Overcoming challenges in the palladium-catalyzed synthesis of electron deficient ortho-substituted aryl acetonitriles†
Molly C.
Brannock
,
William J.
Behof
,
Gregory
Morrison
and
Christopher B.
Gorman
*
Box 8204, Department of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: cbgorman@ncsu.edu; Fax: +1-919-515-8920; Tel: +1-919-515-4252
First published on 2nd March 2011
Abstract
Highly electron deficient monoaryl, di-aryl and bis-diaryl acetonitriles were effectively synthesized using either a nucleophilic aromatic substitution (NAS) or a palladium-mediated coupling pathway. Synthesis of di-aryl acetonitriles most conveniently proceeded via NAS – palladium-mediated coupling was not required. This reaction, however, results in a product that is more acidic than the reactants. Facile deprotonation of the product prevents efficient formation of the bis-diaryl acetonitrile through a NAS pathway. Thus, palladium-mediated coupling is required to prepare the bis-diaryl acetonitrile efficiently. In the palladium-catalyzed coupling, choice of base and solvent (and thus the counter cation for the benzylic anion nucleophile) is important. Also, choice of the supporting ligand is important, indicating the sensitivity of the reaction to steric and ligand electronic effects.
As part of an ongoing research program in the synthesis of cyano-containing polymers, we became interested in synthesizing oligomers and polymers with the general repeat unit shown in Scheme 1. We have recently reported the cascading cyclization of similar aryl and benzyl cyano-containing oligomers to form isoquinoline-type fused-ring molecules (Scheme 2).1 These conjugated materials may have potential for use in organic devices.
 | ||
| Scheme 1 Two possible methods of coupling to form a bis(o-cyanophenyl)acetonitrile (M = metal counterion, LG = leaving group). | ||
 | ||
| Scheme 2 Example of the conversion of a multiple-cyano containing oligomer to the fully cyclized, aromatic product. | ||
The key synthetic step here is the carbon–carbon bond formation of a benzyl/phenyl linkage to form a diaryl methane subunit. Scheme 1 shows the two logical bond deconstructions that could give rise to this linkage. The aryl group could be nucleophilic and the benzylic position would then be the electrophile (Scheme 1, top). Alternatively, these roles could be reversed (Scheme 1, bottom).
Using an aryl organometallic as a nucleophilic equivalent has been useful to prepare aryl–aryl and aryl–alkyl linkages.2–4 The use of aryl nucleophiles with ortho substituents, however, has had mixed results.5 In contrast, deprotonation of RCH2CN or R2CHCN and subsequent use as a nucleophilic equivalent has had several successful precedents. You and Verkade6,7 illustrated coupling of alkyl acetonitrile anions to aryl halides in the presence of a proazaphosphatrane ligand. Hartwig et al., Satoh et al. and Verkade et al. employed, with high efficiency, phenyl acetonitrile for monoarylations and acetonitrile for di-arylations.6–11 Culkin and Hartwig8,10 showed that the anion of alkyl nitriles could undergo a palladium-mediated coupling to aryl halides and studied the mechanism of this transformation in some detail. Wu and Hartwig11 later showed that an α-silyl nitrile was an efficient palladium-mediated coupling partner to aryl halides in the presence of zinc fluoride.
Given these precedents, a cyanobenzyl nucleophile was selected for this coupling. Moreover, o-cyanophenyl acetonitrile is relatively acidic, (19.2 in dimethyl sulfoxide (DMSO))12 and thus easily deprotonated. The resulting carbanion would then be a suitable nucleophile in a carbon–carbon coupling reaction. The pKa determination of similar molecules containing such groups will be reported below. Furthermore, the arene is relatively electron deficient which renders it a good electrophile.
