3-Phosphono-L-alanine as pyrophosphate mimic for DNA synthesis using HIV-1 reverse transcriptase†
Shiqiong
Yang
a,
Mathy
Froeyen
a,
Eveline
Lescrinier
a,
Philippe
Marlière
b and
Piet
Herdewijn
*a
aLaboratory of Medicinal Chemistry, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Minderbroedersstraat 10, 3000 Leuven, Belgium. E-mail: Piet.Herdewijn@rega.kuleuven.be; Fax: +32 16 337340; Tel: +32 16 337387
bIsthmus Sarl, 31 rue de Saint Amand, F-75015, Paris, France
First published on 22nd November 2010
Abstract
A series of sulf(on)ate and phosph(on)ate amino acid phosphoramidate analogues of deoxynucleotides were synthesized as potential substrates for HIV-1 reverse transcriptase. Taurine, L-cysteic acid, 3-phosphono-L-alanine, O-sulfonato-L-serine, and O-phospho-L-serine were investigated as leaving groups in an enzyme catalyzed DNA synthesis protocol. Among these analogues, the phosphonate congener performed best and 3-phosphono-L-alanine can be considered as an excellent mimic of the pyrophosphate (PPi) moiety of deoxyadenosine triphosphate, to be used in enzymatic synthesis of nucleic acids. During a single nucleotide incorporation assay the use of 3-phosphono-L-Ala-dAMP as substrate resulted in 95% conversion to a P + 1 strand in 60 min at 50 μM (a concentration 10 times less than found for L-Asp-dAMP) and with improved incorporation kinetics and less stalling. For the sequences investigated, the efficiency of the incorporation is base dependent and decreases in the order (A ≥ T = G > C). In all cases, the incorporation follows Watson–Crick rules.
Introduction
Nucleoside triphosphates are the building blocks for the enzymatic synthesis of nucleic acids, using polymerases as catalysts. In this reaction, a nucleoside monophosphate is incorporated in a growing nucleic acid chain and pyrophosphate functions as a leaving group. The mechanism of the polymerization reaction and the structural changes involved seem to be conserved between different polymerases.1 Several mimics of nucleoside triphosphates have been tested as substrates for DNA polymerases. Most of these triphosphate mimics are obtained by modifying the bridging O-atoms of the triphosphate moiety2 or using non-standard nucleobases.3 In previous studies, we have demonstrated that the pyrophosphate moiety of nucleoside triphosphate can be replaced by leaving groups whose structures are not based on phosphate chemistry. For example, some selected amino acid derivatives could be considered as mimics of the pyrophosphate moiety of deoxyadenosine triphosphate.4 Among these compounds, L-Asp-dAMP showed efficient incorporation in a primer-template assay and resulted in 90% conversion to a P + 1 strand in 60 min at 500 μM by HIV-1 RT. During this incorporation a P–N bond is cleaved. We have also shown that phosphodiesters are better substrates than phosphoramidates when it comes to polymerization of nucleotides using HIV-1 RT5 and that the structure of the leaving group should not be limited to that of an α-amino acid.5,6 However, the main problem when considering potential applications in biotechnology and medicine is that the kinetics obtained for incorporation of the nucleotide into a growing DNA chain using the alternative building blocks are poor. The Vmax is mostly reasonably good, but the Km value is generally 103–104 times higher when compared with dATP. Besides, chain termination was observed after incorporation of two or three nucleotides.4 As the incorporation efficiency may be influenced by electrostatic interactions between substrate and enzyme, a series of sulf(on)ate and phosph(on)ate analogues (compounds 1–5, Fig. 1) of L-Asp-dAMP have been synthesized as potential substrates for HIV-1 reverse transcriptase.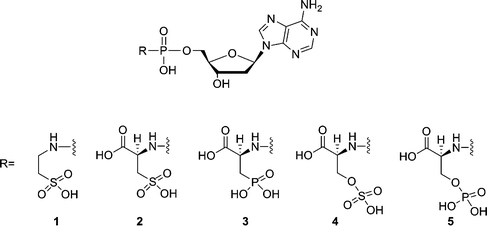 | ||
| Fig. 1 Structures of phosphoramidate analogues of 2′-deoxyadenosine nucleotides. | ||
In a single nucleotide incorporation assay, 2′-deoxyadenosine-5′-(3-phosphono-L-alanine) phosphoramidate (3) is a considerably improved substrate, which resulted in 95% conversion to a P + 1 strand in 60 min at 50 μM (a concentration 10 times less than found for L-Asp-dAMP). It is found that compound 3 has improved incorporation kinetics and elongation capability. For example, the Km for compound 3 is 78-fold higher than for the natural substrate dATP and the measured Vmax is only 1.3-fold lower. Molecular modeling of compound 3 in the active site of HIV-RT revealed that two Mg2+ ions are tightly bound to the phosphonate group, the phosphoramidate group and to three catalytic carboxylate groups (Asp110A, Asp185A and Asp186A). The phosphonate group and the phosphoramidate group seem to mimic the γ- and α-phosphate groups of deoxynucleoside triphosphates, respectively. Lys65A and Arg72A are involved in the interactions with the phosphonate group and the carboxyl group, respectively. The carboxyl group functions as a mimic of the β-phosphate group.
