Combining two-directional synthesis and tandem reactions: a short formal synthesis of halichlorine†
Camille
Gignoux
a,
Annabella F.
Newton
a,
Alexandre
Barthelme
a,
William
Lewis
a,
Marie-Lyne
Alcaraz
b and
Robert A.
Stockman
*a
aSchool of Chemistry, University of Nottingham, Nottingham, UK NG7 2RD. E-mail: Robert.stockman@nottingham.ac.uk
bAstraZeneca, Bakewell Road, Loughborough, Leics, UK LE11 5RH
First published on 1st September 2011
Abstract
A short and efficient synthesis of an advanced intermediate (1) in the Clive route to halichlorine has been achieved in 12 steps and 13.2% yield by a combined two-directional synthesis/tandem reaction strategy.
Halichlorine (Fig. 1) was isolated in 1996 by Uemura and co-workers from the Japanese marine sponge, Halichondria okadai Kadota,1 and was found to selectively inhibit the induced expression of the cell surface protein, VCAM-1 (Vascular Cell Adhesion Molecule-1).2 Increased expression of VCAM-1 has been noted in inflammation,3 tumour metastasis,4angiogenesis5 and microtubule formation.6 Recently it was shown that halichlorine suppresses NF-κB activation.7 The NF-κB transcription factor family plays a key role in inflammation, tumourigenesis and angiogenesis and thus NF-κB is a major chemotherapeutic target for the Pharmaceutical industry.8 A recent study has also shown that halichlorine also inhibits L-type Ca2+ channels in vascular smooth muscle cells and could be targeted as an antihypertensive agent.9 Also in 1996, the two structurally related alkaloids pinnaic acid and tauropinnaic acid, were isolated by the Uemura group from the Okinawan bivalve Pinna muricata.10 Both compounds inhibit the activity of the cytosolic 85-kDa phospholipidase A2 (cPLA2), which is involved in the regulation of inflammatory response.11 Recently a third member of the family, pinnarine, was isolated by Uemura.12
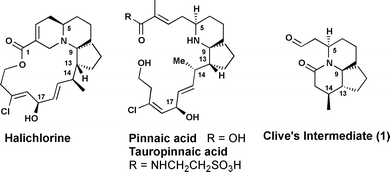 | ||
| Fig. 1 Structure of halichlorine and pinnaic acids. | ||
As a result of their highly promising biological activities and their synthetically challenging, highly functionalised 6-azaspiro[4.5]decane ring, halichlorine and the pinnaic acids have received a high degree of attention from the synthetic community. An excellent review recounts all synthetic studies developed up to 2005,13 with several further studies being published subsequently to this.14 The most recent total synthesis of halichlorine was reported by Clive in 2009.14c
In 2004, we published a concise approach to the azaspirocyclic core structure of halichlorine and pinnaic acid, which combined two-directional synthesis with a tandem cascade strategy.15 The key features of our approach were the use of two-directional synthesis for the formation of a symmetrical ketodiester 2, followed by a tandem cyclisation to yield the tricyclic isoxazolidine 3 (Scheme 1). Further manipulation of 3 then corrected the stereochemistry at C5, and the amine and diol functions were each protected to give 4. Herein, we describe our refinements and advances to this strategy, and the synthesis of azaspirocyclic aldehyde 1 (Fig. 1), which is a late-stage intermediate in the total synthesis of halichlorine by Clive and co-workers.14c
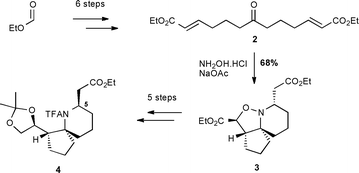 | ||
| Scheme 1 Our 2004 synthesis of spirocyclic core 4. | ||
Initially we concentrated our efforts on shortening the sequence for the synthesis of ketodiester 2 (Scheme 2). We felt particularly that we wished to exclude the requirement for protecting group chemistry. Thus we turned to our recent work on two-directional cross-metathesis,16 which enabled us to convert ketone 5 (available in high yield in 2 steps) directly into keto-diester 2 in 79% yield. This was a significant improvement on our original route, removing three steps), although we did find the cost of the Grubbs–Hoveyda II catalyst to be high for the synthesis of large (>10 g) quantities of 2. We therefore also investigated a two-step approach involving oxidative addition of the alkenes of 5, followed by two-directional Wittig homologation. On a 10 g scale, this gave ketodiester 2 in 47–53% yield after purification, and was significantly cheaper than the cross-metathesis route, albeit one step longer and requiring careful chromatography.
 | ||
| Scheme 2 Optimised route to tricycle 3. | ||
The tandem cyclisation from 2 to yield isoxazolidine 3 (Scheme 2) was optimised by employing a two-stage procedure, whereby after nitrone formation had been observed to be complete by TLC, the reaction was subjected to an aqueous work-up, and evaporated to dryness. The residue was taken up in dichloromethane and any solids removed by filtration, before re-evaporation and redissolving in acetonitrile. The reaction mixture was then heated at reflux to complete the cyclisation. Recrystallisation from hexane then afforded 3 in high yields on 10 g scale.
With large quantities of 3 in hand, we next turned our attention to the installation of the C14 methyl group (Scheme 3). Initially we decided to attempt a Wittig homologation of ketone 9, followed by a hydrogenation. As we previously reported,15 the isoxazolidine ester of 3 can be selectively reduced with sodium borohydride in ethanol, and a subsequent hydrogenation yields a diol, which upon heating in a sealed tube in ethanol, gives the diol 6, in which the stereochemistry at the C5 position has been epimerised to that required for halichlorine. Previously we had used sealed tube conditions for this epimerisation, but we were glad to find that heating in a microwave at 120 °C also yielded the same results. The primary alcohol of 8 was protected using TBDPSCl in acetonitrile with imidazole as base. Surprisingly, it was found that a significant amount of bis-silylated product was formed. This could be deprotected and recycled, but we could not get greater than 65% of the mono-protected compound, despite investigating many conditions. The remaining secondary alcohol was oxidised under Parikh–Doering conditions17 to yield ketone 9. Unfortunately, we found 9 to be unreactive under any olefination conditions – indeed, we were unable even to react 9 with methyl lithium to generate a tertiary alcohol. Thus we decided to convert the alcohol 8 into a leaving group, and try cuprate chemistry to install the required C14 methyl group. Thus 8 was treated with thionyl chloride in dioxane, which resulted not in the expected alkyl chloride with retention of stereochemistry through double displacement, but in fact the chloride 12 through a simple SN2 inversion. It was found not possible to displace this chloride with any cuprate reagent, or indeed with bromide or iodide, and thus we re-thought our strategy.
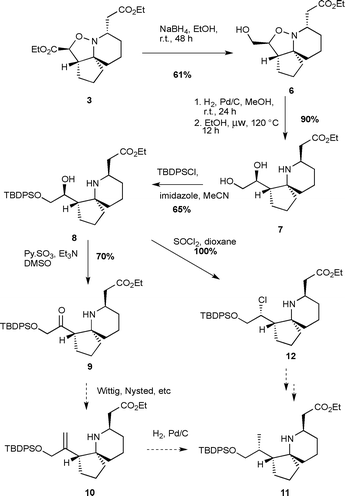 | ||
| Scheme 3 Initial attempts to install the C14 methyl group. | ||
Having failed to install the C14 methyl group by reacting on the open-chain ketone 9 or chloride 12, presumably due to the high steric demands of both the proximal silylated alcohol and the cyclopentane ring, we decided to employ the tactic of using a cyclic α,β-unsaturated lactam to minimize the steric encumbrance for an in-coming nucleophile to the C14 carbon.14c
To this end, we prepared lactam 15 by an oxidative cleavage of the diol 7, followed immediately by an Ando homologation18 to yield Z-enoate 14 (Scheme 4). Although the desired (Z)-conjugated ester was obtained as the major product, variable amounts of the (E)-isomer were also recovered. Using the Still–Gennari reagent19 with KHMDS, 18-crown-6 in THF at −78 °C, gave poor yields. Several bases and additives were screened for the Ando procedure (KHMDS/18-cr-6, DBU/NaI, NaH with or without NaI) and NaH alone in THF was proven to be optimal. The temperature of both deprotonation of phosphonate and aldehyde addition was also varied (room temperature, 0 °C or −78 °C). The best results were obtained when the phosphonate was treated at 0 °C for 15 min with an excess of NaH and 13 was then added at −78 °C to the reaction mixture. (Z)-Ester 14 was prepared in 56% yield from diol 7. Thermal cyclisation of 14 was first attempted in toluene at 100–120 °C over several days, however with moderate success (23–45% yield). Microwave irradiation at higher temperatures or exchanging solvent for chlorobenzene did not show any improvement; furthermore we started observing epimerisation at C5. Ultimately, we found that the reaction proceeded best in toluene at reflux with stoichiometric acetic acid, giving lactam 15 in excellent yield.
 | ||
| Scheme 4 Synthesis of Clive's aldehyde 1 and formal synthesis of halichlorine. | ||
We were now in a position to introduce the required C14 methyl group by conjugate addition to lactam 15. Attack of the methyl anion should occur from the upper face due to the shape of bicyclic lactam and provide the methyl lactam with the desired stereochemistry. Addition of the Gilman reagent20 (lithium dimethylcuprate) to 15 in presence of TMSCl and triethylamine afforded lactam 16 in excellent yield, installing the critical C14 methyl group with complete stereocontrol. It was found that this reaction requires both TMSCl and triethylamine to proceed. With lactam 16 in hand, access to the target aldehyde 1 was initially achieved by reduction of the ester function with LiBH4 in THF in 76% yield, followed by oxidation of resulting alcohol using the Parikh–Doering conditions, however in a poor 38% yield. Other oxidation methods e.g. Swern and TPAP–NMO oxidations were found to give even lower yields. Thus a more efficient strategy was found using DIBAL-H in CH2Cl2 at low temperature and the direct reduction of ester 16 to 1 was achieved in 82% yield.
In conclusion, we have completed a short and efficient synthesis of Clive's aldehyde 1 in 12 steps and 13.2% overall yield from ethyl formate, representing a formal synthesis of halichlorine. Our synthesis of 1 compares favourably with the 27 step synthesis employed by Clive and co-workers. Studies towards the use of this strategy for the synthesis of pinnarine are on-going, and will be reported in due course.
University of Nottingham (CG, AB), AstraZeneca (AN) and EPSRC (RAS, EP/E055346) are acknowledged for funding.
Notes and references
- (a) M. Kuramoto, C. Tong, K. Yamada, T. Chiba, Y. Hayashi and D. Uemura, Tetrahedron Lett., 1996, 37, 3867 CrossRef CAS; (b) H. Arimoto, I. Hayakawa, M. Kuramoto and D. Uemura, Tetrahedron Lett., 1998, 39, 861 CrossRef CAS.
- M. Kuramoto, H. Arimoto and D. Uemura, Mar. Drugs, 2004, 2, 39 CrossRef CAS.
- C. Kummer and M. H. Ginsberg, Biochem. Pharmacol., 2006, 72, 1460 CrossRef CAS.
- A. Tempia-Caliera, L. Z. Horvath, A. Zimmermann, T. T. Tihanyi, M. Korc, H. Friess and M. W. Büchler, J. Surg. Oncol., 2002, 79, 93 CrossRef CAS.
- A. E. Koch, M. M. Halloran, C. J. Haskell, M. R. Shah and P. J. Polverini, Nature, 1995, 376, 517 CrossRef CAS.
- A. Asahina, Y. Tada, K. Nakamura and K. Tamaki, J. Dermatol. Sci., 2001, 25, 1 CrossRef CAS.
- Y. Tsubosaka, T. Murata, K. Yamada, D. Uemura, M. Hori and H. Ozaki, J. Phamacol. Sci., 2010, 113, 208 CAS.
- T. D. Gilmore and M. Herscovitch, Oncogene, 2006, 25, 6887 CrossRef CAS.
- Y. Tsubosaka, T. Murata, K. Kinoshita, K. Yamada, D. Uemura, M. Hori and H. Ozaki, Eur. J. Pharmacol., 2010, 628, 128 CrossRef CAS.
- T. Chou, M. Kuramoto, Y. Otani, M. Shikano, K. Yazawa and D. Uemura, Tetrahedron Lett., 1996, 37, 3871 CrossRef CAS.
- H. van den Bosch, Biochim. Biophys. Acta, Biomembr., 1980, 604, 191 CrossRef CAS; H. Arita, T. Nakano and K. Hanasaki, Prog. Lipid Res., 1989, 28, 273 CrossRef.
- S. Xu, H. Yoshimura, N. Maru, O. Ohno, H. Arimoto and D. Uemura,, J. Nat. Prod., 2010, 74, 1323 CrossRef.
- D. L. J. Clive, M. L. Yu, J. Wang, V. S. C. Yeh and S. Z. Kang, Chem. Rev., 2005, 105, 4483 CrossRef CAS.
- (a) S. Xu, H. Arimoto and D. Uemura, Angew. Chem., Int. Ed., 2007, 46, 5746 CrossRef CAS; (b) H. Wu, H. G. Zhang and G. Zhao, Tetrahedron, 2007, 63, 6454 CrossRef CAS; (c) D. Z. Liu, H. P. Acharya, M. L. Yu, J. Wang, V. S. C. Yeh, S. Z. Kang, C. Chiruta, S. M. Jachak and D. L. J. Clive, J. Org. Chem., 2009, 74, 7417 CrossRef CAS; (d) H. Kim, J. H. Seo, K. J. Shin, D. J. Kim and D. Kim, Heterocycles, 2006, 70, 143 CrossRef CAS; (e) V. Caprio, Synlett, 2007, 219 Search PubMed; (f) D. L. J. Clive, Z. Y. Li and M. L. Yu, J. Org. Chem., 2007, 72, 5608 CrossRef CAS; (g) P. B. Hurley and G. R. Dake, J. Org. Chem., 2008, 73, 4131 CrossRef CAS; (h) G. E. Keck and S. A. Heumann, Org. Lett., 2008, 10, 4783 CrossRef CAS; (i) L. H. Wang, B. Prabhudas and D. L. J. Clive, J. Am. Chem. Soc., 2009, 131, 6003 CrossRef CAS; (j) Y. Yamamoto, Y. Yasuda, H. Nasu and K. Tomioka, Org. Lett., 2009, 11, 2007 CrossRef CAS; (k) S. H. Yang, G. R. Clark and V. Caprio, Org. Biomol. Chem., 2009, 7, 2981 RSC; (l) S. M. Amordea, I. T. Jewetta and S. F. Martin, Tetrahedron, 2009, 65, 3222 CrossRef.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371 CrossRef CAS.
- A. F. Newton, S. J. Roe, J. C. Legeay, P. Aggarwal, C. Gignoux, N. J. Birch, R. Nixon, M. L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2009, 7, 2274 CAS.
- J. R. Parikh and W. V. E. Doering, J. Am. Chem. Soc., 2002, 89, 5505 CrossRef.
- K. Ando, Tetrahedron Lett., 1995, 24, 4105 CrossRef.
- W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405 CrossRef CAS.
- H. Gilman, R. G. Jones and L. A. Woods, J. Org. Chem., 1952, 17, 1630 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details. CCDC reference number 849762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ob06380d |
| This journal is © The Royal Society of Chemistry 2012 |