Cationic nucleolipids as efficient siRNA carriers†
Hye Won
Yang
,
Jeong Wu
Yi
,
Eun-Kyoung
Bang
,
Eun Mi
Jeon
and
Byeang Hyean
Kim
*
Department of Chemistry, BK School of Molecular Science, Pohang University of Science and Technology, Pohang, 790-784 Korea. E-mail: bhkim@postech.ac.kr; Fax: 82 54 279 3399; Tel: 82 54 279 2115
First published on 17th November 2010
Abstract
We synthesized five novel uridine-based cationic nucleolipids, introducing basic amino acid residues at the 5′ position of uridine, through 1,3-dipolar cycloaddition, and hydrophobic alkyl moieties at the 2′ and 3′ positions, through carbamate linkages. Their lipoplexes delivered siRNAs efficiently to cells, in vitro, without any severe toxicity.
Introduction
With the completion of the human genome project, gene therapy is expected to usher in a new medical revolution.1 Among the established gene therapies, RNAi has recently attracted a great deal of worldwide attention.2 This approach employs siRNA to suppress the translation of mRNA by destroying it along with RNAi machinery, RISC, in the body. Although RNAi provides many potential benefits for the treatment of various diseases, it can be difficult to deliver siRNA into cells. Much research effort has been devoted to developing viral and non-viral delivery agents to overcome the intrinsically low cell permeability of siRNA itself. Although viral delivery agents can be efficient, there is a recent trend toward non-viral delivery methods because of the potential risks of viral carriers.3–6Since Felgner and coworkers first reported the use of cationic lipids and lipoplexes as a non-viral delivery method,7 several cationic lipid compounds have been investigated because their self-assembled structures resemble the cellular membrane and because their cationic properties attract both anionic oligonucleotides and negatively charged cellular membranes.8,9 Almost 20 years after the first development of lipid carriers, Yanagawa and colleagues synthesized nucleoside-based lipids (nucleolipids);10 several improved nucleolipids have been reported thereafter.11–13 Most nucleolipids have a polar head group at the 5′ position and hydrophobic groups at both the 2′ and 3′ positions, patterned after natural glycerophospholipids, which are key components of the cellular membrane. Nucleolipids interact with genes through hydrogen bonding, π–π stacking, and nucleobase recognition, as well as electrostatic and hydrophobic interactions.
In this paper, we report the synthesis of several novel cationic nucleolipids based on uridine and amino acid units. We conjugated the hydrophilic moieties—lysine, arginine, and guanidine groups—to the 5′ position of uridine through 1,3-dipolar cycloaddition. These basic amino acids all exhibit net positive charge under physiological conditions. We linked the hydrophobic moieties—octyl, dodecyl, and oleyl chains—to the 2′ and 3′ positions of the uridine sugar unit through chemically stable but biodegradable carbamate linkages.5,14 Unlike the ester linkage, the carbamate linkage was not hydrolyzed only at neutral pH (7.4) but also at mild acidic pH (5.0), which corresponds to the endosomal pH.15 We changed the ester linkage in previous nucleolipids to the carbamate linkage to improve the endosomal escape of lipoplexes. According to this approach, we synthesized five cationic nucleolipids featuring various cationic moieties and alkyl chains and studied their physical and biological properties as delivery agents.
Result and discussion
Scheme 1 outlines the syntheses of the uridine dipoles 4a–c. To introduce hydrophobic units to the 2′ and 3′ positions of uridine selectively, we used a DMTr unit to protect the 5′-OH group16 and then treated the protected sugar with alkylamines, CDI, and DMAP. Because the alkylamines did not couple directly to both the 2′ and 3′ positions of the uridine derivative, we obtained singly substituted compounds that we then subjected to the same conditions to obtain the doubly substituted 2a–c. Next, we deprotected the DMT unit under acidic conditions and then subjected the resulting alcohol to mesylation and azidation.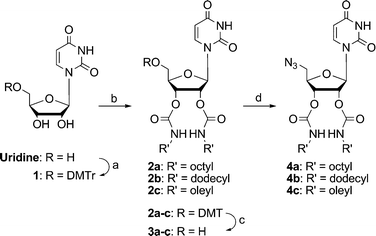 | ||
| Scheme 1 Synthesis of uridine dipoles: (a) 4,4′-dimethoxytrityl chloride, pyridine, r.t., 12 h, 88%; (b) i) CDI, DMAP, DMF, alkylamine, r.t., 12 h, ii) CDI, DMAP, DMF, alkylamine, r.t., 12 h, 56–70%; (c) CF3CO2H, CH2Cl2, −15 to 0 °C, 87–100%; (d) i) DIPEA, DMAP, DMF, r.t., 30 min, ii) MeSO2Cl, −15 °C, 3 h, iii) NaN3, r.t., 12 h, 65–84%. | ||
The amino acid dipolarophiles were synthesized through simple amide coupling with propargylamine (Scheme 2). We then coupled the uridine dipoles and the amino acid dipolarophiles through 1,3-dipolar cycloaddition (“click chemistry”)17 to obtain five different nucleolipids 7–9 (Scheme 3), isolated as trifluoroacetate salts, which we characterized using 1H and 13C NMR spectroscopy and mass spectrometry.