The examples given above suggest several challenges and raise several questions regarding the carbon–carbon bond forming reaction under study here. First, since our proposed arene is so electron deficient, can simple nucleophilic aromatic substitution compete with palladium-mediated coupling? This question may also be relevant in some of the examples shown above. Second, in the work above, limitations were observed when orthocyano groups were present on the arene electrophile.13 The presence of o-cyano groups in our target might thus be an issue. Third, since the methine proton in the product (e.g.Ph2(CN)CH) should be more acidic than the proton that must be removed in the starting material (e.g.Ph(CN)CH2), can the reaction be driven forward in a reaction solution in which a less acidic proton must be removed in the presence of a more acidic proton? This issue is illustrated in Scheme 3.
 | ||
| Scheme 3 Equilibrium between benzylic anions. | ||
In this paper, nucleophilic aromatic substitution and palladium-mediated coupling reactions will be explored to determine how efficiently the reaction illustrated in the bottom half of Scheme 1 can occur. The relative efficacy of these two pathways will be compared. Choice of base, solvent, and catalyst/ligand will be shown to be key parameters in optimizing the palladium-catalyzed coupling. Finally, an optimal, high yielding route will be illustrated.
In the work of Hartwig et al.8,10,11 and You and Verkade6,7 discussed above, both electron rich and electron deficient aryl halide substrates were explored. In the coupling of interest here, the aryl halide is electron deficient, begging the question as to whether, in this case, a nucleophilic aromatic substitution (NAS) reaction would be applicable. We tested this in three ways. First, a reaction from the literature was repeated under NAS conditions. Then from this, two new coupling reactions were explored.
Initially, the NAS pathway was investigated in the case of an electron deficient arene. Culkin and Hartwig reported formation of 2 from p-bromobenzonitrile (1a) in 99% yield in the presence of palladium acetate (Pd(OAc)2) and 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP),8 and You and Verkade reported a similar reaction using p-chlorobenzonitrile (1b) to achieve 2 in 92% yield in the presence of Pd(OAc)2 and a proazaphosphatrane ligand (Scheme 4).6 We repeated these reactions in the absence of palladium/ligand under the same conditions and obtained 42% (X = Br) and 72% (X = Cl) yield, respectively. Thus, palladium-mediated coupling does result in a higher yield. However, in this case, the results provide evidence for competition between NAS and the palladium-mediated pathway.
 | ||
| Scheme 4 NAS reactions run to compare with previously reported, Pd-mediated couplings. Conditions: (CH3)2CHCN, sodium hexamethyldisilazide (NaHMDS), toluene (i) 1a, 100 °C, 1 h (ii) 1b, 90 °C, 2 h. | ||
We then turned to the NAS of 2,6-dichlorobenzonitrile (3a) with o-cyanophenyl acetonitrile (4). When two equivalents of 4 were reacted with 3a in the presence of sodium tert-butoxide (NaOtBu) (Scheme 5), only the mono-coupled product 5a was obtained in 91% yield. When 5a was isolated and reacted with 4 for a longer period of time (72 h) and in excess (3.2 equivalents) base, only a small amount (13%) of bis-coupled product 6a was isolated. This reaction was clearly inefficient for the formation of 6.
 | ||
| Scheme 5 Attempted formation of 6avia NAS. | ||
Our strategy then shifted toward a palladium-mediated coupling given the poor results when attempting to prepare 6via NAS. Culkin and Hartwig8 showed that Pd(OAc)2/BINAP is an efficient palladium/ligand combination to couple phenyl acetonitrile anions with p-tBu-bromobenzene. These conditions were used as the starting point to optimize the reaction for the formation of 6. Because it is the second coupling that is challenging, the reaction of 5 with 4 was explored. Since palladium-mediated couplings typically are more efficient on aryl bromides than chlorides, 3a was replaced with 3b, and 5b was synthesized in 93% yield using NAS in dimethyl formamide (DMF) (Scheme 6).