To further investigate the potential of 3-phosphono-L-alanine as a leaving group, compounds 6–8 (Fig. 2) were synthesized and evaluated. Among the three analogues, 3-phosphono-L-Ala-dGMP (6) and 3-phosphono-L-Ala-dTMP (8) are also good substrates resulting in 77% and 81% conversion to a P + 1 strand in 60 min at 50 μM in a single nucleotide incorporation assay. Nonetheless, the analogue 7 with a cytosine moiety is only incorporated at higher concentration. Apparently the efficiency of the leaving group is base-dependent. In all cases evaluated, the Watson–Crick base pairing rules are respected.
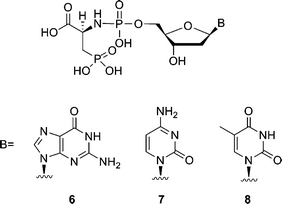 | ||
| Fig. 2 Structures of phosphoramidate analogues of 2′-deoxynucleotides. | ||
Results and discussion
The synthesis of Tau-dAMP (1) or L-Cys-dAMP (2) is shown in Scheme 1. Tau-dAMP (1) or L-Cys-dAMP (2) were synthesized from deoxyadenosine monophosphate (dAMP) and taurine or L-cysteic acid ethyl ester by an N-ethyl-N′-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDAC) mediated coupling, followed by a deprotection with base. The coupling and deprotection of taurine and L-cysteic acid was done in a one-pot reaction.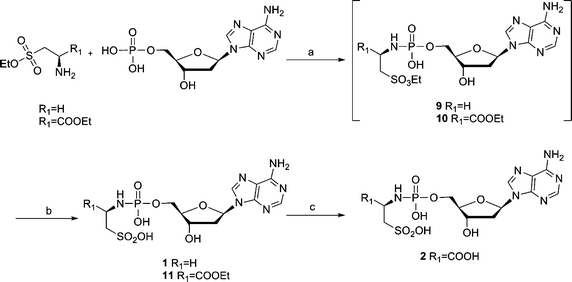 | ||
Scheme 1 (a) EDAC, H2O, r.t., 2–5 h; (b) 1.4 M K2CO3 in MeOH–H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, r.t., 2 h; (c) 0.4 M NaOH in MeOH–H2O 1:1, r.t., overnight. :1, r.t., 2 h; (c) 0.4 M NaOH in MeOH–H2O 1:1, r.t., overnight. | ||
This coupling reaction was described before by F.-Q. Huang et al.,7 in which they performed the EDAC-mediated coupling of adenosine 5′-monophosphate to diamines at room temperature in water. The DCC method (t-BuOH–H2O 5:1, reflux, 4 h)8 to synthesize the phosphoramidates needs more vigorous conditions than the EDAC coupling (H2O, r.t., 2 h). Furthermore, because the protected sulfonic ester of taurine and L-cysteic acid (9 and 10) substituted dAMP were less stable than the carboxylic ester of protected L-Asp-dAMP, the initial attempts to separate and purify the sulfonic ester intermediates (9 and 10) failed and only trace amount of compounds were obtained. In addition, a sulfonamide side product (12) was obtained (Fig. 3). Apparently, the hydrolyzed sulfonic acid group could couple with another protected taurine in the presence of excess EDAC. Hydrolysis of compound 12 further generated compound 13, which could be, likewise, isolated from the same reaction mixture. The formation of side products and the problem obtained with the purification of the sulfonic ester intermediates (9 and 10) can be avoided by carrying out this reaction in a one-pot process using a ratio of 3.3:1 (EDACvs.dAMP). The yield of the desired compounds increases considerably when using this one-pot procedure.
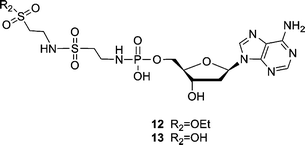 | ||
| Fig. 3 Sulfonamide side products (12 and 13) obtained during the coupling reaction of dAMP and taurine ethyl ester. | ||
The synthesis of compounds 3–8 is described in Scheme 2. As it could be a cumbersome procedure to selectively protect and deprotect the sulfate and phosph(on)ate amino acid derivatives during the preparation of 3–8, these compounds were synthesized in a one-step reaction as described before by H. Sawai9 for the formation of phosphodiesters. In this scheme, adenosine-5′-phosphorimidazolide is reacted with glycolic acid or lactic acid in 0.2 M N-ethylmorpholine buffer, pH 7.0 or 8.0, catalyzed by Pb(NO3)2 or ZnCl2. After 1–10 d, 0.25 M EDTA disodium quench buffer is used to break down the nucleotide-metal complex. The reaction mixtures are subjected to purification by HPLC. It was postulated that divalent metal ions such as Pb2+, Zn2+, Mn2+ and Mg2+ neutralize the charge of the phosphate group and promote the nucleophilicity of the hydroxyl group of the substrate by coordination. When we tried similar reaction conditions for the synthesis of the phosphoramidates (3–5), the obtained compounds have an Rf value (TLC, i-PrOH–NH3–H2O 7:1:2) which is very close to that of EDTA and the compounds were difficult to purify. As the nucleophilicity of a primary amino group is stronger than the nucleophilicity of a hydroxyl group, we modified the reaction conditions by using more N-ethylmorpholine as base without adding any metal ions (the use of EDTA in the quenching procedure can be avoided). However, in the case of compounds 6–8, ZnCl2 was added to increase the reactivity of the nucleoside phosphorimidazolides10 and the compounds were purified by column chromatography followed by preparative HPLC to remove the EDTA–Zn2+ complex.
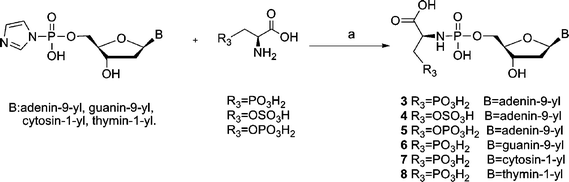 | ||
| Scheme 2 Reagents and conditions: (a) N-ethylmorpholine, H2O, (ZnCl2), r.t., 1–3 d. | ||
The ability of the phosphoramidate analogues (1–5) to function as substrate for HIV-1 RT was investigated by the gel-based single nucleotide incorporation assay (Fig. 4). The natural deoxyadenosine triphosphate (dATP) was used as reference.
![Single incorporation of the primer of P1T1 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 1, B: incorporation of 2, C and D: incorporation of 3, dATP (10 μM) is used as reference.](/image/article/2011/OB/c0ob00554a/c0ob00554a-f4.gif) | ||
| Fig. 4 Single incorporation of the primer of P1T1 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 1, B: incorporation of 2, C and D: incorporation of 3, dATP (10 μM) is used as reference. | ||
Using identical conditions, Tau-dAMP (1), L-Cys-dAMP (2), O-sulfonato-L-Ser-dAMP (4), O-phospho-L-Ser-dAMP (5) were not good substrates for HIV-RT, showing 50%, 63%, 8%, 19% conversion to a P + 1 strand in 60 min at 1 mM, respectively. However, the use of 3-phosphono-L-Ala-dAMP (3) as substrate resulted in 95% conversion to a P + 1 strand in 60 min at 50 μM. When the 3-phosphono-L-Ala-dAMP (3) concentration was decreased to 10 μM 29% of the P + 1 strand was still formed at 10 min. The phosphonate group has an additional charge when compared with L-Asp and L-Cys, which may have a beneficial effect on metal chelation and catalysis. In contrast, O-sulfonato-L-Ser-dAMP (4) is a very poor substrate for HIV-1 RT in the polymerase reaction. The sulfate residue of O-sulfonato-L-Ser-dAMP (4) is unstable and is easily hydrolyzed to L-Ser-dAMP which has already been proven to be a poor substrate.4 Although 3-phosphono-L-Ala-dAMP (3) and O-phospho-L-Ser-dAMP (5) both have four negative charges, the leaving group ability of 3-phosphono-L-alanine is superior. This suggests that the phosphonate group is better accommodated in the active site of HIV-RT leading to a more productive complex than the O-phosphate group. These observations indicate that small changes4 in the chemical constitution of the amino acid derivative can have a profound effect on its biochemical behavior.
The incorporation of more than one nucleotide was tested in a template dependent incorporation assay using HIV-1 RT (Fig. 5). Primer P1 and template T2 containing an overhang of seven thymidine residues and additional four nucleobases (GGAC) at the 5′-end were used.11
![Elongation of the primer of P1T2 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 2 and 3, B: incorporation of 3; blank: 125 nM primer/template (P1T2), [HIV-1 RT] = 0.025 U μL−1 and no nucleotide substrate, dATP (50 μM) is used as reference.](/image/article/2011/OB/c0ob00554a/c0ob00554a-f5.gif) | ||
| Fig. 5 Elongation of the primer of P1T2 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 2 and 3, B: incorporation of 3; blank: 125 nM primer/template (P1T2), [HIV-1 RT] = 0.025 U μL−1 and no nucleotide substrate, dATP (50 μM) is used as reference. | ||
The gel electrophoresis experiments showed that L-Cys-dAMP (2) could extend a primer with one and two adenine nucleotides (P + 1 and P + 2 products). The use of 3-phosphono-L-Ala-dAMP (3), however, leads to full elongation. After 60 min of polymerase reaction, 3-phosphono-L-Ala-dAMP (3) leads to strand elongation to a P + 7 product (24%) at 1 mM, P + 7 (16%) at 500 μM, P + 7 (7%) at 200 μM, respectively. Although the primer has been elongated, the stalling effect is still present, demonstrated by the slow-down of the incorporation reaction resulting predominantly in the formation of P + 2 and P + 3 products. In the case of L-Asp-dAMP incorporation at 500 μM, only a trace amount of P + 6 products was obtained and the major product is the P + 2 elongated oligonucleotide. It is not clear how these non-canonical leaving groups influence the translocation step for processive DNA synthesis. A chemical footprinting assay of PPi analogues revealed that phosphonoformic acid can stabilize the 3′ end complex in a pre-translocated configuration leading to inhibition of DNA synthesis.12a High concentration of PPi (10 mM) also blocked DNA synthesis and results in the formation of P + 1 and P + 2 as major products.12a After formation of the first and the second templated oligonucleotides (i.e. P + 1 and P + 2), the 3′ end of the blocked primer could be shifted to an unproductive configuration.12
To investigate the influence of the leaving group on the incorporation reaction, we carried out a product inhibition experiment. The incorporation of 3-phosphono-L-Ala-dAMP (3) (50 μM, figure in the ESI†) was carried out in the presence of a 2-, 10- and 20-fold higher concentration of 3-phosphono-L-alanine. After 120 min of polymerase reaction, 8–11% less P + 1 product was obtained. The result indicates that 3-phosphono-L-alanine formed in the polymerization reaction may serve as an inhibitory agent, although the effect would be minor.
The presence of an excess of 3-phosphono-L-alanine also has an influence on the chain elongation reaction. When adding 1 mM of 3-phosphono-L-alanine to the reaction mixture containing 150 μM of compound 3, P1T2 (125 nM) and [HIV-1 RT] = 0.025 U μL−1 for 120 min, 9% more P, 10% more P + 1 and 11% less P + 2 product was obtained (data not shown).
The efficiency of 3-phosphono-L-Ala-dAMP (3) was investigated by determining the kinetic parameters.13 Identical enzyme concentration [0.0125 U μL−1] was used for both dATP and 3-phosphono-L-Ala-dAMP (3). Steady-state kinetic analysis (Table 1) indicates that the Km for 3-phosphono-L-Ala-dAMP (3) is 78-fold higher than for the natural substrate dATP, the measured Vmax is only 1.3-fold lower than for dATP and the Vmax/Km ratio is 99 times lower than for dATP. These data indicate efficient nucleophilic displacement of 3-phosphono-L-alanine when the phosphoramidate is bound in the active site. The Vmax/Km value is superior to the value previously observed for L-Asp-dAMP, for which the Vmax/Km ratio is 1292 times lower than for dATP.4
| Substrate | V max/nM min−1 | K m/μM | V max/Km (×10−3 min−1) |
|---|---|---|---|
| dATP | 12.50 ± 0.2623 | 1.010 ± 0.1107 | 12.37 |
| 3-Phosphono-L-Ala-dAMP (3) | 9.842 ± 0.2572 | 78.90 ± 6.699 | 0.125 |
To further investigate the property of 3-phosphono-L-Ala-dAMP (3), it was tested as a substrate for Taq DNA polymerase (Fig. 6). The primer extension by Taq DNA polymerase was less efficient than using HIV-1 RT.
![Incorporation of the primer of P1T1 (125 nM) by Taq DNA polymerase with 3 as substrate; phosphoramidate concentrations and time intervals (min) are indicated; [Taq] = 0.025 U μL−1; dATP (10 μM) incorporation is used as reference.](/image/article/2011/OB/c0ob00554a/c0ob00554a-f6.gif) | ||
| Fig. 6 Incorporation of the primer of P1T1 (125 nM) by Taq DNA polymerase with 3 as substrate; phosphoramidate concentrations and time intervals (min) are indicated; [Taq] = 0.025 U μL−1; dATP (10 μM) incorporation is used as reference. | ||
To investigate the influence of the base moiety on the incorporation efficiency, we have evaluated the leaving group ability of 3-phosphono-L-alanine using the phosphoramidate nucleotides with the other three nucleobases. Single nucleotide incorporation assays (Fig. 7) were carried out for compounds 6–8. Efficient incorporation was observed for 3-phosphono-L-Ala-dGMP (6) and 3-phosphono-L-Ala-dTMP (8), which resulted in 77% and 81% conversion to a P + 1 strand in 60 min at 50 μM, respectively. In contrast, 3-phosphono-L-Ala-dCMP (7) was less efficient. A concentration of 1 mM was needed to reach 50% incorporation at 30 min. In agreement with this observation, the incorporation of dCTP is likewise less efficient than of dATP, dGTP and dTTP. It is not clear by now if this is a sequence specific effect of RT/primer-template interactions on substrate selection14 or a general observation, and this phenomenon needs further investigation. Using 3-phosphono-L-alanine as a leaving group, the incorporation efficiency decreases in the order A ≥ T = G > C.
![Single incorporation by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 6 with P1T3, dGTP (10 μM) is used as reference; B: incorporation of 7 with P2T4, dCTP (50 μM) is used as reference; C: incorporation of 8 with P1T5, dTTP (10 μM) is used as reference.](/image/article/2011/OB/c0ob00554a/c0ob00554a-f7.gif) | ||
| Fig. 7 Single incorporation by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1; A: incorporation of 6 with P1T3, dGTP (10 μM) is used as reference; B: incorporation of 7 with P2T4, dCTP (50 μM) is used as reference; C: incorporation of 8 with P1T5, dTTP (10 μM) is used as reference. | ||
To confirm that the observed incorporation profile was due to true base pairing following canonical Watson–Crick rules, we carried out control experiments (figure in ESI†) with compounds 3, 6 and 8 and mismatch sequences (A against A, C, G; G against G, T, A and T against C, T, G). After 120 min of the polymerase reaction at 1 mM substrate concentration, primer elongation was not observed in any of these cases.
Molecular modeling of 3-phosphono-L-Ala-dAMP in the active site of HIV-RT
The way in which 3-phosphono-L-Ala-dAMP (3), 3-phosphono-L-Ala-dGMP (6) and 3-phosphono-L-Ala-dTMP (8) could be accommodated in the active site of HIV-RT was investigated using molecular modeling and the Amber software.15Fig. 8 shows the entering 3-phosphono-L-Ala-dAMP (3) in complex with the RT enzyme. The α-phosphate and leaving group are in the same position as the triphosphate in the original substrate thymidine triphosphate (TTP) complex, close to the 3′-OH group of the terminal primer residue. Also the 2 Mg2+ ions are comparable in position to their localization in the original TTP complex. They are tightly bound to both the phosphonate group of 3-phosphono-L-Ala-dAMP (3) (mimicking the γ phosphate group), the phosphoramidate group and to three Asp groups in the enzyme: Asp110A and Asp185A, which are widely conserved in polymerases, and Asp186A.17 In the original X-ray structure, Asp186A is not connected to one of the Mg2+ ions. The side chain of Lys65A forms a salt bridge with the phosphonate group. Arg72A stabilizes the carboxyl function of the leaving group. Also Lys65A may interact with this carboxyl groupvia the formation of a salt bridge. Although the Lys65A.NZ carboxyl distance is 4.3 Å in the 3-phosphono-L-Ala-dAMP (3) model, such interaction may be possible. The Lys65A side chain could move near to the carboxyl group because of the flexibility of this side chain and no immediate steric hindrance is present for this group when moving closer. The possible binding mode of 3-phosphono-L-Ala-dNMP (N = G and T, respectively) is very similar and shown in the ESI.†
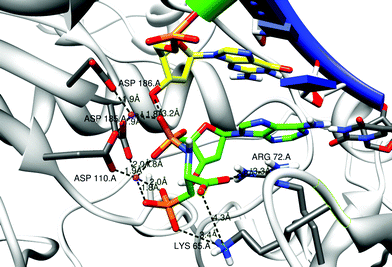 | ||
| Fig. 8 Model structures of 3-phosphono-L-Ala-dAMP in the RT dNTP pocket. The primer strand is drawn with a green ribbon; the template strand has a blue ribbon. The residues Asp 110, 185 and 186 anchor the 2 Mg2+ ions (purple balls). Some distances between charged atoms are indicated. The first nucleic acid of the primer strand (yellow carbons) and the complementary residue to 3-phosphono-L-Ala-dAMP (3) are shown in stick representation. Possible stabilization of the carboxyl function and the phosphonate function in the leaving group by Arg 72 and Lys 65 is indicated. Figures are generated using Chimera.16 | ||
Conclusions
The enzymatic synthesis of DNA uses deoxynucleoside triphosphates as substrates. Little research has been done to investigate the role of the pyrophosphate leaving group in this process. One of the suggested functions of the release of pyrophosphate is that it provides the energy for translocation.18 One way to study the role of the pyrophosphate moiety in the polymerization process is to replace the pyrophosphate group of deoxynucleoside triphosphates by other potential leaving groups, and analyze its influence on the polymerization process. Another important reason to study the substrate specificity of polymerases (such as reverse transcriptase) at the level of the leaving group is to develop analogues of nucleoside triphosphates that are direct substrates for the enzyme. 3-Phosphono-L-alanine is a remarkable leaving group for DNA synthesis catalyzed by HIV-RT. 3-Phosphono-L-Ala-dAMP is a better substrate for the polymerization process than the previously described L-Asp-dAMP (reflected by more successful DNA synthesis at 10-fold lower substrate concentration, and lower Vmax/Km value). However, DNA synthesis using this non-canonical leaving group leads to a stalling effect at P + 2 and P + 3, although at higher concentration the synthesis of P + 7 olignucleotide can be observed. The primer extension is base specific and the efficiency of incorporation for the primer sequence is A ≥ T = G > C. A modeling experiment shows two metal ions tightly binding to the leaving group and the carboxylic residues of the polymerase active site. The phosphonate group and the carboxyl group interact with Lys65A and Arg72A, respectively. As one of our interests is the development of a xenobiology19 based on alternative nucleic acids and their building blocks, an additional level of interest is the potential metabolic accessibility20 of the leaving group. 3-Phosphono-L-alanine is naturally occurring20a in the sea anemone Zoanthus sociatus and the protozoan Tetrahymena pyriformis and is not toxic.20b Biodistribution studies revealed that 3-phosphono-L-alanine is circulated in the human liver, intestine and spleen (no free phosphonic acid was detected and the acid was bound either to lipid or to protein).20c About 19% of 3-phosphono-L-alanine taken up by Tetrahymena pyriformis is accumulated as aminoethylphosphonic acid in the phospholipids.20d The catabolic pathway of 3-phosphono-L-alanine in Tetrahymena is the same as in rats.20e The observations that 3-phosphono-L-alanine is naturally occurring20a and nontoxic,20b further support its potential use as leaving groupin vivo.Experimental section
For all reactions, analytical grade solvents were used. Depending on sample amounts or availability, Bruker 600 MHz, Avance II 500 MHz or Avance 300 MHz spectrometers were used for 1H NMR, 13C NMR and 31P NMR. All chemical shifts are listed in ppm. For sake of clarity of the NMR signal assignment, sugar protons and carbons are numbered with a prime. 31P NMR chemical shifts are referenced to an external 85% H3PO4 standard (δ = 0.000 ppm). UV spectra were recorded on a Varian Cary-300-Bio UV/Vis spectrophotometer and optical rotations were measured at 20 °C on a Perkin–Elmer 341 polarimeter at 589 nm. Mass spectrometry was performed on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Q-Tof-2, Micromass, Manchester, UK) equipped with a standard electrospray probe (Z-spray, Micromass, Manchester, UK). Samples were dissolved in acetonitrile–water (1:1 v/v) and infused with a flow rate of 5 μL min−1. Electrospray capillary voltage was set to 3000 V and cone voltage to 30 V. Nitrogen gas was used for nebulisation and desolvation. Accurate masses were determined by coinfusion of the samples with leucine enkephalin (YGGFL) and recalibration of the spectrum using the peak at m/z 556.2771 as lock mass. Precoated aluminium sheets (MN ALUGRAM SIL G/UV254 20 × 20 cm) were used for TLC; the spots were examined with UV light. Column chromatography was performed on ICN silica gel 63–200, 60 Å. Preparative HPLC was performed on a Waters 1525–2487 system using Prep C18 5 μm column 19 × 150 mm at the flow rate of 3 mL min−1 by a gradient elution of acetonitrile and 50 mM triethylammonium bicarbonate (TEAB) buffer.
General procedure for the synthesis of compounds 1 and 2
The example described is for 2′-deoxyadenosine-5′-taurine phosphoramidate (compound 1).
Taurine ethyl ester was synthesized according to the procedure described previously.21 The crude product was directly used for the amidation reaction, because purification by flash chromatography on silica gel led to hydrolysis. 2′-Deoxyadenosine-5′-monophosphoric acid hydrate (40 mg, 0.11 mmol) and the crude taurine ethyl ester (81 mg, 0.53 mmol) were suspended in 1 mL water and stirred for 5 min under argon. Then, N-ethyl-N′-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDAC, 69 mg, 0.36 mmol) was added to the suspension and the reaction was continued to stir at room temperature. After 2 h, 1.4 M K2CO3 (MeOH–H2O 1:1) 1.2 mL was added to the reaction mixture. The progress of the reaction was monitored by TLC (CHCl3–MeOH–H2O 5:4:0.5) and 31P NMR until the disappearance of ester intermediate. The reaction mixture was neutralized by addition of 1 M triethylammonium acetate (TEAA). The solvent was evaporated to dryness in vacuum. The residue was purified by silica column chromatography eluting with i-PrOH–NH3–H2O (12:1:1, 10:1:1 to 9:1:1) to yield compound 1 (30 mg, 62%) as a white solid. The product was further purified by an ion exchange column (DEAE-Sephadex A-25) with a gradient of 1 M triethylammonium bicarbonate buffer (25 mg, 83.3%).
General procedure for the synthesis of compounds 3–8
The example described is for 2′-deoxyadenosine-5′-(3-phosphono-L-alanine) phosphoramidate (compound 3).
Deoxyadenosine-5′-phosphorimidazolide was synthesized according to the standard procedures.22 In a 25 ml flask, L-alanine-3-phosphono acid HCl salt23 (60 mg, 0.29 mmol) were stirred in 0.5 mL N-ethyl morpholine for 5 min at room temperature, then deoxyadenosine-5′-phosphorimidazolide (240 mg, 0.63 mmol) and 6 mL 0.2 M N-ethyl morpholine buffer (pH 7.5) were added into the flask. The reaction mixture was continued to stir for 3 days at room temperature under argon, while the reaction process was monitored by TLC (i-PrOH–NH3–H2O 7:1:2) and 31P NMR. The reaction mixture was concentrated to dryness in vacuum (30 °C bath) and the residue was purified by silica column chromatography eluting with i-PrOH–NH3–H2O (20:1:1, 9:1:1, 7:1:1 to 4.5:1:1) and yielding crude white solid (32 mg, 22.8%). The product was purified by preparative HPLC with a gradient of CH3CN in 50 mM TEAB buffer (pH = 7.4) to yield compound 3 (10 mg, 31.3%).
:1:2, v/v): Rf 0.62; 1H NMR (500 MHz, D2O): δ 8.42 (s, 1H, H-8), 8.24 (s, 1H, H-2), 6.49 (m, 1H, H-1′), 4.80 (m, 1H, H-3′), 4.25 (m, 1H, H-4′), 4.05 (m, 2H, -OCH2CH3), 4.03 (m, 2H, H-5′ and H-5′′), 3.85 (m, 1H, -CHCOOH), 3.09 (m, 2H, -CH2SO2OH), 2.86 (m, 1H, H-2′), 2.62 (m, 1H, H-2′′); 13C NMR (125 MHz, D2O): δ 173.50 (d, 3J(C, P) = 4.6 Hz, -COOEt), 155.22 (C-6), 152.37 (C-2), 148.40 (C-4), 139.52 (C-8), 118.27 (C-5), 85.60 (d, 3J(C, P) = 16.2 Hz, C-4′), 83.26 (C-1′), 70.90 (C-3′), 63.74 (d, 2J(C, P) = 8.1 Hz, C-5′), 62.02 (-OCH2CH3), 53.22 (d, 3J(C, P) = 8.1 Hz, -CH2SO3H), 51.13 (-CHCOOEt), 38.45 (C-2′), 12.75 (-OCH2CH3); 31P NMR (202 MHz, D2O): δ 5.50; HRMS for C15H23N6O10PS (M-H)− calcd: 509.0861, found: 509.0884.
Oligodeoxyribonucleotides
DNA oligonucleotides P1, P2, T1, T2, T3, T4 and T5 were purchased from Sigma Genosys and Eurogentec. The concentrations were determined with a Varian Cary-300-Bio UV Spectrophotometer. The lyophilized oligonucleotides were dissolved in diethylpyrocarbonate (DEPC)-treated water and stored at −20 °C. The primer oligonucleotides were 5′-labeled with [γ-33P] ATP (Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs) according to the standard procedures. Labeled oligonucleotides were further purified with Illustra™ Microspin™ G-25 columns (GE Healthcare).DNA polymerase reactions
End-labeled primer was annealed to its template by combining primer and template at a molar ratio of 1:2 and heating the mixture to 70 °C for 10 min, followed by slow cooling to room temperature over a period of 2 h. For the single nucleotide incorporation of 1–5, primer P1 was annealed to template T1. For the single nucleotide incorporation of 6, 7 and 8, P1T3, P2T4 and P1T5 were used, respectively. A series of 20 μL batch reactions were performed for the enzyme HIV-1 RT (Ambion, Inc.; 10 U μL−1 stock solution). The final mixture contained 125 nM primer-template complex, RT buffer (50 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 0.5 mM spermidine, 10 mM DTT; pH 8.3), 0.025 U μL−1HIV-1 RT, and different concentrations of phosphoramidate building blocks. In the control reaction with the natural nucleotide, 10 μM or 50 μM dATP, dGTP, dTTP, dCTP were used. Mixtures were incubated at 37 °C, and aliquots (2.5 μL) were removed and quenched after 10, 20, 30, 60, 120 min. Primer elongation study with HIV-1 RT was performed in a fashion similar to the single nucleotide incorporation experiments, in which P1 and T2 were used. The single nucleotide incorporation assay by Taq DNA polymerase (New England Biolabs) was similarly carried out at 75 °C in ThermoPol RB buffer (10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl, 2 mM MgSO4, 0.1% Triton X-100; pH 8.8).
Steady-state kinetics of single nucleotide incorporation
To determine the kinetic parameters for the incorporation of 3-phosphono-L-Ala-dAMP (3) and a natural nucleoside dATP, a steady-state kinetics assay was carried out. The reaction was started by adding HIV-1 RT to P1-T1 complex, buffer, 3-phosphono-L-Ala-dAMP (3) and dATP. The final mixture (20 μL) contained 0.0125 U/μL HIV-1 RT, buffer, 125 nM primer-template complex, and various concentrations of 3-phosphono-L-Ala-dAMP and dATP. The range of concentrations for phosphoramidates was optimized according to a Km value for the incorporation of an individual nucleotide. In the case of HIV-1 RT, reaction mixtures containing the enzyme in concentration (0.0125 U μL−1) to attain 5–25% incorporation and appropriate substrate concentration (10–1000 μM used for 3-phosphono-L-Ala-dAMP (3) and 1–20 μM used for dATP) were incubated at 37 °C and run for 8–10 different time intervals (1–10 min). The incorporation velocities were calculated based on the percentage of single-nucleotide extension product (P + 1 band). The kinetic parameters (Vmax and Km) were determined by plotting V (nM min−1) versus substrate concentration (μM) and fitting the data point to a nonlinear Michaelis–Menten regression using GraphPad Prism software.Electrophoresis
All polymerase reactions (2.5 μL) were quenched by the addition of 10 μL of loading buffer (90% formamide, 0.05% bromophenol blue, 0.05% xylene cyanol, and 50 mM ethylenediaminetetraacetic acid (EDTA)). Samples were heated at 75 °C for 5 min prior to analysis by electrophoresis for 2.5 h at 2000 V on a 0.4 mm 20% denaturing gel in the presence of a 100 mM Tris-borate, 2.5 mM EDTA buffer, pH 8.3. Products were visualized by phosphor imaging. The amount of radioactivity in the bands corresponding to the products of enzymatic reactions was determined with the imaging device Cyclone and the associated Optiquant image analysis software (Perkin–Elmer).Molecular modelling
Acknowledgements
This work was financed by a grant from K. U. Leuven (GOA) and from EC (Orthosome). We are grateful to Dr Jef Rozenski for HRMS and Chantal Biernaux for editorial help. We are also indebted to Luc Baudemprez for running the NMR spectra. Shiqiong Yang is obliged to Chinese Scholarship Council for a Ph.D. fellowship.Notes and references
- A. J. Berdis, Chem. Rev., 2009, 109, 2862–2879 CrossRef CAS; S. G. Sarafianos, K. Das, J.-P. Ding, P. L. Boyer, S. H. Hughes and E. Arnold, Chem. Biol., 1999, 6, R137–146 CrossRef CAS; P. H. Patel, A. Jacobo-Molina, J.-P. Ding, C. Tantillo, A. D. Clark, R. Raag, R. G. Nanni, S. H. Hughes and E. Arnold, Biochemistry, 1995, 34, 5351–5363 CrossRef CAS.
- (a) N. A. Boyle, V. K. Rajwanshi, M. Prhavc, G. Wang, P. Fagan, F. Chen, G. Ewing, J. L. Brooks, T. Hurd, T. W. Bruisce and P. D. Cook, J. Med. Chem., 2005, 48, 2695–2700 CrossRef CAS; (b) B. A. Mulder, S. Anaya, P. Yu, K. W. Lee, A. Nguyen, J. Murphy, R. Willson, J. M. Briggs, X.-L. Gao and S. H. Hardin, Nucleic Acids Res., 2005, 33, 4865–4873 CrossRef CAS.
- H. Strobel, L. Dugué, P. Marlière and S. Pochet, Nucleic Acids Res., 2002, 30, 1869–1878 CrossRef CAS; M. J. Lutz, H. A. Held, M. Hottiger, U. Hübscher and S. A. Benner, Nucleic Acids Res., 1996, 24, 1308–1313 CrossRef CAS.
- (a) O. Adelfinskaya and P. Herdewijn, Angew. Chem., Int. Ed., 2007, 46, 4356–4358 CrossRef CAS; (b) O. Adelfinskaya, M. Terrazas, M. Froeyen, P. Marlière, K. Nauwelaerts and P. Herdewijn, Nucleic Acids Res., 2007, 35, 5060–5072 CrossRef CAS; (c) M. Terrazas, P. Marlière and P. Herdewijn, Chem. Biodiversity, 2008, 5, 31–39 CrossRef CAS.
- A. Giraut, N. Dyubankova, X.-P. Song and P. Herdewijn, ChemBioChem, 2009, 10, 2246–2252 CrossRef CAS.
- I. Zlatev, A. Giraut, F. Morvan, P. Herdewijn and J-J. Vasseur, Bioorg. Med. Chem., 2009, 17, 7008–7014 CrossRef CAS.
- F.-Q. Huang, G.-C. Wang, T. Coleman and N. Li, RNA, 2003, 9, 1562–1570 CrossRef CAS.
- T. W. Abraham, T. I. Kalman, E. J. McIntee and C. R. Wagner, J. Med. Chem., 1996, 39, 4569–4575 CrossRef CAS.
- H. Sawai, Bull. Chem. Soc. Jpn., 1990, 63, 692–696 CrossRef CAS.
- (a) A. R. Kore, M. Shanmugasundaram, I. Charles, A. V. Vlassov and T. J. Barta, J. Am. Chem. Soc., 2009, 131, 6364–6365 CrossRef CAS; (b) K. Huang and P. A. Frey, J. Am. Chem. Soc., 2004, 126, 9548–9549 CrossRef CAS.
- A. Giraut, X.-P. Song, M. Froeyen, P. Marlière and P. Herdewijn, Nucleic Acids Res., 2010, 38, 2541–2550 CrossRef CAS.
- (a) B. Marchand, E. P. Tchesnokov and M. Götte, J. Biol. Chem., 2007, 282, 3337–3346 CAS; (b) L. Menéndez-Arias, Virus Res., 2008, 134, 124–146 CrossRef CAS.
- (a) M. S. Boosalis, J. Petruska and M. F. Goodman, J. Biol. Chem., 1987, 262, 14689–14696 CAS; (b) M. F. Goodman, S. Creighton, L. B. Bloom and J. Petruska, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 83–126 CrossRef CAS.
- (a) H. Huang, R. Chopra, G. L. Verdine and S. C. Harrison, Science, 1998, 282, 1669–1675 CrossRef CAS; (b) G. J. Klarmann, R. A. Smith, R. F. Schinazi, T. W. North and B. D. Preston, J. Biol. Chem., 2000, 275, 359–366 CrossRef CAS.
- D. A. Case, T. E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K. M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- (a) T. A. Steitz, Nature, 1998, 391, 231–232 CrossRef CAS; (b) L. Yang, W. A. Beard, S. H. Wilson, S. Broyde and T. Schlick, Biophys. J., 2004, 86, 3392–3408 CrossRef CAS.
- Y. W. Yin and T. A. Steitz, Cell, 2004, 116, 393–404 CrossRef CAS.
- P. Herdewijn and P. Marlière, Chem. Biodiversity, 2009, 6, 791–808 CrossRef CAS.
- (a) J. S. Kittredge and R. R. Hughes, Biochemistry, 1964, 3, 991–996 CrossRef CAS; (b) N. G. Ternan, J. W. McGrath and J. P Quinn, Appl. Environ. Microbiol., 1998, 64, 2291–2294 CAS; (c) S. A. Tan and L. G. Tan, Clin. Physiol. Biochem., 1989, 7, 303–309 Search PubMed; (d) J. D. Smith and J. H. Law, Biochemistry, 1970, 9, 2152–2157 CrossRef CAS; (e) A. Horigane, M. Horiguchi and T. Matsumoto, Biochim. Biophys. Acta., 1979, 572, 385–394 CAS.
- T. J. Lehmann and J. W. Engels, Bioorg. Med. Chem., 2001, 9, 1827–1835 CrossRef CAS.
- J. Imai and P. F. Torrence, J. Org. Chem., 1985, 50, 1418–1426 CrossRef CAS.
- E. C. R. Smith, L. A. McQuaid, J. W. Paschal and J. DeHoniesto, J. Org. Chem., 1990, 55, 4472–4474 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, Jr., J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- Quatfit, CCL software archives, http://www.ccl.net/(accessed January 15, 2010).
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. E. Cheatham, III, J. L. Miller, T. Fox, T. A. Darden and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 4193–4194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the sulf(on)ate and phosph(on)ate phosphoramidate analogues (compounds 1–8). See DOI: 10.1039/c0ob00554a |
| This journal is © The Royal Society of Chemistry 2011 |