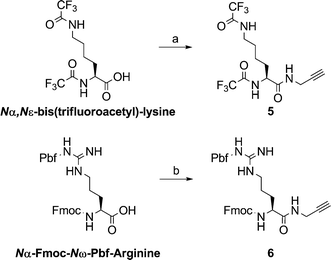 | ||
| Scheme 2 Synthesis of amino acid dipolarophiles: (a) i) EDC, DMAP, DMF, r.t. 30 min, ii) propargylamine, 12 h, 68%; (b) i) TMTU, DIPEA, HOBT, DMF, 30 min, ii) propargylamine, 2 h, 65%. | ||
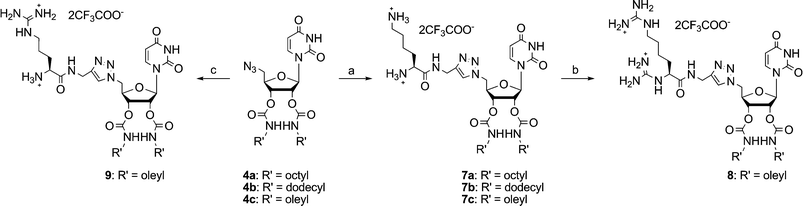 | ||
Scheme 3 Synthesis of nucleolipids 7–9: (a) i) 5, sodium ascorbate, Cu(OAc)2, CH2Cl2–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), r.t., 65–79%; ii) 28% aq. NH4OH, 40–50 °C, 24 h, 88–100%; (b) i) 1,3-bis(BOC)-2-(trifluoromethylsulfonyl)guanidine, CH2Cl2–MeOH (9:1, v/v), Et3N, r.t., 1 h, 79%; ii) CF3CO2H/CH2Cl2 (1:1, v/v), quant.; (c) i) 6, sodium ascorbate, Cu(OAc)2, CH2Cl2–H2O (1:1, v/v), r.t., 82%; ii) piperidine/CH2Cl2 (3:7, v/v), r.t., 78%; iii) CF3CO2H/CH2Cl2 (1:1, v/v), 78%. :1, v/v), r.t., 65–79%; ii) 28% aq. NH4OH, 40–50 °C, 24 h, 88–100%; (b) i) 1,3-bis(BOC)-2-(trifluoromethylsulfonyl)guanidine, CH2Cl2–MeOH (9:1, v/v), Et3N, r.t., 1 h, 79%; ii) CF3CO2H/CH2Cl2 (1:1, v/v), quant.; (c) i) 6, sodium ascorbate, Cu(OAc)2, CH2Cl2–H2O (1:1, v/v), r.t., 82%; ii) piperidine/CH2Cl2 (3:7, v/v), r.t., 78%; iii) CF3CO2H/CH2Cl2 (1:1, v/v), 78%. | ||
Prior to investigating our uridine-based cationic nucleolipids as gene delivery agents, we tested their zeta potentials. Table 1 reveals that all nucleolipids displayed positive surface charges; therefore, we expected them to readily interact with siRNA through electrostatic interactions. Next, through gel retardation assays, we found that our nucleolipids formed well-constructed lipoplexes with siRNAs at different molar ratios, given in Table 1. Upon interaction with a lipid, the band for the siRNA was retarded in the gel; therefore, the original intensity of the siRNA band diminished after lipoplex formation (Fig. 1).
| Compound | ζ potential/mVa | Molar ratiob | N/P |
|---|---|---|---|
|
a Mean values from five experiments; standard deviation is given in parentheses; each solution of nucleolipid [100 mM in 0.1% aqueous CF3CO2H and t-BuOH (1:1, v/v)] was diluted with water 20 times for determination.
b Number of moles of nucleolipid divided by the number of moles of the siRNA.
|
|||
| 7a | 75.0 (± 0.9) | 40 | 2.0 |
| 7b | 55.8 (± 1.6) | 50 | 2.5 |
| 7c | 60.0 (± 2.6) | 100 | 5.0 |
| 8 | 75.8 (± 3.5) | 60 | 3.0 |
| 9 | 82.2 (± 2.7) | 40 | 2.0 |
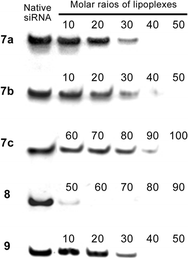 | ||
| Fig. 1 Gel retardation assays performed using the lipoplexes and siRNA. The numbers represent the molar ratios given in Table 1. | ||
Although cationic gene carriers generally have low toxicity relative to that of viral carriers, they can still induce cell toxicity through several pathways. To protect against such side effects, protective molecules [poly(ethylene glycol) or anti-inflammatory agents] are commonly introduced into lipid-mediated delivery systems.18 We found, however, that, as for other nucleoside derivatives,19 our nucleolipids themselves did not induce cell death to any significant extent. Their effects on cell viability did not depend on the type of nucleolipid, but rather on the amount used to form the lipoplex. Although a greater amount of nucleolipid resulted in fewer surviving cells, we found that 80% of cells survived after incubation for 24 h in each case (Fig. 2).
![Cell viability after treatment with lipoplexes. The cells were treated with 50 nM siRNA. Control (CTL): no treatment with a lipoplex; positive control [CTL (+)]: treatment with lipofectamine. The numbers “10,” “50,” and “100” indicate the molar ratios given in Table 1.](/image/article/2011/OB/c0ob00580k/c0ob00580k-f2.gif) | ||
| Fig. 2 Cell viability after treatment with lipoplexes. The cells were treated with 50 nM siRNA. Control (CTL): no treatment with a lipoplex; positive control [CTL (+)]: treatment with lipofectamine. The numbers “10,” “50,” and “100” indicate the molar ratios given in Table 1. | ||
Next, we tested our lipoplexes, complexes of nucleolipids and anti-VEGF siRNA, for their transfection into HeLa cells (Fig. 3). Upon increasing the molar ratio, we detected less VEGF; i.e., siRNA was delivered into the cells and the RNAi machinery worked effectively. Among our lysine family of nucleolipids (7a–c), the oleyl-linked nucleolipid 7c was the most effective siRNA carrier. We suspect that the cis olefinic bond in the oleyl chain might have induced a looser, more fluid structure for the lipoplex, thereby aiding the release of siRNA. Among the oleyl family of compounds (7c, 8, 9), the arginine-linked nucleolipid 9 was the best siRNA-delivering agent. Indeed, the efficacy of the lipoplex formed from 9 nearly matched that of lipofectamine, a commercial transfection agent. Guanidine groups are common components of delivery systems; the presence of several guanidine groups often provides more efficient delivery.20 Interestingly, among our nucleolipids, arginine units provided superior delivery efficiency than did bisguanidine moieties.
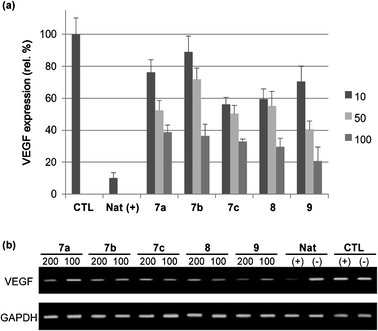 | ||
| Fig. 3 VEGF expression monitored using (a) ELISA and (b) PCR assays. Natural siRNA (Nat) and control (CTL, no treatment with siRNA) in the presence (+) and absence (−) of lipofectamine. HeLa cells were treated with 50 nM siRNA solutions. The numbers “100” and “200” indicate molar ratios, as defined in Table 1. | ||
Conclusion
In summary, we have synthesized five cationic lipids—uridine derivatives presenting basic amino acid residues and long alkyl chains—and tested them as agents for the delivery of siRNA into cells. We used click chemistry to conjugate the amino acid units to the 5′ positions of the uridine moieties. To form a diverse set of nucleolipids in terms of alkyl group lengths and conformations, we linked octyl-, dodecyl-, and oleylamine to the 2′ and 3′ positions of the uridine derivatives through carbamate linkages. All of these synthesized nucleolipids formed lipoplexes with anti-VEGF siRNA; we used gel retardation assays to determine the minimal ratios of cationic nucleolipid to siRNA for each lipoplex. None of our synthesized cationic nucleolipids displayed severe cytotoxicity; their lipoplexes all delivered the siRNA into cells in vitro, down-regulating target mRNA and protein. Notably, our arginine- and oleyl-linked nucleolipid 9 functioned as an efficient siRNA carrier, almost equal to that of the commercial transfection agent lipofectamine.Experimental
2′,3′-Di-O-alkylcarbamyl-5′-azido-5′-deoxyuridine (4a–c)
A solution of 3a–c in dry pyridine was evaporated and dried in vacuo to remove moisture. After adding dry DMF (0.1 M) and DIPEA (6 equiv.) and DMAP (0.1 equiv.), the mixture was stirred for 30 min. at room temperature under Ar (g) conditions. After the mixture was cooled in ice–acetone bath (−15 °C) for 10 min, methanesulfonyl chloride (3 equiv.) was added. After stirring for 3 h, methanesulfonylation at the 5′-OH in uridine was confirmed by TLC and sodium azide (15 equiv.) was added in situ. The mixture was stirred for 12 h following addition of CH2Cl2 and washed with sat. NaHCO3 (aq.), water, and brine. The organic layer was dried (Na2SO4), evaporated and purified through flash column chromatography (SiO2, CH2Cl2–ethyl acetate, 9:1 to 1:1, v/v), then, yellowish solid 4a (84%), 4b (65%), and 4c (75%) was obtained
Compound 4a: M.p.: 61.9–62.5 °C; 1H NMR (300 MHz, DMSO-d6): δ 9.61(s, 1H), 6.39 (d, J = 7.2 Hz, 1H), 6.08–5.71 (m, 2H), 4.76 (d, J = 5.8 Hz, 1H), 4.56 (d, J = 7.1 Hz, 1H), 4.19 (t, J = 5.5 Hz, 1H), 4.06–4.03 (m, 1H), 3.14–3.13 (m, 1H), 2.79–2.75 (m, 2H), 2.14–2.12 (m, 4H), 0.74–0.61 (m, 24H), 0.31–0.27 (m, 6H); 13C NMR (75 MHz, DMSO-d6): δ 162.9, 154.7, 154.5, 150.5, 141.2, 102.7, 86.5, 80.7, 71.6, 70.6, 51.3, 31.3, 29.5, 29.3, 29.2, 28.8, 28.7, 26.3, 26.2, 22.1, 13.9; IR (neat): 3339, 2956, 2927, 2856, 2106, 1708, 1541, 1463, 1383, 1262 cm−1; HRMS-FAB (m/z): calcd for C27H45N7NaO7+ [M+Na]+, 602.3273; found, 602.3276.
Compound 4b: M.p.: 136.4–137.4 °C; 1H NMR (300 MHz, CDCl3): δ 8.75(br, 1H), 7.54 (d, J = 8.2 Hz, 1H), 6.13 (d, J = 6.7 Hz, 1H), 5.82 (d, J = 8.2 Hz, 1H), 5.20–5.17 (m, 2H), 4.97–4.94 (m, 2H), 4.22–4.21 (m, 1H), 3.75 (br, 2H, 5′-H), 3.18–3.09 (m, 4H), 1.50–1.25 (m, 40H), 0.90–0.85 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 162.8, 155.3, 154.8, 150.9, 139.9, 104.0, 86.1, 81.9, 81.6, 73.2, 72.2, 52.7, 41.7, 41.6, 32.3, 31.8, 30.2, 30.1, 30.0, 29.9, 29.7, 29.6, 27.1, 23.0, 14.5; IR (neat): 3341, 2924, 2853, 2107, 1706, 1541, 1463, 1383, 1262 cm−1; HRMS-FAB (m/z): calcd for C35H61N7NaO7+ [M+Na]+, 714.4525; found, 714.4525.
Compound 4c: M.p.: 126.8–127.1 °C; 1H NMR (300 MHz, CDCl3): δ 7.55–7.53 (m, 1H), 6.13 (d, J = 6.7 Hz, 1H), 5.82 (d, J = 8.2 Hz, 1H), 5.38–5.28 (m, 4H), 5.22–5.17 (m, 2H), 4.97 (br, 1H), 4.22–4.21 (m, 1H), 3.75–3.74 (m, 2H), 3.15–3.11 (m, 4H), 2.01–1.99 (m, 8H), 1.47–1.25 (m, 48H), 0.90–0.85 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 165.7, 162.9, 155.3, 154.8, 150.9, 139.9, 131.3, 130.8, 130.6, 130.5, 130.4, 130.1, 129.2, 104.0, 86.1, 81.9, 73.5, 73.2, 72.2, 66.5, 66.2, 52.7, 41.7, 41.6, 32.9, 32.2, 30.2, 30.1, 30.0, 29.9, 29.8, 29.7, 29.6, 27.6, 27.5, 27.1, 23.0, 14.5; IR (neat): 3345, 2925, 2854, 2107, 1708, 1542, 1464, 1384, 1263, 1173, 1090, 1061 cm−1; HRMS-FAB (m/z): calcd for C47H81N7NaO7+ [M+Na]+, 878.6090; found, 878.6092.
N α,Nε-Bis(trifluoroacetyl)-L-lysine-N-propargylamide (5)
EDC (3.4 g, 17.7 mmol) and DMAP (2.17 g, 17.7 mmol) were added to a solution of Nα,Nε-bis(trifluoroacetyl)-L-lysine (3.0 g, 8.9 mmol) in DMF (45 ml). After stirring at room temperature for 30 min, propargyl amine (0.91 ml, 13.3 mmol) was added and the mixture was stirred for an additional 12 h at room temperature. After, ethyl acetate was added to the mixture and it was washed with 1 N HCl (aq.), sat. NaHCO3 (aq.), water, and brine sequentially. The organic layer was dried (Na2SO4) and evaporated and purified through re-crystallization in ether, then, a white solid 5 was isolated (2.25 g, 68%).M.p.: 140.9–142.1 °C; 1H NMR (300 MHz, MeOD): δ 4.37 (dd, J = 8.89, 5.78 Hz, 1H), 3.99–3.95 (m, 2H), 3.28–3.26 (m, 2H), 2.59 (t, J = 2.58 Hz, 1H), 1.83–1.74 (m, 2H), 1.62–1.55 (m, 2H), 1.43–1.37 (m, 2H); 13C NMR (75 MHz, MeOD): δ 172.5, 91.5, 80.2, 72.4, 54.9, 40.4, 32.2, 29.5, 29.4, 26.7, 24.0; 19F NMR (282.38 MHz, MeOD): δ 0.004, −0.39; IR (KBr): 3308, 3289, 3098, 2955, 2932, 1707, 1660, 1557, 1451, 1351, 1188, 1167 cm−1; HRMS-FAB (m/z): calcd for C13H15F6N3NaO3+ [M+Na]+, 398.0910; found, 398.0917.
N α-Fmoc-Nω-Pbf-L-arginine-N-propargylamide (6)
After drying the mixture of Nα-Fmoc-Nω-Pbf-L-arginine (2.0 g, 3.1 mmol), TBTU (1.2 g, 3.72 mmol), and HOBT (0.5 g, 3.7 mmol) in vacuo, the mixture was dissolved in dry DMF (25 mL) under Ar (g) conditions. Adding DIPEA (1.3 ml, 7.4 mmol) and stirring for 30 min at room temperature, propargyl amine (0.26 ml, 3.7 mmol) was added to the mixture and the mixture was stirred for an additional 2 h. After, ethyl acetate was added to the mixture and washed with 1 N HCl (aq.), sat. NaHCO3 (aq.), water, and brine sequentially. The organic layer was dried (Na2SO4), evaporated and purified through flash column chromatography (SiO2, hexane–ethyl acetate, 3:2 to ethyl acetate only, v/v), then, a white solid 6 (1.4 g, 65%) was isolated.
M.p.: 128.3–128.9 °C; 1H NMR (300 MHz, CDCl3): δ 7.74–7.22 (m, 8H), 6.24 (s, 2H), 6.02–6.00 (m, 1H), 4.35–4.32 (m, 3H), 4.15–4.11 (m, 1H), 3.98 (br, 2H), 3.27 (br, 2H), 2.90 (s, 2H), 2.54 (d, J = 22.17 Hz, 6H), 2.13 (s, 1H), 2.06 (s, 3H), 1.87 ∼ 1.58 (br, 6H), 1.42 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 172.1, 159.1, 156.8, 156.6, 144.0, 143.9, 141.5, 138.6, 132.7, 132.5, 128.0, 127.3, 125.3, 125.0, 120.2, 117.9, 86.7, 79.7, 71.6, 67.4, 54.3, 47.3, 43.4, 40.8, 30.8, 29.4, 28.8, 25.5, 19.5, 18.2, 12.7; IR (neat): 3649, 3436, 3309, 2970, 2929, 1718, 1667, 1619, 1549, 1451, 1245, 1104, 1092 cm−1; HRMS-FAB (m/z): calcd for C37H44N5O6S+ [M+H]+, 686.3007; found, 686.3015.
2′,3′-Di-O-(alkylcarbamyl)-5′-deoxy-5′-(4-lysylaminomethyl-1H-1,2,3-triazol-1-yl)uridine (7a–c)
A solution of compound 5, sodium ascorbate (0.2 equiv.), Cu(OAc)2 (0.2 equiv.), and compound 4a–c (2 equiv.) in CH2Cl2–H2O (1:1, v/v) (0.1 M) was stirred at room temperature for 12 h. The mixture was extracted with CH2Cl2 and washed with water and brine. The organic layer was dried (Na2SO4) and evaporated and purified through flash column chromatography (SiO2, chloroform–MeOH, 30:1 to 10:1, v/v), then, a yellowish solid (65–79%) was isolated.
The isolated compound was dissolved in ammonia solution (28%, aq.) (0.01 M) and the mixture was stirred at 40–45 °C for 1 d. After quenching with methanol, the solution was lyophilized and the oilic residue was washed repeatedly and precipitated with ether, then, a yellowish solid 7a (88%), 7b (quant.), and 7c (quant.) was isolated.
Compound 7a: M.p. 101.7–102.5 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.96 (br, 1H), 7.96 (br, 2H), 7.75 (d, J = 8.0 Hz, 1H), 7.47–7.04 (m, 2H), 5.91 (d, J = 6.3 Hz, 1H), 5.73 (d, J = 7.9 Hz, 1H), 5.30–5.28 (m, 1H), 5.23–5.19 (m, 1H), 4.78 (d, J = 5.3 Hz, 2H), 4.45–4.35 (m, 3H), 3.74 (t, J = 6.1 Hz, 1H), 2.98–2.89 (m, 4H), 2.77–2.73 (m, 2H), 1.75–1.67 (m, 2H), 1.52–1.47 (m, 2H), 1.37–1.22 (m, 28H), 0.86–0.82 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ 168.4, 162.9, 158.6, 158.2, 154.6, 154.5, 150.5, 143.8, 123.9, 102.6, 86.9, 80.2, 71.4, 70.6, 52.0, 50.9, 34.9, 31.3, 29.3, 28.8, 28.7, 26.4, 26.3, 22.1, 21.0, 13.9; IR (neat) 3325, 3077, 2956, 2927, 2855, 1678, 1542, 1464, 1266, 1202, 1134 cm−1; HRMS-FAB (m/z) calcd for C36H63N10O8+ [M + H]+, 763.4825; found, 763.4833.
Compound 7b: M.p. 208.0–209.0 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.96 (br, 2H), 7.75 (d, J = 8.0 Hz, 1H), 7,47–7.04 (m, 2H), 5.91 (d, J = 6.3 Hz, 1H), 5.73 (d, J = 8.0 Hz, 1H), 5.29–5.27 (m, 1H), 5.23–5.21 (m, 1H), 4.78 (d, J = 5.3 Hz, 2H), 4.40–4.35 (m, 3H), 3.72 (t, J = 6.0 Hz, 1H), 2.94 (br, 4H), 2.73 (t, J = 7.5 Hz, 2H), 1.71–1.68 (m, 2H), 1.52–1.47 (m, 2H), 1.37–1.23 (m, 44H), 0.87–0.82 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ 168.4, 162.8, 158.6, 158.2, 154.6, 150.4, 143.7, 123.8, 115.3, 102.6, 86.9, 80.1, 71.4, 70.5, 52.0, 50.9, 34.5, 31.3, 30.1, 29.0, 28.8, 28.7, 26.4, 22.0, 21.1, 13.9; IR (neat) 3321, 3066, 2923, 2853, 1679, 1540, 1464, 1267, 1202, 1133 cm−1; HRMS-FAB (m/z) calcd for C44H79N10O8+ [M + H]+, 875.6077; found, 875.6080.
Compound 7c: M.p. 205.8–206.2 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.87 (br, 1H), 7.94 (br, 2H), 7.75 (d, J = 7.9 Hz, 1H), 7.47–7.04 (m, 2H), 5.92 (d, J = 6.3 Hz, 1H), 5.73 (d, J = 7.9 Hz, 1H), 5.33–5.30 (m, 5H), 5.23–5.21 (m, 1H), 4.78 (d, J = 5.4 Hz, 2H), 4.40–4.35 (m, 3H), 3.68 (br, 1H), 2.96–2.89 (m, 4H), 2.76–2.71 (m, 2H), 1.98–1.96 (m, 8H), 1.67 (br, 2H), 1.51–1.49 (m, 2H), 1.37–1.23 (m, 52H), 0.86–0.82 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ 162.9, 154.4, 150.4, 143.7, 141.3, 130.0, 129.6, 123.9, 107.1, 102.4, 86.8, 80.1, 71.4, 70.3, 52.8, 50.4, 46.0, 34.7, 31.9, 31.3, 30.0, 29.2, 29.1, 28.9, 28.8, 28.7, 18.6, 26.6, 26.5, 22.1, 21.5, 13.9; IR (neat) 3452, 3332, 2925, 2854, 1684, 1529, 1463, 1259, 1214, 1137 cm−1; HRMS-FAB (m/z) calcd for C56H99N10O8+ [M + H]+, 1039.7642; found, 1039.7654.
2′,3′-Di-O-(oleylcarbamyl)-5′-deoxy-5′-(4-(2S-2,6-diguanidinohexanyl)aminomethyl-1H-1,2,3-triazol-1-yl)uridine (8)
Compound 7c (200 mg, 0.19 mmol) and 1,3-di-BOC-2-(trifluoromethylsulfonyl)guanidine (223 mg, 0.57 mmol) were dried in vacuo and dissolved in CH2Cl2–MeOH (9:1, v/v) (4 ml). Triethylamine (80 μL, 0.57 mmol) was added to the solution and the mixture was stirred for 1 h. After evaporation under reduced pressure, the mixture was purified through flash column chromatography (SiO2, CH2Cl2–MeOH, 20:1, v/v), then, a yellowish compound (227 g, 79%) was isolated.
The isolated compound (170 mg, 0.1 mmol) was dissolved in 50% CF3CO2H in CH2Cl2 (3 ml) and the mixture was stirred at the room temperature for 4 h. After evaporation under reduced pressure, the mixture was dissolved in 0.1% CF3CO2H in H2O–MeOH, (1:1, v/v) and purified through HPLC (Xterra® PrepRP8 5 μm 10 × 50 mm, 40 °C, flow rate 3.0 ml min−1, UV: λ = 254 nm). The gradient of the HPLC mobile phase was increased linearly over 20 min from 5% MeOH–0.1% CF3CO2H in H2O (v/v) to 100% MeOH at a flow rate of 3 mL min−1. Then, the mobile phase was held isocratically for 5 min with 100% MeOH and the gradient was decreased linearly over 5 min to 5% MeOH–0.1% CF3CO2H in H2O (v/v) at the same flow rate. The fraction was collected and lyophilized, then, yellow oilic compound 8 (140 mg, quant.) was isolated.
1H NMR (300 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.61 (br, 1H), 7.90 (s, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.71–7.60 (m, 2H), 7.48–6.98 (m, 6H), 5.90 (d, J = 6.2 Hz, 1H), 5.72 (d, J = 8.0 Hz, 1H), 5.34–5.29 (m, 4H), 5.22–5.20 (m, 1H), 4.77 (d, J = 5.2 Hz, 2H), 4.42–4.28 (m, 3H), 4.13–4.07 (m, 1H), 3.09–3.02 (m, 2H), 2.73 (br, 4H), 2.25 (t, J = 7.4 Hz, 1H), 1.97–1.93 (m, 8H), 1.72 (br, 1H), 1.61 (br, 2H), 1.44–1.23 (m, 52H), 0.86–0.82 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ 174.5, 169.7, 162.9, 158.7, 158.3, 156.8, 156.6, 154.6, 154.4, 150.5, 144.1, 130.0, 129.6, 123.8, 118.7, 114.8, 102.7, 86.2, 80.2, 71.5, 70.5, 67.3, 54.2, 51.4, 48.6, 34.8, 34.1, 32.1, 31.3, 29.2, 29.1, 29.0, 28.9, 28.7, 28.6, 26.7, 26.6, 26.5, 24.6, 22.1, 13.9, 10.1; HRMS-FAB (m/z) calcd for C58H102N14NaO8+ [M + Na]+, 1145.7897; found, 1145.7899.
2′,3′-Di-O-(oleylcarbamyl)-5′-deoxy-5′-(4-arginylaminomethyl-1H-1,2,3-triazol-1-yl)uridine (9)
A solution of compound 6 (800 mg, 1.2 mmol), sodium ascorbate (39 mg, 0.2 mmol), Cu(OAc)2 (4 mg, 0.02 mmol), and compound 7c (840 mg, 0.89 mmol) in CH2Cl2–H2O (1:1, v/v) (10 mL) was stirred at room temperature for 12 h. The mixture was extracted with CH2Cl2 and washed with water and brine. The organic layer was dried (Na2SO4), evaporated and purified through flash column chromatography (SiO2, CH2Cl2–MeOH, 100:1 to 30:1, v/v), then, a yellowish solid (1.24 g, 82%) was isolated.
The isolated compound (1.1 g, 0.87 mmol) was dissolved in 30% piperidine in CH2Cl2 (30 mL) and the mixture was stirred at room temperature for 10–15 min. After evaporation under reduced pressure, the mixture was purified through flash column chromatography (SiO2, CH2Cl2–MeOH, 25:1 to 10:1, v/v), then, a yellowish olid (0.9 g, 78%) was isolated.
This compound (200 mg, 0.15 mmol) was dissolved in 50% CF3CO2H in CH2Cl2 (4 ml) and stirred at the room temperature for 1.5 h. After evaporation, the mixture was dissolved in 0.1% CF3CO2H in t-BuOH–H2O (4:1, v/v) and purified through HPLC (Xterra® PrepRP8 5 μm 10 × 50 mm, 40 °C, UV: λ = 254 nm). The gradient of the HPLC mobile phase was increased linearly over 20 min from 5% MeOH–0.1% CF3CO2H in H2O (v/v) to 100% MeOH at a flow rate of 3 mL min−1. Then, the mobile phase was held isocratically for 5 min with 100% MeOH and the gradient was decreased linearly over 5 min to 5% MeOH–0.1% CF3CO2H in H2O (v/v) at the same flow rate. The fraction was collected and lyophilized, then, a yellowish oily compound 9 (89 mg, 56%) was finally isolated.
1H NMR (300 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.92 (t, J = 5.2 Hz, 1H), 8.18 (br, 2H), 7.94 (s, 1H), 7.74–7.76 (m, 2H), 7.49–7.01 (m, 4H), 5.90 (d, J = 6.3 Hz, 1H), 5.72 (d, J = 8.0 Hz, 1H), 5.33–5.26 (m, 4H), 5.21–5.18 (m, 1H), 4.77 (d, J = 5.6 Hz, 2H), 4.48–4.28 (m, 3H), 3.73 (br, 2H), 3.10–3.08 (m, 2H), 2.95–2.92 (m, 4H), 1.97–1.93 (m, 8H), 1.71–1.68 (m, 2H), 1.47–1.17 (m, 50H), 0.84 (t, J = 6.5 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 206.7, 168.3, 163.0, 159.0, 158.7, 158.2, 157.8, 156.8, 154.7, 154.5, 150.5, 143.7, 141.4, 130.1, 129.7, 129.6, 124.0, 118.3, 114.4, 102.7, 86.5, 80.2, 71.5, 70.5, 67.1, 52.0, 51.0, 34.9, 31.7, 31.3, 30.7, 29.1, 28.9, 28.7, 28.6, 26.7, 26.6, 24.7, 22.1, 14.0; HRMS-FAB (m/z) calcd for C56H99N12O8+ [M + H]+, 1067.7631; found, 1067.7709.
Lipoplex preparation
Cationic lipids stock solutions were prepared by solubilizing the compounds in 0.1% aqueous CF3CO2H and t-BuOH (1:1, v/v). The lipid solution was vortexed for 30 s and stirred for 30 min at 45 °C to make it clear. After cooling to room temperature, 10 volumes of nuclease free water were added and the solution was vortexed and incubated at 4 °C before adding the siRNAs. Lipoplexes were then prepared by mixing appropriate amounts of siRNAs and cationic nucleolipids based on their N/P ratios. Finally, the formulations were mixed and incubated for 30 min at room temperature before use.
MTT assay
Lipids were complexed with VEGF siRNA and added to the cells. Cells were incubated with complexes at 37 °C and 5% CO2 for 24 h. After incubation, the media was removed, the cells washed with 100 mL of DPBS buffer (2 times) and then 100 μL of MTT solution (1 mg mL−1 of MTT dissolved in phenol red free medium, Sigma) was added to each well and incubated at 37 °C for 4 h. After incubation, 100 μL DMSO was added to each well and absorbances were measured at a wavelength of 570 nm using a microplate reader (Asys) and converted to percentage of cell viability (relative to control cells).Cell culture and transfection
HeLa cells were cultured in DMEM (HyClone), supplemented with 10% FBS (HyClone), 100 μg mL−1 of streptomycin, and 100 U mL−1 of penicillin at 37 °C in 5% CO2 incubator. Cells were split, using trypsin/EDTA medium when almost confluent. HeLa cells were seeded at a density of 2.5 × 104 cells per well, each well of which contained 2 mL of 10% FBS supplemented DMEM, and incubated for 4 h. HeLa cells were transfected in the absence of serum with VEGF siRNAs using lipid (7–9), Lipofectamine™ 2000 (Invitrogen), or without any transfection reagent. The cells were allowed to incubate at 37 °C for 6 h in a CO2 incubator followed by replacement of 2 mL of DMEM containing 10% FBS. After more 18 h incubation, cell media were collected and analyzed for the VEGF expression level by using VEGF ELISA kit (QIA51 VEGF ELISA Kit, Human, Calbiochem) following the manufacturer's instructions.RT-PCR experiment
VEGF siRNA-treated cells were washed with PBS and lyzed in 1.0 ml of TRIzol®-reagent (Invitrogen) and total RNA was isolated. A 1 μg sample of RNA was used in reverse transcription with Improm-II™ Reverse Transcription System (Promega) and the procedures were performed by the manufacturer's protocols. The reverse transcription reaction was carried out at 25 °C (5 min), 42 °C (60 min) and 70 °C for 15 min followed by PCR: 1 cycle, 95 °C, 5 min; 30 cycles, 95 °C, 30 s; 55 °C, 30 s; 72 °C, 30 s; 1 cycle, 72 °C, 5 min. For VEGF mRNA amplification, forward and reverse primers were 5′-ATGAACTTTCTGCTGTCTTGGGT-3′ and 5′-TCACCGCCTCGGCTTGTCACA-3′, respectively. And, for the control mRNA of GAPDH, forward and reverse primers were 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-TTGATTTTGGAGGGATCTCG-3′, respectively. The PCR products were analyzed by electrophoresis on a 1.5% agarose gel (80 V, 50 min) stained with ethidium bromide.Abbreviations
BOC—tert-butyloxycarbonyl, CDI—carbonyldiimidazole, DIPEA—N,N-diisopropylethylamine, DMF—N,N-dimethylformamide, DMAP—N,N-dimethylaminopyridine, DMEM—Dulbecco's modified Eagle's medium, DPBS—Dulbecco's Phosphate buffered saline, EDC—N-(3-dimethylaminopropyl-N′-ethylcarbodiimide hydrochloride, ELISA—Enzyme-linked immunosorbent assay, FBS—fetal bovine serum, Fmoc—9-fluorenylmethyloxycarbonyl, HOBT—N-hydroxybenzotriazole, Pbf—2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl, PBS—Phosphate buffered saline, RNAi—RNA interference, RISC—RNA-induced silencing complex, siRNA—small interfering RNA, TMTU—O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate, VEGF—Vascular endothelial growth factor.Acknowledgements
This study was supported by grants (no. 20090065415 and 20100000763) from the National Research Foundation (NRF) and EPB center, funded by the Korean government (MEST). We thank Prof. Sungjee Kim for assistance with the zeta potential measurements.References
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280 CAS.
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806 CrossRef CAS.
- S.-D. Li and L. Huang, J. Controlled Release, 2007, 123, 18.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 347 CrossRef.
- P. P. Karmali and A. Chaudhuri, Med. Res. Rev., 2007, 27, 696 CrossRef CAS.
- M. Z. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413.
- A. D. Miller, Angew. Chem., Int. Ed., 1998, 37, 1768 CrossRef.
- R. Srinivas, S. Samanta and A. Chaudhuri, Chem. Soc. Rev., 2009, 38, 3326 RSC.
- H. Yanagawa, Y. Ogawa, H. Furuta and K. Tsuno, Chem. Lett., 1988, 269 CAS; H. Yanagawa, Y. Ogawa, H. Furuta and K. Tsuno, J. Am. Chem. Soc., 1989, 111, 4567 CrossRef CAS.
- S. Bonaccio, M. Wessicken, D. Berti, P. Walde and L. Luisi, Langmuir, 1996, 12, 4976 CrossRef CAS.
- L. Moreau, P. Barthélémy, M. E. Maataoui and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 7533 CrossRef CAS; A. Gissot, M. Cample, M. W. Grinstaff and P. Barthélémy, Org. Biomol. Chem., 2008, 6, 1324 RSC; C. Ceballos, C. A. H. Prata, S. Giorgio, F. Garzino, D. Payet, P. Barthélémy, M. W. Grinstaff and M. Camplo, Bioconjugate Chem., 2009, 20, 193 CrossRef CAS; P. Barthélémy, C. R. Chimie, 2009, 12, 171 CrossRef CAS.
- S. K. Choi, T. K. Vu, J. M. Jung, S. J. Kim, H. R. Jung, T. Chang and B. H. Kim, ChemBioChem, 2005, 6, 432 CrossRef CAS; H. R. Jung, T. K. Vu, S. K. Choi, S. M. Park and B. H. Kim, Bull. Korean Chem. Soc., 2010, 31, 549 CrossRef CAS; S. M. Park, J. E. Woo, E. M. Jeon and B. H. Kim, Chem. Commun., 2010, 46, 1523 RSC.
- D. Liu, W. Qiao, Z. Li, X. Cui, K. Li, L. Yu, K. Yan, L. Zhu and L. Cheng, Bioorg. Med. Chem., 2008, 16, 995 CrossRef CAS.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141 CrossRef CAS; E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmaceutics, 2005, 2, 129 CrossRef CAS.
- M. Smith, D. H. Rammler, I. H. Goldberg and H. G. Khorana, J. Am. Chem. Soc., 1962, 84, 430 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- Y.-C. Tseng, S. Mozumdar and L. Huang, Adv. Drug Delivery Rev., 2009, 61, 721 CrossRef CAS.
- S. M. Park, Y. S. Lee and B. H. Kim, Chem. Commun., 2003, 2912 RSC; S. M. Park, Y. Shen and B. H. Kim, Org. Biomol. Chem., 2007, 5, 610 RSC; Y. S. Lee, S. M. Park and B. H. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 1126 CrossRef CAS.
- S. Futaki, I. Nakase, T. Suzuki, Z. Youjun and Y. Sugiura, Biochemistry, 2002, 41, 7925 CrossRef CAS; H. H. Chung, G. Harms, C. M. Seong, B. H. Choi, C. Min, J. P. Taulane and M. Goodman, Biopolymers, 2004, 76, 83 CrossRef CAS; B. Wu, Y. Li, P. A. Morcos, T. J. Doran, P. Lu and Q. L. Lu, Mol. Ther., 2009, 17, 864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of 1–3, NMR spectra of synthesized compound and HPLC profiles and MALDI-MS spectra of RNA strands. See DOI: 10.1039/c0ob00580k |
| This journal is © The Royal Society of Chemistry 2011 |