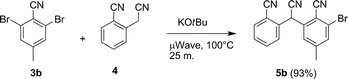 | ||
| Scheme 6 Preparation of 5b. | ||
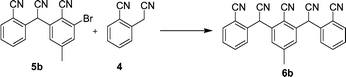
Reaction of 4 and 5 under palladium-mediated conditions was then explored. The results are shown in Table 1. A maximum yield of 62% was obtained. Comparison of entries 1 and 2 in Table 1 indicates that use of the aryl bromide indeed does result in a higher yield. Comparison of entries 2 and 3 indicates that NAS is less efficient compared to palladium-mediated coupling. Comparison of entries 3 and 4 in Table 1 indicates that microwave heating was more efficient than thermal heating and gave a much higher yield of product. Moreover, extending the reaction time under thermal heating showed no further increase in yield when the reaction time was increased from 4 h to 6 h. For these reasons, microwave heating was used in all subsequent reactions. Also, poor yields were obtained when fewer than 3 equiv. of base were added. This point is treated below.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Eq. KOtBu | Heat Source | T/°C | Time | % 6ba |
| Conditions: 0.3 M THF, 1.2 eq. 4, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP.a Values obtained from HPLC with an estimated error of ± 5%.b Molecule 5a was used instead of molecule 5b.c No Pd catalyst or BINAP was added. | |||||
| 1 | 3.2 | Microwave | 130 | 5 min | 0b |
| 2 | 3.2 | Microwave | 130 | 5 min | 33c |
| 3 | 3.2 | Microwave | 130 | 5 min | 62 |
| 4a | 3.2 | Thermal | 80 | 2 h | 17 |
| 4b | 3.2 | Thermal | 80 | 4 h | 37 |
| 4c | 3.2 | Thermal | 80 | 6 h | 36 |
| 5 | 2.1 | Microwave | 130 | 5 min | 29 |
| 6 | 1.1 | Microwave | 130 | 5 min | 5 |
Palladium-catalyzed coupling can be greatly influenced by the base employed. Furthermore, the data above indicate that excess base is required. This requirement likely results because (1) both 5 and 6 are deprotonated preferentially to 4 and (2) any equilibrium between 5− or 6− and 4 favors 5− or 6−. At first, we speculated that the opposite might be true. You and Verkade showed that the reaction between bromobenzene and benzyl cyanide to form diphenyl acetonitrile could be accomplished in 93% yield using 1.4 equivalents of sodium hexamethyl disilazide.6 The pKa values of benzyl cyanide and diphenyl acetonitrile are 21.9 and 17.5, respectively.14 Thus, we speculate that there likely was some equilibrium between the anion of the product and that of the starting material in this case. However, in our case, the o-cyano groups likely have an important influence on the pKa (and more importantly, the relative pKa) values of our starting materials and product.
To determine the relative acidity of the protons on 4, 5 and 6, pKa measurements in DMSO were performed using the procedure developed by Bordwell et al.15 The pKa of 4 was measured to be 19.2, consistent with that reported in the literature.12 Molecule 5b had a pKa of 13.6 and molecule 6b had pKa1 and pKa2 values of 13.2. The similarity of the two pKa values for 6b is consistent with the findings of Streitwieser where unfused diprotic structures were determined to have indistinguishable pKa values.16 Thus, if the anion of 4 is produced, it is likely in equilibrium with the anion of 5 (cf.Scheme 3) and the (di)anion of 6. Given the values, this equilibrium is likely to be unfavorable, resulting in the need to use more than one, or even two, equivalents of base. This need is in contrast to the results reported by You and Verkade above.
The pKa values may be only partially relevant, however. There are reports that suggest weak bases such as potassium carbonate (K2CO3)17 and dimethylamino pyridine (DMAP)18 are able to deprotonate 4. However, in THF the relative acidities of 4–6 might be quite different. Based on computations, Ding et al. suggest that neutral acids are typically eleven orders less acidic in THF than in DMSO.19 Furthermore, even if the anions of 5 and 6 are produced, they may be innocent or unreactive. Their reactivity particularly depends on the solvent and counter-cation present.20 Thus, some variation of base, solvent and counter-cation was explored.
To explore variation of base in an efficient manner, reactions were conducted under otherwise identical conditions and analyzed by HPLC. Both the percentage of unreacted 5 and the percentage of 6 are given in Table 2. Weak bases (entries 1–5) clearly were ineffective. Potassium tert-butoxide yielded the greatest conversion. Addition of 18-crown-6, however, resulted in little recovered starting material or product. Thus, use of the larger potassium counterion favors product formation, but complexation of the potassium tends to produce side products (e.g. loss of starting material without formation of product).
|
|
|||
|---|---|---|---|
| Yield% ofa | |||
| Entry | Base | Recovered 5b | 6b |
| Conditions: 0.3 M in THF, 1.2 eq. 4, 3.2 eq. base, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP, 130 °C, μW, 100 W, 5 min.a Values obtained from HPLC with an estimated error of ± 5%.b Not detected by HPLC. | |||
| 1 | CsF | 93 | 4 |
| 2 | NEt3 | 100 | ndb |
| 3 | Pyridine | 97 | nd |
| 4 | Ph2NH | 98 | 1 |
| 5 | Cs2CO3 | 87 | 8 |
| 6 | NaOMe | 47 | 22 |
| 7 | NaOiPr | 53 | 1 |
| 8 | LiOtBu | 100 | nd |
| 9 | NaOtBu | 74 | 26 |
| 10 | KOtBu | 15 | 62 |
| 11 | KOtBu/0.1 eq. 18-crown-6 | 3 | 37 |

As this reaction generates anions that must transmetallate to palladium, and as metalated nitriles form both complex aggregates with counterions21 and also complex to palladium,7 the choice of counterion and solvent is likely to have a large influence on the efficiency of this reaction. Thus, several solvents were explored for further optimization of the coupling. The results are presented in Table 3. The best conditions found above are reproduced as Entry 1. Inohet al. used Cs2CO3/DMF to deprotonate p-nitro toluenes (pKa of 20.4 in DMSO),22 yet entries 2 and 3 in Table 3 indicate poor conversion and loss of starting material when DMF was used. When 6b was heated in DMF briefly, decomposition was observed indicating that DMF is not a suitable solvent for this reaction. N-Methylpyrrolidone (NMP) was tried as an alternative polar, aprotic solvent (Table 3, entry 4). The desired product was obtained in 20% yield with no starting material recovered. However, when mixed NMP/THF was used (Table 3, entries 5–9), excellent results were obtained. In 90/10 THF/NMP, in the presence of 0.1 eq. 18-crown-6, an 83% yield of product was obtained. Note that in the presence of NMP, 18-crown-6 increased the reaction yield. This behavior was not the case in pure THF.
| Yield% ofa | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Ratio | Base | Recovered 5b | 6b |
| Conditions: 1.2 eq. 4, 3.2 eq. base, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP, 130 °C, μW, 100 W, 5 min.a Obtained from HPLC with an estimated error of ± 5%.b Not detected by HPLC.c Entry 10 of Table 2 repeated for ease of comparison. | |||||
| 1c | THF | — | KOtBu | 15 | 62 |
| 2 | DMF | — | KOtBu | 14 | 31 |
| 3 | DMF | — | Cs2CO3 | 28 | 26 |
| 4 | NMP | — | KOtBu | ndb | 20 |
| 5 | THF/NMP | 90/10 | KOtBu | 5 | 71 |
| 6 | THF/NMP | 85/15 | KOtBu | 24 | 65 |
| 7 | THF/NMP | 80/20 | KOtBu | ndb | 71 |
| 8 | THF/NMP | 50/50 | KOtBu | ndb | 49 |
| 9 | THF/NMP | 90/10 | KOtBu + 0.1 eq. 18-crown-6 | ndb | 83 |
| 10 | THF/NMP | 90/10 | KOH | 84 | 18 |
| 11 | THF/NMP | 90/10 | Cs2CO3 | 64 | 9 |
| 12 | THF/NMP | 90/10 | K2CO3 | 93 | 1 |
| 13 | THF/NMP | 90/10 | K2CO3 + 0.1 eq. 18-crown-6 | 94 | 10 |
| 14 | THF/NMP | 90/10 | KOCH3 | 4 | 47 |
| 15 | THF/NMP | 90/10 | KOiPr | 76 | 16 |
| 16 | THF/HOtBu | 75/25 | KOtBu | 6 | 75 |
| 17 | THF/HOtBu | 50/50 | KOtBu | 18 | 63 |
| 18 | THF/HOtBu | 25/75 | KOtBu | 21 | 61 |
To test the efficiency of a milder base in this solvent system, (Table 3, entries 10–13) several other, weaker bases than KOtBu were explored. The moderate conversion to 6b using potassium hydroxide (KOH) (Table 3, entry 10) indicates that KOH was strong enough for deprotonation, but this base was not efficient. It was suspected that the formation of water upon the protonation of hydroxide anion might be hindering the further formation of product by inactivating the palladium. To test the effect of water on the yield, reactions containing 0.22, 0.44, and 1.1 equivalents of water were run. Yields were found to change minimally from 71% to 76% when 0.22 eq. were added, 70% when 0.44 equivalents were added, but decreased dramatically from 71% to 39% when 1.1 eq. were added. Carbonate bases were also explored but gave poor yields. The slightly less basic potassium methoxide and potassium iso-propoxide gave little advantage in yield. Furthermore, addition of tert-butyl alcohol (as a buffer) systematically decreased the yield of the desired product. Thus the relatively strong potassium tert-butoxide base was employed.
Next, additional monodentate and bidentate phosphine ligands were tested for coupling efficiency (Table 4). The bidentate bis-diphenylphosphinoferrocene (dppf) was used both in the presence and absence of 18-crown-6 (entries 2 and 3 respectively). A slight increase in yield was observed in the absence of 18-crown-6, so subsequent reactions were run without this additive. Use of the bidentate bis-diphenylphosphinobutane (dppb) and bis-diphenylphosphinoethane (dppe) resulted in good yields (entries 4 and 5). Three monodentate ligands were then explored (entries 6, 7, and 8). Of those tested, tri-cyclohexylphosphine (P(Cy)3) provided the best results. The comparably poor yield obtained with P(tBu)3 indicates a large sensitivity to the steric bulk of the supporting ligand.
| Yield% ofa | |||
|---|---|---|---|
| Entry | Ligand | Recovered 5b | 6b |
| Conditions: 0.3 M THF/NMP 90/10 solvent, 1.2 eq. 4, 3.2 eq. KOtBu, 0.1 eq. Pd(OAc)2, 0.2 eq. ligand, 130 °C, μW, 100 W, 5 min.a Values obtained from HPLC with an estimated error of ± 5%.b Data from entry 9 of Table 3 repeated for ease of comparison.c 0.1 eq. 18-crown-6. | |||
| 1 | BINAP | 0 | 83b,c |
| 2 | dppf | 1 | 75c |
| 3 | dppf | 2 | 87 |
| 4 | dppb | 1 | 89 |
| 5 | dppe | 50 | 53 |
| 6 | P(tBu)3 | 18 | 36 |
| 7 | P(Cy)3 | 7 | 90 |
| 8 | P(Ph)3 | 33 | 64 |
Conclusions
In palladium-mediated coupling to form diaryl acetonitriles, the reaction yield varied substantially with the choice of base, solvent and supporting ligand. In the absence of palladium, NAS could form the products, but not as efficiently as under palladium-mediated coupling conditions. Microwave heating also contributed substantially to the increased yield when compared to thermal heating. KOtBu was found to be the best base in a 90/10 ratio of THF/NMP with P(Cy)3 as the supporting ligand.Experimental section
Instrumental analysis
HPLC was performed on a Grace Nucleosil C18 (5 micron, 4.6 mm ID, 250 mm length) column. The mobile phase was a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 acetonitrile/H2O solvent system at a flow rate of 1.0 mL min−1. Solvents were filtered HPLC-grade, and the H2O was adjusted to a pH of 2.88 using glacial acetic acid. 3,5-Dimethylanisole served as the internal standard. UV-vis spectra were recorded on a JASCO V-550 spectrophotometer. LCMS data were collected from an Agilent Technologies 6210 LC-TOF mass spectrometer equipped with an Agilent SB-C18 1.8 μm 2.1 × 50 mm column. Samples were diluted in methanol and analyzed via a 1 μL injection at 400 μL min−1 in a water–methanol gradient with 0.1% formic acid. The mass spectrometer was operated in positive-ion mode with a capillary voltage of 4 kV, nebulizer pressure of 30 psig, and a drying gas flow rate of 12 L min−1 at 350 °C. The fragmentor and skimmer voltages were 210 and 65 V, respectively.
:30 acetonitrile/H2O solvent system at a flow rate of 1.0 mL min−1. Solvents were filtered HPLC-grade, and the H2O was adjusted to a pH of 2.88 using glacial acetic acid. 3,5-Dimethylanisole served as the internal standard. UV-vis spectra were recorded on a JASCO V-550 spectrophotometer. LCMS data were collected from an Agilent Technologies 6210 LC-TOF mass spectrometer equipped with an Agilent SB-C18 1.8 μm 2.1 × 50 mm column. Samples were diluted in methanol and analyzed via a 1 μL injection at 400 μL min−1 in a water–methanol gradient with 0.1% formic acid. The mass spectrometer was operated in positive-ion mode with a capillary voltage of 4 kV, nebulizer pressure of 30 psig, and a drying gas flow rate of 12 L min−1 at 350 °C. The fragmentor and skimmer voltages were 210 and 65 V, respectively.
pKa measurements
The procedure of Bordwell15 was followed for the pKa determination of mono-protic acids 4 and 5. DMSO was distilled under reduced pressure from sodium amide and a few milligrams of triphenylmethane without first being dried over molecular sieves. All solvents and stock solutions were made and stored in the nitrogen drybox until removed for spectral analysis in a quartz cuvette capped with a rubber septum and parafilm to minimize exposure to air and moisture. Unknown acids were first added (via syringe) to the potassium dimsyl solutions and then titrated with indicator acids. All indicators were purified as described in the literature.23Toluene and tetrahydrofuran were distilled from sodium and benzophenone and were stored in a nitrogen filled dry box. The pKa value for the diprotic acid 6 was obtained in the same way, assuming that only the dianionic species was formed (e.g. no spectral signature for the intermediate, mono-anionic species was observed during the titration).Synthesis of 4-(cyano-dimethyl-methyl)-benzonitrile (2)
NaHMDS (256 mg, 1.4 mmol) and toluene (2 mL) were added to a Schlenk flask containing 4-chlorobenzonitrile (137 mg, 1 mmol). Under nitrogen, isobutyronitrile (83 mg, 1.2 mmol) was added dropwise and allowed to react at 90 °C for 2 h. The reaction was quenched using dilute HCl, extracted with EtOAc, and purified by column chromatography on silica gel (1:4 EtOAc/hexanes) to give the desired product (123 mg, 72%) as a pale yellow solid. All spectral data matched reported values.6,8 Mp: 86–88 °C; 1H NMR (CD2Cl2): δ = 1.75 (s, 6H), 7.63 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H); 13C NMR (CD2Cl2): δ = 29.0, 37.7, 112.2, 118.4, 123.5, 126.3, 133.0, 146.7; IR (KBr): 3040, 2985, 2920, 2225, 1608, 1505, 1475, 1468, 1454, 1404, 1390, 1368, 1288, 1237, 1198, 1181, 1102, 1021, 930, 766, 734, 631, 569, 551, 541 cm−1.
Synthesis of 2-chloro-6-[cyano-(2-cyano-phenyl)-methyl]-benzonitrile (5a)
To a stirring solution of KOtBu (246 mg, 2.2 mmol) in THF (4 mL) was added 4 (340 mg, 2.4 mmol) dropwise. Upon anion formation, the solution was added dropwise to a Schlenk flask containing a stirring solution of 2,6-dichlorobenzonitrile (172 mg, 1.0 mmol) in THF (1 mL). The combined solution was allowed to react at 80 °C for 16 h. The reaction was quenched using dilute HCl, extracted with EtOAc, and purified by column chromatography on silica gel (1:2 EtOAc/hexanes) to give the desired product (253 mg, 91%) as a yellow solid: Mp: 138–142 °C; 1H NMR (CDCl3): δ = 5.87 (s, 1H), 7.51–7.76 (m, 7H); 13C NMR (CDCl3): δ = 39.9, 112.8, 113.6, 113.7, 116.1, 116.4, 127.9, 129.8, 130.2, 130.8, 134.2, 134.4, 134.6, 135.9, 138.9, 139.0; IR (KBr): 2917, 2229, 1589, 1483, 1446, 1204, 1172, 1138, 889, 768, 632 cm−1; Anal. Calcd for C16H18ClN3 (277.04): C, 69.20; H, 2.90; N, 15.13. Found: C, 69.35; H, 2.87; N, 15.03.
Synthesis of 2-bromo-6-[cyano-(2-cyano-phenyl)-methyl]-4-methyl-benzonitrile (5b)
To a stirring solution of KOtBu (1.38 g, 12.3 mmol) in DMF (15.5 mL) was added 4 (1.91 g, 13.4 mmol). The solution was stirred for 30 min then added dropwise to a stirring solution of 3b (1.55 g, 5.6 mmol) in DMF (7.8 mL) in a Schlenk flask. The solution was heated to 85 °C and allowed to react under nitrogen for 12 h. The reaction was quenched using 2 M HCl, extracted with EtOAc, rinsed with brine, and purified by column chromatography on silica gel (1:2 EtOAc/hexanes) to give the desired product (1.75 g, 93%) as a white solid: Mp: 141–144 °C; 1H NMR (CDCl3): δ = 2.43 (s, 1H), 5.83 (s, 1H), 7.36–7.75 (m, 7H); 13C NMR (CDCl3): δ = 21.8, 39.7, 112.6, 112.7, 114.9, 116.0, 116.2, 127.1, 129.1, 129.5, 129.9, 133.9, 134.2, 134.3, 135.8, 138.4, 146.2; IR (KBr): 3072, 2921, 2229, 1598, 1551, 1485, 1451, 1290, 1255, 1213, 1099, 911, 863, 766, 732, 652 cm−1; Anal. Calcd for C17H10BrN3 (335.01): C, 60.73; H, 3.00; N, 12.50. Found: C, 60.68; H, 2.98; N, 12.33.
Synthesis of 2,6-bis-[cyano-(2-cyano-phenyl)-methyl]-benzonitrile (6a)
To 5a (80 mg, 0.29 mmol), 4 (50 mg, 0.35 mmol), and NaOtBu (104 mg, 0.93 mmol) was added THF (1.5 mL) in the drybox. The solution was allowed to react at reflux for 72 h. The reaction was quenched using dilute HCl, extracted with EtOAc, and purified by column chromatography on silica gel (1:1 EtOAc/hexanes) to give the desired product (14.4 mg, 13%) as a white solid: Mp. > 240° (dec.); UV-vis (THF) λmax (log ε): 225 (4.7), 278 (3.7) nm; 1H NMR (CD2Cl2): δ = 5.88 and 5.89 (s, 2H, diastereotopic), 7.51–7.59 (m, 4H), 7.66–7.87 (m, 7H); 13C NMR (CD2Cl2): δ = 40.3, 40.3 (diastereotopic), 113.2, 113.3, 113.5, 113.8, 114.4, 116.4, 116.5, 116.7, 116.8, 130.1, 130.5, 130.5, 130.7, 130.7, 134.4, 134.5, 134.8, 135.0, 136.5, 136.5, 139.2; IR (KBr): 3076, 2924, 2227, 1595, 1450, 1266, 763 cm−1; ESI-MS (210 V, MeOH–0.1% formic acid) m/z (%): 406 ([MH+Na]+, 100), 384 (49), 385 (12), 407 (26). HRMS (ESI) for C25H13N5 [M+H]+ calcd 383.1171, found 383.1167.
Synthesis of 2,6-bis-[cyano-(2-cyano-phenyl)-methyl]-4-methyl-benzonitrile (6b)
To a microwave vial was added 5b (84 mg, 0.25 mmol), 4 (43 mg, 0.3 mmol), KOtBu (90 mg, 0.8 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), P(Cy)3 (16.8 mg, 0.050 mmol), THF (450 μL), NMP (50 μL), and 3,5-dimethylanisole as an internal standard (350 μL). The mixture was allowed to react in the microwave reactor at 130 °C, 100 W, for a run time of 3 min, hold time of 5 min, and pressure limit of 150 psi. An aliquot was removed and diluted in a 70:30 acetonitrile/acidified H2O (pH = 2.88) solution, filtered, and analyzed by HPLC to yield converted product and recovered starting material. Mp. > 240 °C (dec.); UV-vis (THF) λmax (log ε): 225 (4.8), 250 (4.1), 278 (3.8) nm; 1H NMR (CDCl3): δ = 2.51–2.56 (s, 3H, diastereotopic), 5.81–5.82 (s, 2H, diastereotopic), 7.49–7.61 (m, 6H), 7.68–7.79 (m, 4H); 13C NMR (CDCl3): δ = 22.4, 22.5 (diastereotopic), 39.8, 39.9 (diastereotopic), 110.0, 110.5, 112.9, 113.1, 114.1, 114.1, 116.0, 116.1, 116.3, 116.3, 129.7, 129.7, 130.0, 130.1, 131.0, 134.0, 134.0, 134.4, 136.1, 136.2, 138.5, 146.3, 146.4; IR (KBr): 3061, 2920, 2225, 1604, 1447, 1265, 1199, 1114, 873, 760, 736, 700 cm−1; ESI-MS (210 V, MeOH–0.1% formic acid) m/z (%): 420 ([MH+Na]+, 100), 398 (43), 399 (11), 421 (27). HRMS (ESI) for C26H15N5 [M+H]+ calcd 397.1327, found 397.1320.
Acknowledgements
This work was supported by the Department of Energy (Grant DE-FG02-05ER46238). Mass spectra were obtained at the NCSU Department of Chemistry Mass Spectrometry Facility. Funding was obtained from the North Carolina Biotechnology Center and the NCSU Department of Chemistry.References
- W. J. Behof, D. Wang, W. Niu and C. B. Gorman, Org. Lett., 2010, 9, 2146 CrossRef.
- S. M. Nobre and A. L. Monteiro, Tetrahedron Lett., 2004, 45, 8225 CrossRef CAS.
- R. J. de Lang, M. van Hooijdonk, L. Brandsma, H. Kramer and W. Seinen, Tetrahedron, 1998, 54, 2953 CrossRef CAS.
- M. C. Pirrung, M. Wedel and Y. Zhao, Synlett, 2002, 143 CrossRef CAS.
- M. Amatore and C. Gosmini, Chem. Commun., 2008, 5019 RSC.
- J. S. You and J. G. Verkade, Angew. Chem., Int. Ed., 2003, 42, 5051 CrossRef CAS.
- J. S. You and J. G. Verkade, J. Org. Chem., 2003, 68, 8003 CrossRef CAS.
- D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9330 CrossRef CAS.
- T. Satoh, J. Inoh, Y. Kawamura, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 2239 CrossRef CAS.
- D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS.
- L. Y. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824 CrossRef CAS.
- J. H. Atherton, M. R. Crampton, G. L. Duffield and J. A. Stevens, J. Chem. Soc., Perkin Trans. 2, 1995, 443 RSC.
- M. R. Netherton and G. C. Fu, Top. Organomet. Chem., 2005, 14, 85 CAS.
- F. G. Bordwell, J. C. Branca, J. E. Bares and R. Filler, J. Org. Chem., 1988, 53, 780 CrossRef CAS.
- W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006 CrossRef CAS.
- A. Streitwieser, Acc. Chem. Res., 1984, 17, 353 CrossRef CAS.
- P. Langer, J. T. Anders, K. Weisz and J. Jähnchen, Chem.–Eur. J., 2003, 9, 3951 CrossRef CAS.
- L. W. Deady, A. M. Ganakas and B. H. Ong, Aust. J. Chem., 1989, 42, 1029 CrossRef CAS.
- F. Ding, J. M. Smith and H. Wang, J. Org. Chem., 2009, 74, 2679 CrossRef CAS.
- E. Buncel and T. Durst, Comprehensive Carbanion Chemistry; Elsevier Scientific: Amsterdam; New York, 1980 Search PubMed.
- F. F. Fleming, Z. Y. Zhang and P. Knochel, Org. Lett., 2004, 6, 501 CrossRef CAS.
- J. Inoh, T. Satoh, S. Pivsa-Art, M. Miura and M. Nomura, Tetrahedron Lett., 1998, 39, 4673 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd edn; Pergamon Press: Oxford; New York, 1988 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0ob00903b |
| This journal is © The Royal Society of Chemistry 2011 |