Selective partial hydrolysis of amphiphilic copoly(2-oxazoline)s as basis for temperature and pH responsive micelles†
Hanneke M. L.
Lambermont-Thijs
ab,
Johan P. A.
Heuts
c,
Stephanie
Hoeppener
d,
Richard
Hoogenboom
*ae and
Ulrich S.
Schubert
abd
aLaboratory of Macromolecular Chemistry and Nanoscience, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
bDutch Polymer Institute (DPI), John F. Kennedylaan 2, 5612 AB, Eindhoven, The Netherlands
cLaboratory of Polymer Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
dLaboratory of Organic and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldtstr. 10, 07743, Jena, Germany
eInstitute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: r.hoogenboom@tue.nl
First published on 21st May 2010
Abstract
The acidic and basic hydrolyses of gradient and diblock copolymers based on 2-methyl-2-oxazoline (MeOx) and 2-phenyl-2-oxazoline (PhOx) were investigated. Various reaction times were examined revealing polymers with varying ratios of PMeOx, PPhOx and poly(ethylene imine) (PEI). It could be shown that under acidic conditions, PMeOx as well as PPhOx are readily cleaved while under basic conditions PPhOx was almost not hydrolyzed leading to higher selectivity. However, partial degradation of the polymers occurred under basic conditions as evidenced by SEC. Thermal investigations of the polymers cleaved under acidic conditions revealed that most obtained copolymers exhibited a melting temperature due to the large PEI content. Self-assembly studies revealed that the partially hydrolyzed copolymers formed micelles at both ambient and elevated temperatures in acidic medium due to protonation of the ethylene imine units leading to good solubility. In contrast, the copolymers were insoluble at ambient temperature in water or basic medium, but self-assembled into spherical micelles at elevated temperatures as evidenced by transmission electron microscopy and dynamic light scattering. As such, these novel PEI–PPhOx copolymers exhibit thermoresponsive micellization behavior based on the crystallization induced phase separation of linear PEI at lower temperatures. Moreover, the copolymer consisting of 84 ethylene imine repeat units, 16 PPhOx groups also revealed pH responsive micellization due to increased solubility of the ethylene imine units upon protonation in acidic solution.
Introduction
The formation of polymeric aggregates such as micelles or vesicles, based on amphiphilic block copolymers, has received increasing attention for possible applications in, e.g., aqueous self-assembly, micellar catalysis, drug delivery and hydrogels.1–13 Poly(2-oxazoline)s represent an excellent class of biocompatible functional polymers. The cationic ring opening polymerization of 2-oxazolines leads to well-defined polymers with narrow molar mass distribution.14–16 Due to the ease and versatility to vary the side group of poly(2-oxazoline)s, the properties of amphipilic diblock and triblock poly(2-oxazoline)s in solution have been studied in detail.17–19 Short alkyl side groups, like methyl or ethyl, result in water soluble poly(2-oxazoline)s while longer or aromatic side groups are more hydrophobic and lead to water insoluble polymers.20,21 Moreover, a large number of studies discuss structure–property relationships for poly(2-oxazoline)s using systematic variations in monomer composition.22–24It is well known that poly(2-oxazoline)s can be used as precursor for the synthesis of linear poly(ethylene imine) (PEI) via acidic25 or basic26hydrolysis. Linear PEI is known to exhibit interesting solubility properties since it is soluble in water at elevated temperatures and insoluble in water at room temperature.27 This specific behavior is ascribed to a phase separation induced by crystallization of linear PEI.28 At low temperatures, the unsubstituted flexible polymer chains favor ordered packing in hydrated crystals while at elevated temperature the increased chain mobility causes a melting of the crystals and, therefore, dissolution.29 In literature, several block copolymers are reported combining these interesting properties of PEI with other common polymers. Litt and Lin, e.g., reported the synthesis of diblock and triblock copolymers containing poly(N-butylbenzoyl ethylene imine) or poly(N-lauryl ethylene imine) as hydrophobic block and PEI as hydrophilic block by selective hydrolysis of poly(N-propionyl ethylene imine).30 Other examples include block copolymers consisting of poly(ethylene oxide) (PEO) and PEI,31 which were studied for their surfactant properties and clay dispersing ability. Furthermore, a porphyrin-centered cationic star block copolymer composed of cationic PEI and hydrophobic poly(2-phenyl-2-oxazoline) was studied for its micellization behavior.32 However, none of these studies addressed the thermoresponsive properties of the resulting micelles based on the crystallization induced phase separation of PEI.
Recently, we reported a detailed investigation on the kinetics of cleaving the side groups of water soluble poly(2-oxazoline)s via acidic hydrolysis.29 It was found that the methyl side groups of poly(2-methyl-2-oxazoline) (PMeOx) could be cleaved easier in comparison to the ethyl side groups of poly(2-ethyl-2-oxazoline). It is anticipated that a more bulky hydrophobic group, for example phenyl, as present in poly(2-phenyl-2-oxazoline) (PhOx), is even more difficult to cleave under similar conditions. Therefore, the selective hydrolysis of PMeOx–PPhOx copolymers is believed to result in PEI–PPhOx copolymers that might exhibit thermoresponsive micellization behavior in water.
Here, we report the synthesis of PMeOx–PPhOx gradient and diblock copolymers as well as subsequent acidic and basic hydrolyses. The structures, thermal properties and solubility behavior of the resulting (partially) hydrolyzed polymer structures were investigated in detail. Furthermore, the aggregation behavior of these PEI-containing copolymers was studied at different temperatures and pH values.
Experimental section
Materials and instrumentation
All chemicals were purchased from Aldrich. The 2-oxazoline monomers and methyl tosylate were distilled over barium oxide (BaO) and stored under argon. Acetonitrile (CH3CN, Aldrich) was dried over molecular sieves (3 Å). Polymerizations were carried out in the single-mode microwave reactor Emrys Liberator (Biotage) with capped reaction vials. These vials were heated to 105 °C, allowed to cool to room temperature, and filled with argon before use. All microwave polymerizations were performed under temperature control (IR sensor).1H NMR spectra were recorded on a Varian AM-400 spectrometer. All measurements were performed in deuterated methanol. Chemical shifts are given in part per million with respect to tetramethylsilane or residual solvent signals. Size exclusion chromatography (SEC) was measured on a system equipped with a Waters 1515 Isocratic HPLC pump, a Waters 2414 refractive index detector (40 °C), a Waters 2707 autosampler, and a PSS PFG guard column followed by 2 PFG-linear-XL (7 µm, 8 × 300 mm) columns in series at 40 °C. Hexafluoroisopropanol (HFIP, Apollo Scientific Limited) with potassium trifluoroacetate (3 g L−1) was used as eluent at a flow rate of 0.8 mL min−1. The molar masses were calculated against polystyrene standards. Thermal transitions were determined on a DSC Q100 from TA Instruments. Sample weights were in the range of 3 to 10 mg. The samples were measured in aluminium cups with a heating and cooling rate of 20 K min−1 for both the glass transition temperatures and the melting points (three measurements per sample after an initial first heating run that was not considered for the determination of the thermal transitions). DLS investigations were measured on a Malvern Zetasizer Nano. A sample concentration of 1 mg mL−1 was utilized in acidic medium (deionized water with 16.8 wt% HCl), pure deionized water (Laborpure, Behr Labor Technik) or basic medium (deionized water with 2.5 M NaOH). The polymer samples were added to the medium of choice and heated to elevated temperature to prepare the micellar solution. Prior to measuring, the samples were equilibrated at room temperature or 75 °C.
The cloud point temperatures were measured by heating the polymer (5.0 ± 0.2 mg) in acidic medium (deionized water with HCl), pure deionized water (Laborpure, Behr Labor Technik) or basic medium (deionized water with NaOH) (1.0 mL). The investigated temperature range was 2 to 105 °C with heating and cooling steps of 1 °C min−1. During these controlled heating and cooling cycles (two cycles per sample), the transmission through the solutions was monitored with red light in a Crystal 16™ from Avantium Technologies.33 Four blocks of four parallel temperature controlled sample holders are connected to a Julabo FP40 cryostat allowing 16 simultaneous measurements. All vials were visually inspected after the heating program to facilitate the interpretation of the observed transmission profiles. The presented cloud point temperatures correspond to the dissolution temperature at 50% transmittance from the second heating run. Transmission electron microscopy measurements were performed on a FEI Tecnai 20, type Sphera TEM operating at 200 kV (LaB6 filament). Images were recorded with a bottom mounted 1k × 1k Gatan CCD camera. TEM grids, 200 mesh carbon coated copper grids were purchased from SPI. Prior to blotting, the grids were made hydrophilic by surface plasma treatment using a Cressington 208 carbon coater operating at 5 mA for 40 seconds. Sample preparation was performed by dispensing 3 µL aliquots onto a 200 mesh carbon coated copper grid and excess liquid was quickly blotted away with filter paper. For the samples that were prepared above 75 °C, the heated solution was dispensed onto a heated grid followed by blotting away excess liquid before cooling to ambient temperature.
Synthesis of PMeOx60-stat-PPhOx40 gradient copolymer
The statistical gradient copolymer consisting of 2-phenyl-2-oxazoline (PhOx) and 2-methyl-2-oxazoline (MeOx) with a ratio of 40 : 60, respectively, and a total DP of 100 was synthesized via a cationic ring-opening polymerization mechanism as reported previously.34 The synthesized copolymer was analyzed by SEC to reveal the molar mass distribution and polydispersity index (PDI) and 1H NMR spectroscopy to determine the composition.Acidic hydrolysis of PMeOx60-stat-PPhOx40 gradient copolymer
For the acidic hydrolysis of PMeOx60-stat-PPhOx40, the copolymer was first dissolved in water and heated to elevated temperature to obtain a micellar solution. To this micellar solution, a 37 wt% aqueous HCl solution was added until a final polymer concentration of 48 g L−1 (4.4 mM) and an acid concentration of 16.8 wt% HCl in water were reached. The hydrolysis reaction was performed for 2 hours or 5 hours at 100 °C in a microwave synthesizer. After cooling the reaction mixture, the solution was neutralized with 2.5 M NaOH solution to pH > 8. The formed precipitate was filtered off, recrystallized from deionized water and, finally dried by freeze drying. The obtained polymers were characterized by 1H NMR spectroscopy to study the degree of hydrolysis and analyzed by SEC to determine the molar mass and PDI.Basic hydrolysis of PMeOx60-stat-PPhOx40 gradient copolymer
PMeOx60-stat-PPhOx40 was also used for basic hydrolysis reactions. Therefore, 0.606 g (0.055 mmol) of the copolymer was dissolved in 4 mL of water, and by heating the solution to elevated temperatures, a micellar solution was obtained. Subsequently, 0.612 g of NaOH dissolved in 3 mL water was added to the micellar solution resulting in 87 g L−1 (7.9 mM) polymer concentration, and the hydrolysis reaction was performed for 2, 5 or 10 hours at 100 °C in the microwave synthesizer. After a reaction time of 2 or 5 hours, the polymers were soluble in the alkaline solution and, therefore, subsequent purification was performed by dialysis to remove the excess of NaOH. The polymerization mixture after 10 hours reaction contained precipitated polymer, which was filtered off, recrystallized from water and, finally dried by freeze drying. The obtained polymers were characterized by 1H NMR spectroscopy to study the degree of hydrolysis and analyzed by SEC to determine the molar mass and PDI.Synthesis of PMeOx60-b-PPhOx40 block copolymer
The polymerization of the PMeOx60-b-PPhOx40 block copolymer was performed by a sequential monomer addition procedure as reported previously.35 The synthesized copolymer was analyzed by SEC to reveal the molar mass and PDI and 1H NMR spectroscopy to determine the composition.Acidic hydrolysis of PMeOx60-b-PPhOx40 block copolymer
Acidic hydrolysis of PMeOx60-b-PPhOx40 was performed in a similar manner as described for the PMeOx60-stat-PPhOx40copolymer, but was performed for 2 hours in an oil bath or 2 hours in a microwave synthesizer. The polymer obtained after hydrolysis in an oil bath was soluble at room temperature and the purification was performed by dialysis. The polymer obtained after 2 hours hydrolysis in the microwave synthesizer was not soluble after neutralization and purification was performed by recrystallization in water. The polymers were isolated by freeze-drying.Basic hydrolysis of PMeOx60-b-PPhOx40 block copolymer
The basic hydrolysis of PMeOx60-b-PPhOx40 was performed for 5 or 10 hours at 100 °C in a microwave synthesizer in a similar manner as described for the gradient copolymer. Both polymers were insoluble after the hydrolysis reactions in the basic medium, and the precipitate was filtered off and recrystallized in water followed by freeze-drying.Results and discussion
Poly(2-oxazoline)s are well known precursors for the synthesis of linear poly(ethylene imine)s via acidic25 or basic26hydrolysis. In a previous study, the influence of the polymer length of linear PEI on specific polymer properties such as thermal- and solubility behavior was investigated in detail.29 Moreover, a kinetic study showed that methyl side chains could be hydrolyzed faster than ethyl side groups. It is therefore hypothesized that PMeOx–PPhOx copolymers can be selectively or at least preferentially cleaved resulting in PEI–PPhOx copolymers, which might exhibit thermoresponsive micellization behavior based on the crystallization induced phase separation of linear PEI at lower temperatures. To investigate this hypothesis, a detailed study was performed on the hydrolysis of a PMeOx-stat-PPhOx gradient copolymer and a PMeOx-b-PPhOx diblock copolymer. Different reaction times for the acidic hydrolysis as well as basic hydrolysis conditions were investigated and selected polymer properties were determined such as thermal-, solubility-, and responsive self-assembly behavior.Synthesis of PMeOx–PPhOx copolymers and partial acidic hydrolysis reactions
A complete series of PMeOx-stat-PPhOx copolymers with compositions ranging from 100 mol% MeOx to 100 mol% PhOx was investigated in a previous study.34 Detailed investigations on the copolymerization parameters revealed that MeOx is first incorporated followed by slow incorporation of PhOx resulting in the formation of a gradient copolymer with a steep monomer gradient, i.e. a quasi-diblock copolymer.34 It was found that the PMeOx60-stat-PPhOx40 gradient polymer with a total DP of 100 self-assembled into coexisting spherical and wormlike micelles in water.19 Increasing the amount of PhOx resulted in water insoluble polymers and a decrease in PhOx content resulted in completely soluble polymers in aqueous media. Therefore, the 60 : 40 ratio of MeOx : PhOx was chosen for the current investigations on the hydrolysis and the properties of the resulting polymers.To be able to perform the hydrolysis reactions in water, a micellar solution was first prepared from the gradient copolymer by heating the copolymer in water. Subsequently, a concentrated aqueous HCl solution was added and the hydrolysis reaction was performed in a microwave synthesizer at 100 °C for 2 or 5 hours. Fig. 1 depicts a schematic overview of all investigated reactions. 1H NMR spectroscopy of the starting gradient copolymer in deuterated methanol revealed a MeOx : PhOx ratio of 64 : 36, calculated from the integration of the PhOx and MeOx signals and assuming a total DP of 100, which corresponds to the initial monomer to initiator ratio and near-quantitative monomer conversion. After 2 hours of acidic hydrolysis, the MeOx : PhOx : EI ratio changed to 0 : 6 : 94 indicating that all PMeOx side groups as well as most of the PPhOx side groups were removed. After 5 hours of acidic cleavage, the amount of PPhOx is even further reduced to only 2 units. A detailed overview of all obtained monomer ratios is provided in Table 1. These results demonstrate that both MeOx and PhOx side groups of the gradient copolymer are readily cleaved by acidic hydrolysis resulting in copolymers mostly consisting of EI units.
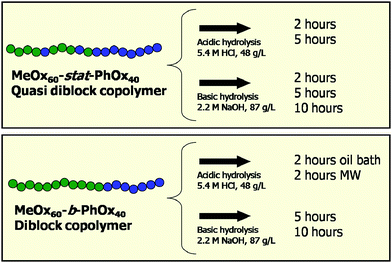 | ||
| Fig. 1 Schematic overview of the investigated hydrolysis reactions of the gradient and diblock copolymers (MW = microwave; 48 g L−1 = 4.4 mM and 87 g L−1 = 7.9 mM). | ||
| Hydrolysis | M n/g mol−1 | PDI | DP MeOx : PhOx : EI | T g/°C | T m/°C | |
|---|---|---|---|---|---|---|
| PMeOx-stat-PhOx | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
1.40 | 64 : 36 : 0 | 91.4 | ||
| 2 hours | Acidic | 19400 |
1.18 | 0 : 6 : 94 | 58.2 | |
| 5 hours | Acidic | 16300 |
1.24 | 0 : 2 : 98 | 62.5 | |
| 2 hours | Basic | 19200 |
1.35 | 54 : 37 : 9 | ||
| 5 hours | Basic | 21800 |
1.30 | 48 : 35 : 17 | ||
| 10 hours | Basic | 17500 |
1.37 | 20 : 21 : 59 | ||
| PMeOx-b-PhOx | 12400 |
1.20 | 60 : 40 : 0 | 91.1 | ||
| 2 hours oil bath | Acidic | 19900 |
1.21 | 22 : 33 : 45 | 60.2 | |
| 2 hours microwave | Acidic | 19500 |
1.13 | 0 : 16 : 84 | 53.0/63.8 | |
| 5 hours | Basic | 18200 |
1.28 | 35 : 40 : 25 | ||
| 10 hours | Basic | 17300 |
1.27 | 23 : 36 : 42 | ||
| Linear PEI | 18100 |
1.14 | 0 : 0 : 100 | 69.0 |
SEC analyses were performed in hexafluoroisopropanol (HFIP), which was found to be the most suitable solvent to suppress column interactions revealing symmetrical molar mass distributions for linear PEIs.29 The molar masses and PDI values of the obtained copolymers are listed in Table 1 and the SEC chromatograms are depicted in Fig. 2. After 2 hours hydrolysis the molar mass is increasing and slightly decreases again after a reaction time of 5 hours. An increase in molar masses was observed after full hydrolysis of PMeOx to PEI due to the different solubility and/or hydrogen bonding ability of PMeOx compared to PEI in the HFIP.29
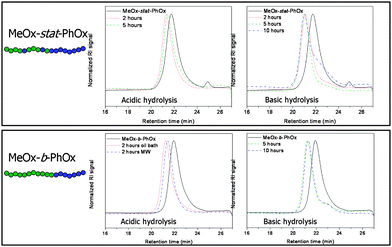 | ||
| Fig. 2 SEC chromatograms of the polymers obtained after acidic and basic hydrolyses of the gradient and block copolymers. | ||
The increase in molar mass after 2 hours hydrolysis can thus also be ascribed to the different hydrodynamic volume of PEI in HFIP. However, the obtained difference in molar mass between 2 and 5 hours reaction time might indicate the occurrence of some degradation reactions as is also indicated by the broadening of the molar mass distribution. Nonetheless, the main peak shape of the molar mass distribution is similar before and after 2 and 5 hours hydrolysis, demonstrating that the majority of the backbone remains intact.
Besides the gradient copolymer, a PMeOx-b-PPhOx block copolymer with a MeOx : PhOx ratio of 60 : 40, determined by 1H NMR spectroscopy, was synthesized by sequential monomer addition. The acidic hydrolysis of this block copolymer was investigated using a similar procedure, i.e. micelles were prepared in water followed by the addition of a concentrated HCl solution. Since almost complete hydrolysis was observed after 5 hours for the gradient copolymer, the hydrolysis of the block copolymer was performed for 2 hours in an oil bath or in a microwave synthesizer at 100 °C (Fig. 1). The block copolymer that was hydrolyzed in an oil bath revealed a MeOx : PhOx : EI ratio of 22 : 33 : 45 while the block copolymer hydrolyzed under microwave irradiation revealed a MeOx : PhOx : EI ratio of 0 : 16 : 84 (Table 1). It should be noted that the resulting hydrolyzed copolymer might not have a pure block copolymer architecture consisting of PEI and PhOx, but might rather consist of a pure PEI block and a second copolymer block consisting of both EI and PhOx units. Although the same reaction time and temperature were used for the hydrolysis reactions of the block copolymer, there is a significant difference between the ratios of the two resulting copolymers indicating more efficient hydrolysis under microwave irradiation. This observed difference might be due to the strong microwave absorption of the acidic solution leading to a significant thermal overshoot during initial heating resulting in a faster reaction. Nonetheless, these results clearly prove that MeOx side groups are indeed easier to remove by acidic hydrolysis than PhOx side chains. Moreover, comparing the hydrolysis of the gradient and the block copolymer revealed that, after a reaction time of 2 hours in the microwave synthesizer at 100 °C, more PhOx groups remained in the block copolymer (16 compared to 6), which can be ascribed to a better phase separation of the block copolymer. The PhOx groups are not soluble in water and form the inner core of the micelle. Compared to the block copolymer, the inner core of the gradient copolymer consists of a smaller PPhOx block and a gradient part which consists of PhOx as well as MeOx groups. This gradient part is partially soluble in water and may therefore be more readily hydrolyzed. SEC measurements revealed an increase in molar mass for both hydrolyzed block copolymers similar to the gradient copolymer (Table 1 and Fig. 2). The molar masses obtained after 2 hours hydrolysis in the oil bath or microwave reactor are similar to the molar mass of the gradient copolymer after 2 hours hydrolysis.
Partial basic hydrolysis reactions
The selective partial hydrolysis of the PMeOx–PPhOx gradient and block copolymers was also investigated under basic conditions. It was reported by Saegusa et al. that poly(N-formylethylene imine) and poly(2-methyl-2-oxazoline) are readily cleaved under basic conditions,26,27 while poly(2-phenyl-2-oxazoline) was found to remain intact after refluxing in 30% NaOH solution for 2 days.25 Therefore, the basic hydrolysis of the PMeOx–PPhOx copolymers was studied and compared to the acidic hydrolysis. The procedure for basic hydrolysis was performed in a similar manner as described in literature27 although a micellar solution was first prepared before adding the NaOH solution. Therefore, the PMeOx–PPhOx copolymers were dissolved in water and heated to elevated temperatures to obtain the micellar solution as visually observed by the translucent appearance. Subsequently, a concentrated NaOH solution was added. A schematic overview of the reactions performed under basic conditions is provided in Fig. 1.After the basic hydrolysis, the gradient copolymer was investigated by 1H NMR spectroscopy in deuterated methanol to determine the composition of the partially hydrolyzed copolymer. The measurements demonstrated that after a hydrolysis time of 2 and 5 hours all PhOx side groups were still present and only part of the MeOx groups were cleaved. After 5 hours, the MeOx : PhOx : EI ratio was found to be 48 : 35 : 17 (Table 1), calculated via the PMeOx, PPhOx and PEI signals and assuming a total DP of 100. An increase of the reaction time to 10 hours resulted in a MeOx : PhOx : EI ratio of 20 : 21 : 59, which means that with longer reaction times the PhOx side groups are also partially cleaved under the applied basic hydrolysis conditions. Compared to the acidic cleavage, the basic cleavage is significantly slower. This is not only caused by a lower NaOH concentration, but might also be explained by the formation of an inhomogeneous solution, i.e. upon addition of the NaOH solution the polymeric micelles precipitated. These results are in agreement with previously reported kinetic investigations revealing higher rates for the acidic hydrolysis compared to basic hydrolysis.36SEC measurements revealed the appearance of a small lower molar mass shoulder after 2 and 5 hours of basic hydrolysis (Fig. 2). After a reaction time of 10 hours, this shoulder becomes a significant peak indicating partial degradation of the copolymer; although this was not observed by 1H NMR spectroscopy. The basic hydrolysis of the block copolymer revealed that the PhOx side groups remained intact, even after a reaction time of 10 hours. The MeOx : PhOx : EI ratio after 10 hours hydrolysis was found to be 23 : 36 : 42 (Table 1). In summary, less PhOx groups of the block copolymer are cleaved compared to the gradient copolymer as also observed for the acidic cleavage, this can again be explained by the stronger phase separation between PMeOx and PPhOx. Moreover, SEC measurements revealed that a low molar mass shoulder also appeared in the SEC traces after basic hydrolysis of the block copolymer (Fig. 2). Even though several examples of basic hydrolysis are reported for the preparation of, e.g. poly(ethylene glycol)-b-poly(ethylene imine)31,37 and poly(styrene)-b-poly(ethylene imine),38,39 successful hydrolysis was only confirmed by 1H NMR spectroscopy but not by SEC due to strong column interactions. However, SEC analysis using HFIP, as developed in this work, clearly demonstrated significant polymer degradation under basic hydrolysis. Since this partial degradation occurred under basic conditions, resulting in a non-homogeneous polymer mixture, further investigations were not performed using these resulting copolymers.
Thermal properties of the partially hydrolyzed PMeOx–PPhOx copolymers
The thermal properties of the PMeOx–PPhOx gradient and block copolymers that were partially hydrolyzed under acidic conditions were investigated by DSC (Fig. 3). All observed thermal transition temperatures are listed in Table 1.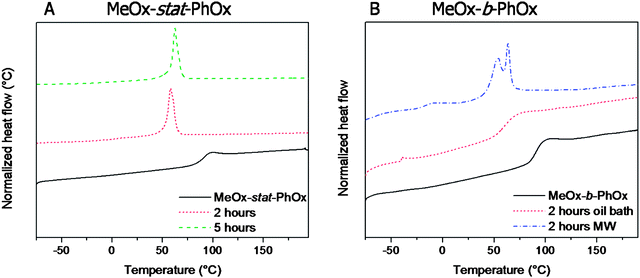 | ||
| Fig. 3 DSC heating curves (heating rate 20 °C min−1). Left: gradient PMeOx-stat-PPhOx copolymer after 2 and 5 hours acidic hydrolysis. Right: PMeOx-b-PPhOx copolymer after 2 hours oil bath and microwave heating (MW) under acidic conditions. | ||
The starting gradient and block copolymers are amorphous and revealed a single Tg at 91 °C, which is in agreement with the previously reported Tg of the gradient PMeOx60-stat-PPhOx40.34 In contrast, the gradient copolymers obtained after 2 and 5 hours acidic hydrolysis are semi-crystalline revealing a melting transition (Fig. 3). This might be expected since most of the MeOx and the PhOx side groups were removed and the resulting linear PEI is semi-crystalline. Furthermore, during neutralization of the hydrolysis reaction mixtures, the polymers precipitated indicative of crystallization induced phase separation. However, the PhOx side groups that are still present have a significant influence on the melting temperature. Linear PEI with a DP of 100 showed a Tm of 69 °C (peak maximum),29 while the melting temperature of the copolymer with only 6 PhOx units has a Tm of 58 °C. Therefore, it can be concluded that the remaining PhOx units disturb the crystallization causing a lower melting temperature. The hydrolyzed block copolymer, on the other hand, behaved differently compared to the gradient copolymer. The block copolymer, which was cleaved in an oil bath, is still amorphous revealing a Tg of 60 °C. The presence of MeOx and PhOx units obstructs the formation of crystalline PEI domains while the higher chain flexibility resulting from removal of some amidic methyl and phenyl side groups lowers the Tg. This is in agreement with previously reported results showing that below 70% deacylation the derivatives of poly(2-ethyl-2-oxazoline) were glassy amorphous thermoplastics while above 70% hydrolysis they became predominantly crystalline.36 The block copolymer that was hydrolyzed in the microwave synthesizer exhibits a small step in the measured heat flow at −15 °C, which might be assigned to the Tg of the amorphous domains. Furthermore, a double melting peak is observed, which indicates the presence of different crystalline phases possibly consisting of pure PEI and PEI with some PhOx that disturb the packing or PEI with different amounts of PhOx. This effect was reported earlier for linear PEI prepared from poly(2-phenyl-2-oxazoline) having a debenzylation degree of 98%, which also revealed a double melting peak at 51 and 58 °C, respectively.25 After multiple heating runs, a single melting peak was observed at 58 °C indicating the formation of only one type of crystalline domains after removal of the thermal history of the polymer. In contrast, the gradient copolymer obtained after 2 hours hydrolysis containing only 6 PhOx units showed only one melting transition already during the second heating run.
