve, P. Williams, D. M. Haddleton, R. M. J. Palmer and S. Missailidis, Bioconjugate Chem., 2010, 21, 169–174 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- J. Liu, H. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899–912 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830–847 CrossRef CAS.
- J. Liu, H. Liu, Z. Jia, V. Bulmus and T. P. Davis, Chem. Commun., 2008, 6582–6584 RSC.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS.
- J. Liu, H. Liu, V. Bulmus, L. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399–1405 CrossRef CAS.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS.
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699–2707 CrossRef CAS.
- Z. Jia, J. Liu, C. Boyer, T. P. Davis and V. Bulmus, Biomacromolecules, 2009, 10, 3253–3258 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- C. L. Duvall, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Mol. Pharmaceutics, 2010, 7, 468–476 CrossRef CAS.
- S. Flanary, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 241–248 CrossRef CAS.
- G. Pound, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Chem. Commun., 2008, 3193–3195 RSC.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS.
- P. J. Roth, M. Haase, T. Basche, P. Theato and R. Zentel, Macromolecules, 2010, 43, 895–902 CrossRef CAS.
- D. Kessler and P. Theato, Langmuir, 2009, 25, 14200–14206 CrossRef CAS.
- D. Kessler, P. J. Roth and P. Theato, Langmuir, 2009, 25, 10068–10076 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860–3863 CrossRef CAS.
- M. Barz, M. Tarantola, K. Fischer, M. Schmidt, R. Luxenhofer, A. Janshoff, P. Theato and R. Zentel, Biomacromolecules, 2008, 9, 3114–3118 CrossRef CAS.
- P. Theato, J.-U. Kim and J.-C. Lee, Macromolecules, 2004, 37, 5475–5478 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- K. Nilles and P. Theato, Eur. Polym. J., 2007, 43, 2901–2912 CrossRef CAS.
- M. Eberhardt, R. Mruk, R. Zentel and P. Theato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- M. Eberhardt and P. Theato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef CAS.
- M. Bathfield, F. D'Agosto, R. Spitz, M.-T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546–2547 CrossRef CAS.
- L. McDowall, G. Chen and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 1666–1671 CrossRef CAS.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- I. Selo, L. Negroni, C. Creminon, J. Grassi and J. M. Wal, J. Immunol. Methods, 1996, 199, 127–138 CrossRef CAS.
Footnote |
| † This article is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |