Protecting-group-free synthesis of a dual CCK1/CCK2 receptor antagonist†
Jing
Liu
,
Xiaohu
Deng
*,
Anne E.
Fitzgerald
,
Zachary S.
Sales
,
Hariharan
Venkatesan
and
Neelakandha S.
Mani
Johnson & Johnson Pharmaceutical Research and Development, L.L.C. 3210 Merryfield Row, San Diego, CA 92121, USA. E-mail: xdeng@its.jnj.com; Fax: +1-858-320-3354; Tel: +1-858-320-3338
First published on 2nd March 2011
Abstract
In our pursuit of an efficient, protecting-group-free synthesis of the dual CCK1/CCK2 receptor antagonist 1, we have developed chemoselective conditions for sulfonamide formation reaction in pure water and a PhNMe2 mediated carboxamide formation, both in the presence of a carboxylic acid. Practical synthesis of an unnatural, chiral β-aryl-α-amino acid is also described.
Introduction
Cholecystokinin (CCK), a 33 amino acid peptide, is a regulatory hormone predominantly found in the gastrointestinal tract as well as in the central nervous system. Two specific G protein coupled receptor subtypes (CCK1 and CCK2) regulate the major biological effects of CCK in the gastrointestinal system, which include motility, pancreatic enzyme secretion, gastric emptying, and gastric acid secretion.1 In the past 20 years, selective inhibition of either of the two receptors has been vigorously pursued both in academia and in the pharmaceutical industry, which has resulted in a steady stream of CCK receptor antagonists into human clinical trials for the treatment of various gastrointestinal diseases.2 It was recently proposed that dual CCK1/CCK2 receptor inhibition might provide a synergistic effect: CCK1 receptor inhibition improving lower esophageal sphincter smooth muscle function and increasing the rate of gastric emptying and the concurrent CCK2 receptor inhibition moderating gastric acid secretion, which might be beneficial for treatment of gastroesophageal reflux disease.3 Our laboratories have been active contributors to the study of selective CCK14 and CCK25receptor inhibition. Recent medicinal chemistry efforts identified compound 1 as a dual CCK1/CCK2 receptor antagonist with desirable pharmacologic properties (Fig. 1).3 For the purpose of further profiling this compound, a large-scale synthesis of compound 1 was required.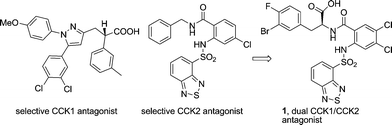 | ||
| Fig. 1 Representative selective CCK1, CCK2 and dual CCK1/CCK2 antagonists identified in our laboratories. | ||
Two routes were utilized in the drug discovery efforts to prepare compound 1. The first-generation synthesis involved an eight step sequence starting from 4,5-dichlorophthalic anhydride (Scheme 1). It was noticed that one of the major contributors to the length of this sequence was the protection/deprotection manipulations of the carboxylic acid groups.
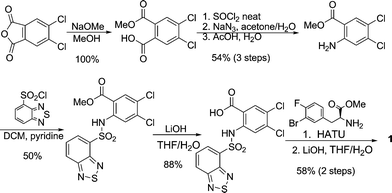 | ||
| Scheme 1 First-generation synthesis of compound 1. | ||
In the second-generation route (Scheme 2), the amide bond was formed prior to the sulfonamide. This obviated the manipulation of the benzoic acid functional group, which significantly shortened the overall sequence to four steps. However, like the first generation synthesis, two chemical steps were committed to the introduction and removal of the acid protection group for the α-amino acid. To streamline the synthesis and avoid the use of protecting groups, we developed a two step route featuring a sulfonamide formation in water followed by a PhNMe2 mediated amide formation, both in the presence of the unmasked acid group. In addition, a practical synthesis of unnatural, chiral β-aryl-α-amino acid 2 was developed featuring a Rh catalyzed asymmetric hydrogenation. Significant efforts were also devoted to the development of “green” conditions to minimize environmental impacts.
 | ||
| Scheme 2 Second-generation synthesis of compound 1. | ||
Results and discussion
Protecting groups have long been utilized by synthetic chemists to mask competitive reactivity from interfering with the desired transformation. This strategy, offering security and predictability, has become a textbook approach for the precise control over the individual reactivity within a complex molecule. However, the use of protecting groups, which adds two steps to the overall synthesis, is against the “ideal synthesis” principle, which states that chemical syntheses should only consist of skeleton-building steps.6 Recently, Hoffmann and Baran have been advocating “protecting-group-free” synthesis,7 not only for achieving higher overall synthetic efficiency, but also as a unique opportunity for inventing novel reactions/conditions by taking advantage of the intrinsic reactivity of the unprotected functionalities.8 We reasoned that a “protecting-group-free” synthesis of 1 might be feasible, if functionality compatibility was judiciously taken into consideration at the route designing stage. Guided by this principle, we started the retro-synthetic analysis exercise (Scheme 3). The natural disconnections of compound 1 are on the amide C–N bond and sulfonamide N–S bond. Depending on the order of constructing these two bonds, two routes were proposed, using three common building blocks. In either case, our challenge was to find suitable chemoselective conditions/strategies that would effect the desired transformation in the presence of the unmasked, competitive functionalities (COOH, NH2). Before these two routes could be investigated, easy access to the three building blocks was needed. Compound 4 was commercially available and compound 3 was easily prepared in two steps in water.9 However, practical multi-gram synthesis of unnatural, chiral β-aryl-α-amino acid 2 was not trivial.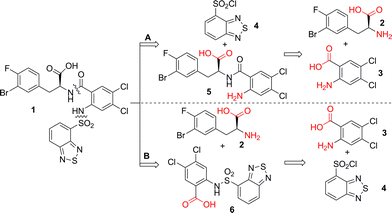 | ||
| Scheme 3 Retro-synthetic analysis of protecting-group-free synthesis of 1. | ||
The initial synthesis of compound 2 and analogs used by our medicinal chemistry colleagues in their SAR work involved the enantioselective alkylation of glycine derivatives with benzyl halides.10 The chirality was established in the alkylation step with the aid of either a pseudoephedrine chiral auxiliary or phase-transfer catalyst derived from a Cinchona alkaloid.11 These methods were useful for providing milligram quantities of material, but scaling-up proved to be difficult due to expensive reagents, low yields, and variable enantioselectivity. It was clear that a practical, scalable route was required.
In line with our pursuit of the “ideal synthesis”, we set a few criteria for the synthesis of 2: 1) the stereogenic center should be established by a catalytic method rather than a chiral auxiliary mediated approach; 2) functional and protecting group manipulations should be minimized; 3) the reactions should be robust, easily scalable, and environmentally friendly. With these considerations in mind, we chose inexpensive 3-bromo-4-fluorobenzaldehyde as the starting point. Two readily scalable steps (Erlenmeyer–Plöchl azlactone synthesis and hydrolysis) provided intermediate 7 in high yield without the need for chromatographic purification (Scheme 4).12 To establish the chirality, we envisioned two potential opportunities: the transition-metal catalyzed asymmetric hydrogenation of compound 7 or an enzymatic enantioselective amide hydrolysis of compound 8.
 | ||
| Scheme 4 Proposed enantioselective syntheses of 2. | ||
Transition-metal catalyzed asymmetric hydrogenation of α-amido cinnamic acids (or esters) for the synthesis of chiral phenylalanine derivatives is an extensively-investigated reaction. Ever since Knowles's landmark industrial scale synthesis of amino acidL-DOPA almost 40 years ago,13 which garnered him the 2001 Nobel prize along with Noyori and Sharpless, numerous chiral ligands have been developed for this transformation.14 However, the efficiency of chiral induction appeared to be highly substrate-dependent. For instance, the versatile ligand Me-DuPhos15 failed to afford the desired product 8 in our case. In contrast, a much less utilized ferrocene-based ligand Me-BoPhoz16 led to the complete conversion with 94% ee under low catalyst loading on a 40 gram scale (eqn (1)). Me-BoPhoz is commercially available, air stable, and does not require special handling, which satisfied our aforementioned criteria for a robust, scalable synthesis.
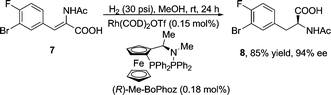 | (1) |
To increase the enantiomeric purity to >99% ee, two efficient strategies were successfully developed (Scheme 5). In the first route, an enzymatic amide hydrolysis was attempted to selectively hydrolyze the desired enantiomer 8 to acid 2. Acylases operate in environmentally friendly conditions and have been widely adapted in the industry for asymmetric hydrolysis because of their unparalleled precision and efficiency.17 Screening a number of commercially available acylases identified that the acylase “Amano”18 provided superb selectivity. Under mild conditions (pH = 8.0, 40 °C, 4 days, with CoCl2 as co-factor), the desired enantiomer 8 was hydrolyzed exclusively whereas the undesired minor enantiomer remained intact. Isolation of the desired product 2 as a single enantiomer (>99% ee) was easily achieved through pH adjustment followed by filtration. Alternatively, a classic recrystallization method was also developed to achieve the requisite enantiomeric enrichment. Systematic solvent screening revealed that a single recrystallization from 2-propanol/water provided a robust process for enantiomeric enrichment (>99% ee). Subsequent amide hydrolysis with 6 M HCl afforded the final product as the HCl salt in excellent yield. Both routes have been carried out on multi-gram scale to provide enantiomerically pure 2.
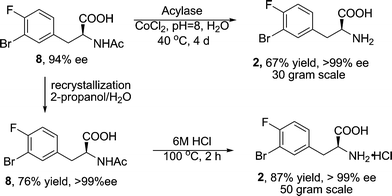 | ||
| Scheme 5 Enantiomeric enrichment and amide hydrolysis. | ||
With all three building blocks in hand, route A from Scheme 3 was first investigated. To construct 5 directly from 2 and 3, the requisite chemoselective amide bond formation would be difficult to achieve because of the existence of reactive amino and carboxylic acid groups in both 2 and 3. To circumvent this problem, isatoic anhydride 9 was chosen as a surrogate with an intrinsically activated acid group (Scheme 6).19 This strategy had been successfully utilized in the second-generation synthesis (Scheme 2), where the chemoselective ring-opening at the benzoic carbonyl (position 1) was achieved with the methyl ester of 2. However, when the same conditions were employed on 2 with an unprotected acid functional group, compound 10 was the only product in 90% isolated yield. It is noteworthy that the ring-opening reaction of isatoic anhydride at the 2 position is only rarely found in the literature, and usually in low yields.20 Apparently, the intrinsic reactivity of the free acid functionality of 2 facilitated the usually more difficult ring-opening reaction. Nevertheless, to access the desired compound 5, new conditions specific for the unprotected amino acid 2 were required. Adding water as co-solvent has been shown to promote the ring opening reaction of isatoic anhydrides at the 1 position;21 unfortunately in our case this did not provide the desired chemoselectivity (Scheme 6, entry 2). In contrast, with water as the sole solvent and pH control at 8.0, the chemoselectivity of this ring-opening reaction completely reversed to afford the desired compound 5 exclusively.22 Simple acidification of the reaction solution with aqueous HCl solution precipitated pure 5 in 83% yield.
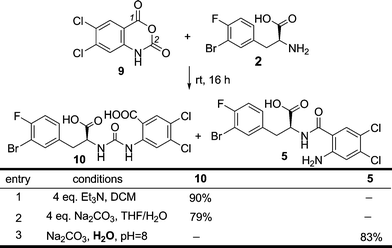 | ||
| Scheme 6 The chemo-selective ring-opening of isatoic anhydride 5. | ||
Unfortunately, the subsequent sulfonamide formation between compound 5 with 4 was surprisingly difficult. In contrast to the 87% yield obtained in the second-generation synthesis with the methyl ester of 5 (Scheme 2), the same sulfonamide formation conditions (pyridine/DCM) failed to produce any desired product 1 (Table 1, entry 1). Employment of stronger base (Et3N) or an increase of the reaction temperature did not promote this reaction (entries 2–4). Schotten–Baumann conditions23 in mixed organic/water solvent systems provided small amounts of 1 (entries 5 and 6). The best yield of 1 (20%) was obtained in pure water with pH control at 8.0 (entry 7).24 Addition of a phase transfer catalyst (Bu4N+Br−) was detrimental to this reaction (entry 8). The difficulty of forming the sulfonamide bond is probably due to the intramolecular hydrogen bonding between the carboxylic acid proton and the free amino group of compound 5. This route was not pursued further.

|
Route B (Scheme 3) was next investigated. Based on methodology we have developed previously,24 direct sulfonamide formation with unprotected 2-amino-4,5-dichloro-benzoic acid and compound 4 was achieved in pure water (Scheme 7). Compound 6 precipitated in 95% yield after simple acidification with concentrated HCl solution. The reaction was exceptionally green, proceeding in high yield and generating no organic waste.25 To avoid the use of expensive coupling reagents and the associated waste disposal problems, we chose to use acid chloride 11 for the subsequent amide bond forming reaction. The acid chloride 11 was readily prepared in quantitative yield under standard conditions.
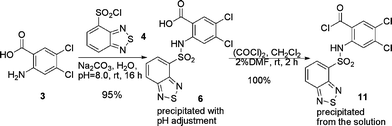 | ||
| Scheme 7 Direct sulfonamide formation in water. | ||
With 11 in hand, direct amide formation with unprotected 2 was investigated. Under classic and modified Schotten–Baumann conditions,26hydrolysis of 11 was simply too fast for the amide formation process to compete (Table 2, entries 1–4). The typical non-aqueous conditions (DCM/Et3N) also afforded the hydrolyzed acid 6 as the major product (entry 5). Switching to THF as the solvent significantly suppressed the hydrolysis reaction (entry 6).27 It appeared that stronger bases (entries 7, 8, 10) tended to promote the hydrolysis process, whereas weaker bases such as pyridine favoured the formation of 1 (entry 9). A weak and sterically hindered base, 2,6-lutidine, provided a synthetically useful 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of compound 1 and 6 (entry 11). Attempts to enhance this ratio with a bulkier base (2,6-di-t-butyl pyridine, entry 12) was counterproductive. Finally, we found that N,N-dimethylaniline provided the best ratio of 1:6 (20:1, entry 13). The reaction profile was rather clean and pure compound 1 was afforded in 70% isolated yield after recrystallization from iPrOAc. Since racemization of amino acids is often observed in amide coupling reactions, it is important to note that no loss of ee was observed during the above process. The same conditions were successfully applied to the synthesis of related analogs as well.
:1 ratio of compound 1 and 6 (entry 11). Attempts to enhance this ratio with a bulkier base (2,6-di-t-butyl pyridine, entry 12) was counterproductive. Finally, we found that N,N-dimethylaniline provided the best ratio of 1:6 (20:1, entry 13). The reaction profile was rather clean and pure compound 1 was afforded in 70% isolated yield after recrystallization from iPrOAc. Since racemization of amino acids is often observed in amide coupling reactions, it is important to note that no loss of ee was observed during the above process. The same conditions were successfully applied to the synthesis of related analogs as well.

|
Conclusions
Two “protecting-group-free” routes were explored for the preparation of compound 1, a dual CCK1/CCK2 receptor antagonist. In the presence of unprotected, competitive functional groups, development of new chemoselective conditions are often required even for the “easy” reactions such as amide and sulfonamide formation. Notable new reactions/conditions include a selective ring-opening reaction of isatoic anhydride in water, a sulfonamide formation in water and a PhNMe2 mediated amide formation reaction. In the end, a highly efficient, two-step route for the synthesis of 1 was developed.Experimental
General techniques
Proton and carbon NMR spectra were recorded on a 500 MHz or a 600 MHz NMR spectrometer. Flash column chromatography was performed using Merck silica gel 60. HRMS (ESI) was performed on a μTof apparatus. Melting points were determined using an electrothermal apparatus and are uncorrected. Unless specified, all the reagents and solvents were purchased from commercial sources and used without further purification. Optical rotation was measured on a Perkin–Elmer 341 polarimeter. Chiral HPLC analysis was performed on a Hewlett Packard 1100.Synthetic procedures
Acknowledgements
We wish to thank Dr Jiejun Wu, Ms. Heather McAllister and Ms. Bita Naderi for analytical support; and Dr Daniel J. Pippel for proof-reading this manuscript.Notes and references
- R. Herranz, Med. Res. Rev., 2003, 23, 559–605 CrossRef CAS.
- (a) M. Tonini, R. De Giorgio and F. De Ponti, Drugs, 2004, 64, 347–361 CrossRef CAS; (b) A. Varnavas and L. Lassiani, Expert Opin. Ther. Pat., 2006, 16, 1193–1213 Search PubMed; (c) J. J. Berna, J. A. Tapia, V. Sancho and R. T. Jensen, Curr. Opin. Pharmacol., 2007, 7, 583–593 CrossRef.
- (a) M. Pippel, B. D. Allison, V. K. Phuong, L. Li, M. F. Morton, C. Prendergast, X. Wu, N. P. Shankley and M. H. Rabinowitz, Bioorg. Med. Chem. Lett., 2009, 19, 6373–6375 CrossRef CAS; (b) M. Pippel, K. Boyce, H. Venkatesan, V. K. Phuong, W. Yan, T. D. Barrett, G. Lagaud, L. Li, M. F. Morton, C. Prendergast, X. Wu, N. P. Shankley and M. H. Rabinowitz, Bioorg. Med. Chem. Lett., 2009, 19, 6376–6378 CrossRef CAS.
- (a) K. McClure, M. Hack, L. Huang, C. Sehon, M. Morton, L. Li, T. D. Barrett, N. Shankley and J. G. Breitenbucher, Bioorg. Med. Chem. Lett., 2006, 16, 72–76 CrossRef CAS; (b) C. Sehon, K. McClure, M. Hack, M. Morton, L. Gomez, L. Li, T. D. Barrett, N. Shankley and J. G. Breitenbucher, Bioorg. Med. Chem. Lett., 2006, 16, 77–80 CrossRef CAS; (c) L. Gomez, M. D. Hack, K. McClure, C. Sehon, L. Huang, M. Morton, L. Li, T. D. Barrett, N. Shankley and J. G. Breitenbucher, Bioorg. Med. Chem. Lett., 2007, 17, 6493–6498 CrossRef CAS; (d) J. T. Liang, N. S. Mani and T. K. Jones, J. Org. Chem., 2007, 72, 8243–8250 CrossRef CAS; (e) C. M. Mapes, N. S. Mani, X. Deng, C. R. Pandit, K. J. McClure, M. C. W. Pippel, C. A. Sehon, L. Gomez, S. Shinde, J. G. Breitenbucher and T. K. Jones, J. Org. Chem., 2010, 75, 7950–7953 CrossRef CAS.
- (a) B. D. Allison, V. K. Phuong, L. C. McAtee, M. Rosen, M. Morton, C. Prendergast, T. Barrett, G. Lagaud, J. Freedman, L. Li, X. Wu, H. Venkatesan, M. Pippel, C. Woods, M. C. Rizzolio, M. Hack, K. Hoey, X. Deng, C. King, N. P. Shankley and M. H. Rabinowitz, J. Med. Chem., 2006, 49, 6371–6390 CrossRef CAS; (b) C. R. Woods, M. D. Hack, B. D. Allison, V. K. Phuong, M. D. Rosen, M. F. Morton, C. E. Prendergast, T. D. Barrett, N. P. Shankley and M. H. Rabinowitz, Bioorg. Med. Chem. Lett., 2007, 17, 6905–6909 CrossRef CAS; (c) M. D. Rosen, M. D. Hack, B. D. Allison, V. K. Phuong, C. R. Woods, M. F. Morton, C. E. Prendergast, T. D. Barrett, C. Schubert, L. Li, X. Wu, J. Wu, J. M. Freedman, N. P. Shankley and M. H. Rabinowitz, Bioorg. Med. Chem., 2008, 16, 3917–3925 CrossRef CAS; (d) M. D. Rosen, Z. M. Simon, K. T. Tarantino, L. X. Zhao and M. H. Rabinowitz, Tetrahedron Lett., 2009, 50, 1219–1221 CrossRef CAS.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS.
- (a) R. W. Hoffmann, Synthesis, 2006, 3531–3541 CrossRef CAS; (b) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS; (c) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS; (d) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- Recent examples: (a) R. M. McFadden and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 7738–7739 CrossRef CAS; (b) X. Li and A. K. Yudin, J. Am. Chem. Soc., 2007, 129, 14152–14153 CrossRef CAS; (c) A. V. Gudmundsdottir and M. Nitz, Org. Lett., 2008, 10, 3461–3463 CrossRef CAS; (d) M. D. Lainchbury, M. I. Medley, P. M. Taylor, P. Hirst, W. Dohle and K. I. Booker-Milburn, J. Org. Chem., 2008, 73, 6497–6505 CrossRef CAS; (e) C. D. Grant and M. J. Krische, Org. Lett., 2009, 11, 4485–4487 CrossRef CAS; (f) K. Lee, H. Kim and J. Hong, Org. Lett., 2009, 11, 5202–5205 CrossRef CAS; (g) P. Chen, L. Cao and C. Li, J. Org. Chem., 2009, 74, 7533–7535 CrossRef CAS.
- M. Teng, M. D. Johnson, C. Thomas, D. Kiel, J. N. Lakis, T. Kercher, S. Aytes, J. Kostrowicki, D. Bhumralkar, L. Truesdale, J. May, U. Sidelman, J. T. Kodra, A. S. Jorgensen, P. H. Olesen, J. C. de Jong, P. Madsen, C. Behrens, I. Pettersson, L. B. Knudsen, J. J. Holst and J. Lau, Bioorg. Med. Chem. Lett., 2007, 17, 5472–5478 CrossRef CAS.
- B. Allison, V. K. Phuong, M. C. W. Pippel, M. H. Rabinowitz, H. Venkatesan, US Patent 0069286, 2006.
- For reviews, see: (a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS; (b) M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef CAS.
- (a) E. Erlenmeyer, Justus Liebigs Ann. Chem., 1904, 337, 205–221 CrossRef CAS; (b) F. Bailly, C. Queffélec, G. Mbemba, J.-F. Mouscadet, N. Pommery, J. Pommery, J.-P. Hénichart and P. Cotelle, Eur. J. Med. Chem., 2008, 43, 1222–1229 CrossRef CAS.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS.
- For a review, see: U. Nagel and J. Albrecht, Top. Catal., 1998, 5, 3–23 Search PubMed.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125–10138 CrossRef CAS.
- (a) N. W. Boaz, S. D. Debenham, E. B. Mackenzie and S. E. Large, Org. Lett., 2002, 4, 2421–2424 CrossRef CAS; (b) N. W. Boaz, S. E. Large, J. A. Ponasik Jr., M. K. Moore, T. Barnette and W. D. Nottingham, Org. Process Res. Dev., 2005, 9, 472–478 Search PubMed.
- H. K. Chenault, J. Dahmer and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 6354–6364 CrossRef CAS.
- M. Bois-Choussy and J. Zhu, J. Org. Chem., 1998, 63, 5662–5665 CrossRef CAS.
- (a) H. A. McManus and P. J. Guiry, J. Org. Chem., 2002, 67, 8566–8573 CrossRef CAS; (b) T. Asano, T. Yoshikawa, H. Nakamura, Y. Uehara and Y. Yamamoto, Bioorg. Med. Chem. Lett., 2004, 14, 2299–2302 CrossRef CAS; (c) A. L. Sabb, R. L. Vogel, G. S. Welmaker, J. E. Sabalski, J. Coupet, J. Dunlop, S. Rosenzweig-Lipson and B. Harrison, Bioorg. Med. Chem. Lett., 2004, 14, 2603–2607 CrossRef CAS; (d) A. Varnavas, L. Lassiani, V. Valenta, L. Mennuni, F. Makovec and D. Hadjipavlou-Litina, Eur. J. Med. Chem., 2005, 40, 563–581 CrossRef CAS; (e) N. Hunter and K. Vaughan, J. Heterocycl. Chem., 2006, 43, 731–738 CrossRef CAS; (f) P. Hradil, M. Grepl, J. Hlavac, M. Soural, M. Malon and V. Bertolasi, J. Org. Chem., 2006, 71, 819–822 CrossRef CAS.
- (a) R P. Staiger and E. C. Wagner, J. Org. Chem., 1953, 18, 1427–1439 CrossRef CAS; (b) R. P. Staiger and E. B. Miller, J. Org. Chem., 1959, 24, 1214–1219 CrossRef CAS; (c) A. M. Felix, L. B. Czyzewski, D. P. Winter and R. I. Fryer, J. Med. Chem., 1969, 12, 384–387 CrossRef CAS; (d) H. L. Yale, J. Heterocycl. Chem., 1977, 14, 1357–1359 CrossRef CAS.
- (a) M. G. Bock, R. M. Dipardo, S. M. Pitzenberger, C. F. Homnick, J. P. Springer and R. M. Freidinger, J. Org. Chem., 1987, 52, 1646–1647 CrossRef; (b) M. Bakavoli, A. Davoodnia and R. Shahnaee, Chin. Chem. Lett., 2008, 19, 12–14 CrossRef CAS; (c) M-C Tseng, C-Y Lai, Y-W Chu and Y-H. Chu, Chem. Commun., 2009, 445–447 RSC.
- B. Maryanoff, J. Matthews, PCT Int. Appl. WO 2006049984 A2.
- C. M. R. Low, H. B. Broughton, S. B. Kalindjian and I. M. McDonald, Bioorg. Med. Chem. Lett., 1992, 2, 325–330 CrossRef CAS.
- X. Deng and N. S. Mani, Green Chem., 2006, 8, 835–838 RSC.
- P. Anastas, J. Warner, Green Chemistry: Theory and Practice, Oxford Univ. Press, 1998 Search PubMed.
- (a) B. S. Jursic and D. Neumann, Synth. Commun., 2001, 31, 555–564 CrossRef CAS; (b) A. G. Godfrey, D. A. Brooks, L. A. Hay, M. Peters, J. R. McCarthy and D. Mitchell, J. Org. Chem., 2003, 68, 2623–2632 CrossRef CAS.
- L. Zhang, X. Wang, J. Wang, N. Grinberg, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2964–2966 CrossRef CAS.
- The ee of the hydrogenation reaction on different batches ranged from 88%–95%.
- Compound 2 was converted back to 8 viaN-acetylation (Ac2O, NaHCO3, H2O, 0 °C) for ee determination.
- The solid contained Et3N·HCl. The amount varied from batch to batch.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, 1H and 13C NMR spectra for compounds 1, 2, 5-8, 10, 11 and chiral HPLC chromatogram of compounds 1 and 8. See DOI: 10.1039/c0ob01004a |
| This journal is © The Royal Society of Chemistry 2011 |