Solution properties of the partially hydrolyzed PMeOx–PPhOx copolymers
Further characterization of the partially hydrolyzed polymers was performed by turbidity measurements. It is known that (partial) protonation significantly influences the solubility of linear PEI.40 Therefore, solubility measurements were performed in acidic (aqueous HCl; pH ≈ 2), neutral (demineralized water) and basic medium (aqueous NaOH; pH ≈ 10). Both cleaved gradient copolymers were found to be soluble in acidic solution once heated to elevated temperatures; this heating is required to melt the polymer crystals as was also observed for linear PEI. However, no precipitation takes place after cooling to ambient temperature due to protonation of the amine groups. In neutral as well as in basic solution, the polymers become soluble at elevated temperatures and precipitate upon cooling indicative of a reversible solubility transition. Fig. 4 depicts the transmission versus temperature profiles.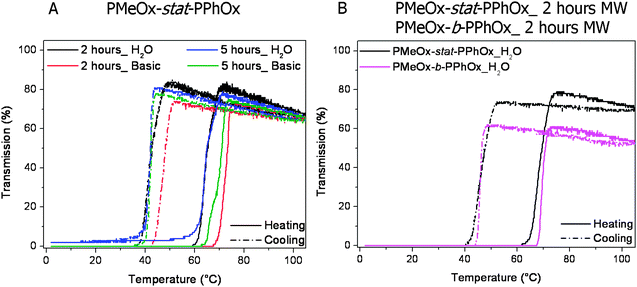 | ||
| Fig. 4 Transmittance versus temperature plots upon heating and cooling (1 K min−1) of the obtained polymers by acidic hydrolysis. Left: gradient PMeOx-stat-PPhOx copolymer after 2 and 5 hours reaction in H2O and basic medium (5 mg mL−1). Right: gradient PMeOx-stat-PPhOx compared to PMeOx-b-PPhOx in H2O after 2 hours reaction in the microwave synthesizer (5 mg mL−1). | ||
The solubility at elevated temperatures was also observed in previous investigations of linear PEI with varying DP and could be explained by the melting of the hydrated crystals which causes dissolution of the polymer in solution.29 Upon cooling, the hydrated crystals form again resulting in precipitation at lower temperatures, i.e.crystallization induced phase separation. The cloud point temperatures upon heating in H2O are in good agreement with the melting temperature as was also the case for linear PEI. The cloud point temperatures in basic solution for both gradient copolymers are slightly higher compared to the cloud point temperature in water due to complete deprotonation. This deprotonation results in a more hydrophobic polymer backbone and therefore a higher cloud point temperature. Concerning the hydrolyzed gradient copolymers, a behavior similar to linear PEI is expected due to the low number of remaining PhOx side groups.
The hydrolyzed block copolymers reveal a slightly different solubility behavior in water. As might be expected, the block copolymer cleaved in an oil bath, which contained still a relatively large amount of PMeOx as well as PPhOx groups, is still soluble in water in the investigated temperature range. Furthermore, this was the only polymer that was amorphous after hydrolysis, and, therefore, no melting of crystals is required to solubilize the copolymer. The block copolymer hydrolyzed in the microwave synthesizer for 2 hours behaves similar to the gradient copolymers; the block copolymer is soluble in acidic medium once heated to elevated temperatures and only soluble at elevated temperatures in aqueous or basic medium. By comparing the cloud point temperatures of the gradient and block copolymer, it is found that the cloud point temperatures in water are similar (see Fig. 4). Apparently, the larger amount of PhOx groups on the block copolymer does not significantly influence the PEI crystallization. However, the transmission of the block copolymer at elevated temperature is not raising above 70% as is the case for the gradient copolymer. Because the PhOx groups are not soluble in water, the higher amount of PPhOx in the block copolymer is expected to induce the formation of larger aggregates. The gradient copolymer, on the other hand, contains only a few PhOx groups probably resulting in smaller aggregates or molecularly dissolved chains.
Self-assembly behavior of the partially hydrolyzed PMeOx–PPhOx copolymers
To study the formation of aggregates, TEM and DLS investigations were performed for the gradient and the block copolymer before hydrolysis. At first, the gradient copolymer was investigated by DLS measurements in acidic, neutral and basic solution to reveal the effect of the chosen medium. The solutions were prepared with a concentration of 1 mg mL−1, heated to elevated temperature and allowed to cool to room temperature prior to measuring. Table 2 summarizes all hydrodynamic diameters (Dh) and polydispersity indices of the particles (PDIp) obtained by DLS measurements. As can be observed, the Dh of the gradient PMeOx-stat-PPhOx copolymer is not influenced by the chosen medium as expected based on the non-ionic nature of the amide groups, and the micelles have a narrow size distribution. TEM investigations of the micellar solution in neutral water revealed a size of ∼13 nm, which is much lower compared to the value found by DLS. This effect is well-known and is based on the fact that DLS determines the radius from the core as well as the corona while the TEM investigations were performed on a dried sample where the corona collapses onto the core causing a smaller diameter.41| Hydrolysis | Solution | D h/nm | PDIp | TEM size/nm | TEM STD/nm | |
|---|---|---|---|---|---|---|
| PMeOx-stat-PhOx | Acidic | 31 | 0.05 | n.d. | n.d. | |
| PMeOx-stat-PhOx | Water | 31 | 0.04 | 13 | 2 | |
| PMeOx-stat-PhOx | Basic | 31 | 0.01 | n.d. | n.d. | |
| 2 hours | Acidic | Water_75 °C | Too broad distribution | 16 | 3 | |
| 5 hours | Acidic | Water_75 °C | Too broad distribution | Micelles: 14, fibers: 7 | Micelles: 3, fibers: 2 | |
| PMeOx-b-PhOx | Acidic | n.d. | n.d. | 16 | 2 | |
| PMeOx-b-PhOx | Water | 51 | 0.18 | n.d. | n.d. | |
| 2 hours oil bath | Acidic | Water | 38 | 0.27 | 14 | 3 |
| 2 hours MW | Acidic | Acidic_20 °C | n.d. | n.d. | 22 | 5 |
| 2 hours MW | Acidic | Acidic_75 °C | n.d. | n.d. | 54 | 11 |
| 2 hours MW | Acidic | Water_75 °C | n.d. | n.d. | 24 | 4 |
Subsequently, the partially hydrolyzed gradient PMeOx-stat-PPhOx copolymers were investigated by DLS measurements in water at elevated temperature indicated a broad size distribution (PDIp > 0.3; no reliable interpretation possible) and, therefore, TEM investigations were performed (Fig. 5). Even though sample preparation was performed at room temperature or above 75 °C followed by quickly blotting away excess liquid to freeze in the self-assembled structures, TEM was performed at room temperature. Therefore, PEI crystallization during cooling cannot be fully excluded and might influence the observed structures. TEM of the gradient copolymer obtained after 2 hours acidic hydrolysis revealed the presence of spherical micelles. Many of these micelles cluster together, which explains the broad size distribution obtained by DLS. This clustering is most likely enhanced by aggregation upon drying.43 However, it is interesting to observe that only 6 PPhOx units are sufficient to form stable micellar aggregates. It is known that linear PEI has a hydrated random coil conformation above the solubility transition temperature and, therefore, the hydrophobic PhOx groups drive the formation of spherical micelles.42TEM revealed that the size of the micelles is comparable to the starting gradient copolymer, which is unexpected since the core should be much smaller due to the removal of most PPhOx side chains. This similar size can be explained by the fact that the PPhOx gradient renders part of the PEI hydrophobic.
 | ||
| Fig. 5 TEM images of the micellar structures obtained from PMeOx-stat-PPhOx, after 2 hours and after 5 hours of acidic hydrolysis prepared from water. | ||
The gradient copolymer that was hydrolyzed for 5 hours revealed spherical micelles as well as long fibers in TEM. The spherical micelles have a slightly smaller diameter compared to the sample after 2 hours acidic hydrolysis. Since this copolymer contains on average 2 PhOx units per chain it will consist of pure PEI as well as PEI with multiple PhOx units. Therefore, it is proposed that the fibers are formed by crystallization of pure linear PEI as has been reported in literature43 while the spherical micelles are based on the PhOx containing copolymers.
The self-assembly of the block copolymer and the corresponding hydrolyzed polymers was first investigated by DLS in water. Compared to the gradient copolymer, the block copolymer revealed larger micellar structures in solution (51 nm compared to 31 nm, respectively), which can be explained by a stronger phase separation between the PMeOx and PPhOx. The block copolymer has a more hydrophilic corona due to the absence of the gradient middle part, which can cause increased stretching of the corona while the more hydrophobic core may result in a larger aggregation number resulting in larger micelles. TEM investigations revealed only a small difference between the micellar size of the gradient and the block copolymer indicating that stretching of the coronal chains causes the large size observed by DLS. Furthermore, the block copolymer obtained after 2 hours acidic hydrolysis in an oil bath was found to be soluble in water at room temperature. DLS measurements at room temperature revealed that the starting MeOx-b-PhOx copolymer had a Dh of 51 nm with a PDIp of 0.18 while this hydrolyzed copolymer revealed a Dh of 38 nm and a PDIp of 0.27 (Table 2). Although the PDIp is rather high, it might be anticipated that the formation of smaller micelles of the partially cleaved copolymer could be due to a more hydrophilic corona resulting from the presence of the ethylene imine units as well as the repulsion of the cationic ethylene imine groups in the corona, both causing a lower aggregation number. Comparison of the TEM images (Fig. 6) revealed spherical micelles for the starting block copolymer as well as the copolymer that was hydrolyzed in an oil bath. The size of the micellar structures determined by TEM is slightly smaller for the hydrolyzed copolymer indeed indicating a lower aggregation number.
 | ||
| Fig. 6 TEM images of PMeOx-b-PPhOx micelles after 2 hours acidic cleavage in an oil bath and a microwave synthesizer. | ||
Finally, the self-assembly of the block copolymer that was hydrolyzed for 2 hours in the microwave was investigated. This polymer is soluble in acidic solution once heated to elevated temperatures to melt the polymer crystals. TEM samples were prepared at elevated temperature as well as after cooling to room temperature (Fig. 6). At room temperature, the copolymer revealed spherical micelles that cluster together, comparable to the gradient copolymer after 2 hours of hydrolysis probably caused by drying effects. The size of these micelles is larger compared to the starting block copolymer as well as the copolymer after 2 hours of acidic cleavage in an oil bath due to stretching of the protonated coronal chains. Moreover, micelles expand upon increasing the temperature due to increased chain mobility resulting in stretching of the coronal chains.
Furthermore, this block polymer was found to be pH responsive. In acidic solution, the polymer is soluble at room temperature and forms spherical micelles while in water or basic solution this polymer is insoluble at room temperature. Upon heating in acidic solution, the micelle size increases. In water and basic solution, the PEI crystals melt upon heating resulting in the formation of solubilized spherical micelles. The combined temperature and pH responsiveness of this PEI84-b-PPhOx16 block copolymer are schematically depicted in Fig. 7.
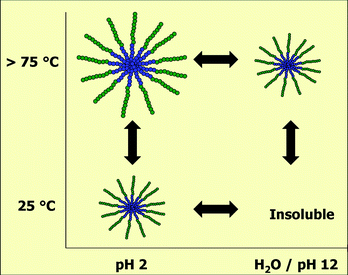 | ||
| Fig. 7 Schematic overview of the pH and temperature responsiveness of the PEI84-b-PPhOx16 block copolymer obtained by acidic hydrolysis of the PMeOx60-b-PPhOx40 block copolymer under microwave irradiation. | ||
Conclusions
In summary, a detailed study was performed on acidic and basic hydrolyses of PMeOx–PPhOx gradient and diblock copolymers. Under acidic hydrolysis conditions, both MeOx as well as PhOx side groups are readily cleaved, although it was found that MeOx side groups are preferentially hydrolyzed. A comparison between the gradient and block copolymer revealed that the stronger phase separation of the block copolymer causes a slower cleavage of the PPhOx groups, resulting in more selective hydrolysis. The hydrolysis rate for basic hydrolysis was much slower and the PhOx groups were even more difficult to remove resulting in selective hydrolysis. However, partial degradation was observed after basic hydrolysis by SEC using HFIP as eluent. Therefore, further investigations were only performed for the polymers obtained by acidic cleavage. Thermal investigations revealed that both hydrolyzed gradient copolymers exhibited a melting temperature, which was expected since most of the MeOx as well as the PhOx side groups were removed. The hydrolyzed block copolymer with a MeOx : PhOx : EI ratio of 22 : 33 : 45 was still amorphous, although the Tg decreased due to a higher chain flexibility by removal of the amidic side groups. The second hydrolyzed block copolymer, i.e.PEI84-b-PPhOx16, showed a double melting peak. Solubility measurements revealed that polymers, which were hydrolyzed for at least 84%, were soluble in acidic solution and insoluble in water or basic solution at room temperature. Upon heating, the polymers became soluble in water and basic solution due to melting of the PEI crystals. Moreover, TEM and DLS investigations revealed the formation of spherical micelles in water at elevated temperatures for these copolymers indicating thermoresponsive micellization behavior based on the crystallization induced phase separation of linear PEI at lower temperatures. Moreover, the PEI84-b-PPhOx16 block copolymer was found to be pH responsive due to the solubility of the ethylene imine upon protonation in acidic solution resulting in temperature and pH responsive micellization behavior.Acknowledgements
This work forms part of the research program of the Dutch Polymer Institute (DPI), project no. 449. Ing. Martin Fijten is acknowledged for the SEC measurements and Dr Stephanie Hornig for helpful comments. RH (Veni-grant) and USS (Vici-award) are grateful to the Netherlands Scientific Organization (NWO) for financial support. This research has been carried out with the support of the Soft Matter cryo-TEM Research Unit, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology.Notes and references
- T. Saegusa and H. Ikeda, Macromolecules, 1973, 6, 808 CrossRef CAS.
- T. Saegusa, H. Ikeda and H. Fujii, Macromolecules, 1972, 5, 359 CrossRef CAS.
- H. Huang, R. Hoogenboom, M. A. M. Leenen, P. Guillet, A. M. Jonas, U. S. Schubert and J.-F. Gohy, J. Am. Chem. Soc., 2006, 128, 3784 CrossRef CAS.
- R.-H. Jin, J. Mater. Chem., 2004, 14, 320 RSC.
- V. Percec, T. K. Bera and R. J. Butera, Biomacromolecules, 2002, 3, 272 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1993, 26, 6315 CrossRef CAS.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504 CrossRef CAS.
- O. Nuyken, R. Weberskirch, M. Bortenschlager and D. Schönfelder, Macromol. Symp., 2004, 215, 215 CrossRef CAS.
- O. Nuyken, P. Persigehl and R. Weberskirch, Macromol. Symp., 2002, 177, 163 CrossRef CAS.
- T. B. Bonne, K. Luedtke, R. Jordan, P. Stepanek and C. M. Papadakis, Colloid Polym. Sci., 2004, 282, 833 CrossRef CAS.
- T. B. Bonne, K. Luedtke, R. Jordan and C. M. Papadakis, Macromol. Chem. Phys., 2007, 208, 1402 CrossRef CAS.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622 CrossRef CAS.
- A. Gress, A. Voelkel and H. Schlaad, Macromolecules, 2007, 40, 7928 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., 1966, 78, 913 CAS.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441 CrossRef CAS.
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871 CrossRef.
- H. M. L. Lambermont-Thijs, R. Hoogenboom, C.-A. Fustin, C. Bomal-D'Haese, J.-F. Gohy and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 515 CrossRef CAS.
- R. Hoogenboom, H. M. L. Lambermont-Thijs, M. J. H. C. Jochems, S. Hoeppener, C. Guerlain, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 3590 RSC.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103 RSC.
- S. Kobayashi, T. Igarashi, Y. Moriuchi and T. Saegusa, Macromolecules, 1986, 19, 535 CrossRef CAS.
- R.-H. Jin, Adv. Mater., 2002, 14, 889 CrossRef CAS.
- R. Hoogenboom, Macromol. Chem. Phys., 2007, 208, 18 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978 CrossRef CAS.
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849 CrossRef CAS.
- T. Saegusa, S. Kobayashi and A. Yamada, Macromolecules, 1975, 8, 390 CrossRef.
- T. Saegusa, H. Ikeda and H. Fujii, Macromolecules, 1972, 5, 108 CrossRef CAS.
- Y. Chatani, H. Tadokoro, T. Saegusa and H. Ikeda, Macromolecules, 1981, 14, 315 CrossRef CAS.
- H. M. L. Lambermont-Thijs, F. S. Van der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 927 CrossRef CAS.
- M. H. Litt and C. S. Lin, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 779 CrossRef CAS.
- J. Zhang and B. Lonnie, Chin. J. Chem., 2003, 21, 460 CAS.
- R.-H. Jin, ChemPhysChem, 2003, 4, 1118 CrossRef CAS.
- M. Birch, S. J. Fussel, P. D. Higginson, N. McDowall and I. Marziano, Org. Process Res. Dev., 2005, 9, 360 Search PubMed.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten, B. M. Van Lankvelt and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 416 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, S. F. G. M. Van Nispen, M. Van der Loop, C. H. Abeln, A. M. J. Van den Berg and U. S. Schubert, Macromolecules, 2005, 38, 7957 CrossRef CAS.
- K. M. Kem, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 1977 CrossRef CAS.
- Y. Akiyama, A. Harada, Y. Nagasaki and K. Kataoka, Macromolecules, 2000, 33, 5841 CrossRef CAS.
- S. Ibrahim and B. Voit, Macromol. Symp., 2009, 275–276, 59 CrossRef CAS.
- K. Ishizu, T. Fukutomi and T. Kakurai, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 405 CrossRef CAS.
- K. F. Weyts and E. J. Goethals, Makromol. Chem. Rapid Commun., 1989, 10, 299 CrossRef CAS.
- C. Guerrero-Sanchez, D. Wouters, C.-A. Fustin, J.-F. Gohy, B. G. G. Lohmeijer and U. S. Schubert, Macromolecules, 2005, 38, 10185 CrossRef CAS.
- H. Kakuda, T. Okada and T. Hasegawa, J. Phys. Chem. B, 2009, 113, 13910 CrossRef CAS.
- J.-J. Yuan and R.-H. Jin, Langmuir, 2005, 21, 3136 CrossRef CAS.
Footnote |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |