A novel synthesis of oxazolidine-2,4-diones via an efficient fixation of CO2 with 3-aryl-2-alkynamides†
Received
7th August 2010
, Accepted 29th September 2010
First published on 17th November 2010
Abstract
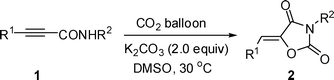
A very mild protocol for fixation of carbon dioxide with 2-alkynamides in DMSO at 30 °C using a CO2 balloon in the presence of K2CO3 has been developed, which leads to an efficient assembly of oxazolidine-2,4-diones. It is observed that the regioselectivity was controlled by the aryl group.
Introduction
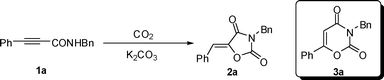
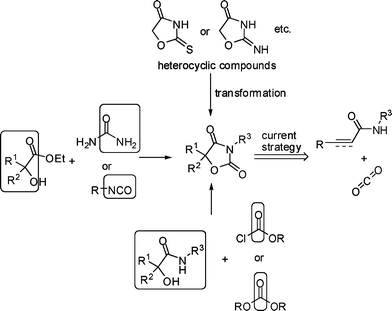
Recently much attention has been paid to the chemistry of carbon dioxide as an attractive carbon resource in organic synthesis. More and more methods for transformations of CO2 into useful organic compounds have been reported.1 In this area, the reaction of propargyl alcohols and amines with CO2 yielding cyclic carbonates and urethanes has already been well described in the literature.2 However, due to the low nucleophilicity of amide, the fixation of CO2 with amide has seldom been reported.3 On the other hand, we noticed that oxazolidine-2,4-diones are widely used in medicine and agriculture as antiepileptic agents,4a anti-inflammatory agents,4b–c herbicides,4f–g and the synthesis of oxazolidine-2,4-diones is usually lengthy (Fig. 1).4 Besides transformations from heterocyclic intermediates, the most general methods are the cyclizations of α-hydroxy esters with urea or isocyanates and α-hydroxy amides with chloroformates or carbonates (Scheme 1).4a,5 However, the most efficient route to oxazolidine-2,4-diones would be from the reactions of 2-alkenamide or 2-alkynamide with CO2. Recently, we have reported the fixation of CO2 with 2,3-allenamides under mild conditions to synthesize 1,3-oxazine-2,4-diones (eqn (1)).6 Herein, we wish to report a simple chemical fixation of CO2 with 3-aryl-2-alkynamides for unexpected synthesis of oxazolidine-2,4-diones. | |  | (1) |
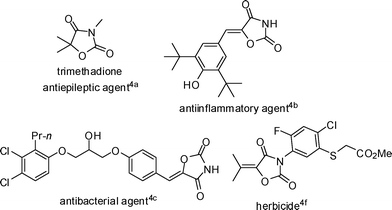 |
| | Fig. 1 Some biologically active oxazolidine-2,4-diones. | |
 |
| | Scheme 1 Synthesis of oxazolidine-2,4-diones. | |
Results and discussion
We used N-benzyl-3-phenylpropiolamide 1a as the substrate to test the reaction under the CO2 transformation conditions for the reaction of 2,3-allenamides with CO2.6 Fortunately, we observed the formation of a new solid compound in 16% yield determined by 1H NMR spectroscopy (entry 1, Table 1). With the X-ray diffraction study, we learned that the solid compound was oxazolidine-2,4-dione 2a with a single Z configuration but not 1,3-oxazine-2,4-dione 3a as we were expecting (Fig. 2).7 Encouraged by this result, we optimized the reaction conditions for the chemical fixation of CO2 with 1a. Some typical reaction conditions are summarized in Table 1. Due to the possible instability of product 2a, we lowered the reaction temperature to 30 °C, and 2a was formed in 75% yield determined by 1H NMR spectroscopy with 9% recovery of starting material 1a within 11 h (entry 2, Table 1). No reaction occurred with 100% recovery of starting material 1a when commercial DMSO was used, which shows that a trace amount of water in DMSO may stop this transformation in the deprotonation step (entry 3, Table 1). When 2.0 equiv of K2CO3 were used, 2a was obtained in 75% yield determined by 1H NMR spectroscopy with complete conversion of the starting material 1a within 11 h (entry 6, Table 1). Increasing the concentration of K2CO3 to 3.0 equiv did not improve the yield of 2a very much (entry 7, Table 1). Without the CO2 balloon, the yield of 2a dropped to 66% determined by 1H NMR spectroscopy under the CO2 atmosphere (entry 8, Table 1). When the reaction was carried out in the presence of K2CO3 under N2 atmosphere, the formation of oxazolidine-2,4-dione 2a was not observed with 93% recovery of the starting material 1a, which indicated that the CO2 unit in the product 2a was from the CO2 gas, not the carbonate base K2CO3 (entry 9, Table 1).
Table 1 Optimization of reaction conditions for the reaction of N-benzyl-3-phenylpropiolamide 1a with carbon dioxidea
| entry |
solvent |
base (equiv) |
temp. (°C) |
time (h) |
yield (%) of 2a |
recovery of 1a |
|
The reaction was carried out using 0.2 mmol of 1a in dried solvent with a CO2 balloon, and the yields were determined by 1H NMR analysis with CH2Br2 as the internal standard.
DMSO was commercially available and used without treatment.
The reaction was carried out under CO2 atmosphere.
The reaction was carried out under N2 atmosphere.
|
| 1 |
DMSO |
1.0 |
70 |
3 |
16 |
0 |
| 2 |
DMSO |
1.0 |
30 |
11 |
75 |
9 |
| 3b |
DMSO |
1.0 |
30 |
11 |
0 |
100 |
| 4 |
DMF |
2.0 |
30 |
11 |
65 |
22 |
| 5 |
DMA |
2.0 |
30 |
11 |
40 |
30 |
| 6 |
DMSO
|
2.0
|
30
|
11
|
75
|
0
|
| 7 |
DMSO |
3.0 |
30 |
11 |
77 |
0 |
| 8c |
DMSO |
2.0 |
30 |
11 |
66 |
0 |
| 9d |
DMSO |
2.0 |
30 |
11 |
0 |
93 |
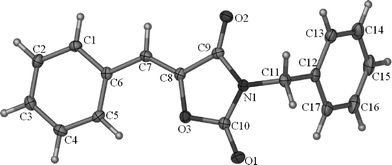 |
| | Fig. 2 ORTEP representation of 2a. | |
Some typical results of different 3-aryl-2-alkynamides under the optimized conditions are listed in Table 2. The substituent R2 on the nitrogen atom of 2-alkynamides can be alkyl, benzyl and allyl groups (entries 1–4, Table 2). When the i-propyl group substituted 2-alkynamide 1e was applied, the product 2e was afforded in 68% yield with 16% recovery of 1e even using 3.0 equiv of K2CO3 due to the steric effect (entry 5, Table 2). Using a stronger base such as Cs2CO3, the yield of 2e was 46% though the starting material 1e was consumed completely (entry 6, Table 2). However, when 3-phenylpropiolamide 1f was applied, no reaction occurred with 64% recovery of the starting material (entry 7, Table 2). The substituent R1 can be phenyl groups bearing both electron-donating and electron-withdrawing groups (entries 8–12, Table 2). Heterocyclic aryl groups such as 2- or 3-thienyl group substituted 2-alkynamides could also be applied to the reaction with slightly lower yields (entries 13 and 14, Table 2). It should be noted that the reaction of cyclohexenyl substituted propiolamides 1n and n-butyl substituted propiolamides 1o under the same reaction conditions did not occur with recovery of starting materials, which shows the importance of the aryl group moiety for this transformation (entries 15–16, Table 2).
Table 2 Reaction of different 2-alkynamides with carbon dioxidea
| entry |
R1 |
R2 |
time (h) |
isolated yield (%) of 2 |
|
The reaction was carried out using 0.2 mmol of 1 and 2.0 equiv of K2CO3 in DMSO (distilled from CaH2) with a CO2 balloon at 30 °C unless otherwise stated.
K2CO3 (3.0 equiv) was used.
Compound 1e was recovered in 16% yield.
Cs2CO3 (2.0 equiv) was used.
Compound 1f was recovered in 64% yield.
Compound 1n was recovered in 92% yield.
Compound 1o was recovered in 100% yield.
|
| 1 |
Ph |
Bn (1a) |
11 |
62 (2a) |
| 2 |
Ph |
n-Bu (1b) |
11 |
80 (2b) |
| 3 |
Ph |
Et (1c) |
11 |
66 (2c) |
| 4 |
Ph |
allyl (1d) |
11 |
72 (2d) |
| 5b,c |
Ph |
i-Pr (1e) |
11 |
68 (2e) |
| 6d |
Ph |
i-Pr (1e) |
11 |
46 (2e) |
| 7e |
Ph |
H (1f) |
11 |
0 |
| 8 |
4-PrC6H4 |
n-Bu (1g) |
12 |
69 (2g) |
| 9 |
4-MeC6H4 |
n-Bu (1h) |
16 |
69 (2h) |
| 10 |
4-MeOC6H4 |
n-Bu (1i) |
11 |
67 (2i) |
| 11 |
4-MeOC6H4 |
Bn (1j) |
11 |
68 (2j) |
| 12 |
4-FC6H4 |
n-Bu (1k) |
11 |
72 (2k) |
| 13 |
3-thienyl |
Bn (1l) |
11 |
61 (2l) |
| 14b |
2-thienyl |
Bn (1m) |
11 |
55 (2m) |
| 15f |
cyclohexenyl |
Bn (1n) |
11 |
0 |
| 16g |
n-Bu |
Bn (1o) |
11 |
0 |
Based on these facts, a rational mechanism for the formation of 2 is depicted in Scheme 2. The amide 1 would lose a proton under the basic conditions with K2CO3 to form anionic intermediate M1, which may attack the carbon atom in carbon dioxide to form the intermediate M2. There is an issue of regioselectivity (α vs. β). When the R′ is an allenyl group, the oxygen anion would attack the central carbon atom in the allene moiety (β carbon atom) to form six-membered product 4, which was controlled by the carbonyl group. However, when the amide was changed to 3-aryl-2-alkynamide, although both 5-exo-dig and 6-endo-dig are favored according to the Baldwin's rule, the oxygen anion in the intermediate M2 would attack α carbon atom in the C–C triple bond to form five-membered product 2, which was obviously directed by the aryl group. This also explains why the aryl group is so important in this reaction and why the alkyl substituted 2-alkynamides fail for this reaction.
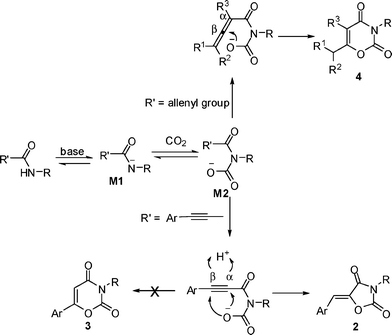 |
| | Scheme 2 Reaction mechanism. | |
Conclusions
In conclusion, we have developed a very mild protocol for fixation of carbon dioxide with 2-alkynamides in DMSO at 30 °C using a CO2 balloon in the presence of K2CO3, which leads to an efficient assembly of oxazolidine-2,4-diones. The regioselectivity of this reaction was controlled by the aryl group which is different from the reaction of 2,3-allenamides with carbon dioxide. As a result of the easy availability of starting materials, the usefulness of the products and the efficient fixation of carbon dioxide, this reaction may have potentials in organic synthesis. Further studies including expanding the substrate scope in this area are being pursued in our laboratory.
Experimental
Materials
DMSO was distilled from CaH2. THF was distilled from Na/benzophenone. Et3N was distilled from KOH. The other commercially available chemicals were purchased and used without additional purification unless noted otherwise.
Synthesis of starting materials
Known compounds 1a,8a1b,8b1c,8c1d,8d1e,8c1f,8a1j,8e1o8f and new compounds 1g–1i, 1k–1n were prepared following the known procedure.8a
N-(n-Butyl)-3-(4-n-propylphenyl)propiolamide (1g).
To the reaction vessel containing ethyl 3-(4-n-propylphenyl)propiolate (1.0808 g, 5.00 mmol) were added 2 mL of H2O and 2 mL of n-BuNH2 sequentially, Then the resulting solution was stirred at room temperature. After 15 h, the reaction was diluted with 25 mL of CH2Cl2, washed with water twice, and dried over anhydrous Na2SO4. After filtration and evaporation, chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5/1) of the crude product afforded 1g (1.1213 g, 92%) as a solid, m.p.: 48.0–49.0 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.51–7.39 (m, 2H, Ar-H), 7.22–7.10 (m, 2H, Ar-H), [5.92 (bs), 5.76 (bs), 1H, NH], [3.49 (q, J = 6.8 Hz), 3.35 (q, J = 6.7 Hz), 2H, N-CH2], 2.70–2.50 (m, 2H, Ar-CH2), 1.71–1.47 (m, 4H, 2×MeCH2), 1.46–1.30 (m, 2H, CH2), 1.01–0.83 (m, 6H, 2×Me); MS (m/z): 244 (M+ + 1, 2.47), 243 (M+, 14.26), 171 (100); IR (KBr, cm−1): 3285, 2959, 2931, 2866, 2226, 1629, 1537, 1464, 1433, 1410, 1375, 1307, 1223, 1179, 1151, 1112; Anal. Calcd. for C16H21NO: C 78.97, H 8.70, N 5.76; Found: C 79.16, H 8.72, N 6.08%.
N-(n-Butyl)-3-(p-tolyl)propiolamide (1h).
Following the procedure for the preparation of 1g, the reaction of 1.0657 g (5.67 mmol) of ethyl 3-(p-tolyl)propiolate, 2 mL of H2O and 2 mL of n-BuNH2 afforded 1.0541 g (86%) of 1h (eluent: petroleum ether/ethyl acetate = 10/1–5/1) as a solid, m.p.: 56.9–57.4 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.47–7.37 (m, 2H, Ar-H), 7.22–7.10 (m, 2H, Ar-H), [5.95 (bs), 5.81 (bs), 1H, NH], [3.48 (q, J = 6.7 Hz), 3.35 (q, J = 6.7 Hz), 2H, N-CH2], [2.38 (s), 2.36 (s), 3H, Ar-CH3], 1.63–1.48 (m, 2H, CH2), 1.48–1.30 (m, 2H, MeCH2), 1.01–0.88 (m, 3H, Me); MS (m/z): 216 (M+ + 1, 1.61), 215 (M+, 10.30), 143 (100); IR (KBr, cm−1): 3271, 2961, 2932, 2872, 2216, 1630, 1534, 1464, 1377, 1353, 1303, 1222, 1209, 1182, 1023; Anal. Calcd. for C14H17NO: C 78.10, H 7.96, N 6.51; Found: C 77.99, H 7.81, N 6.63%.
N-(n-Butyl)-3-(4-methoxyphenyl)propiolamide (1i).
Following the procedure for the preparation of 1g, the reaction of 1.0130 g (4.97 mmol) of ethyl 3-(4-methoxyphenyl)propiolate, 2 mL of H2O and 2 mL of n-BuNH2 afforded 1.1276 g (98%) of 1ias a solid, m.p.: 66.2–67.0 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.53–7.42 (m, 2H, Ar-H), 6.92–6.80 (m, 2H, Ar-H), 6.02 (bs, 1H, NH), [3.82 (s), 3.81 (s), 3H, OMe], [3.47 (q, J = 7.0 Hz), 3.34 (q, J = 6.8 Hz), 2H, N-CH2], 1.65–1.48 (m, 2H, CH2), 1.48–1.27 (m, 2H, MeCH2), 1.00–0.87 (m, 3H, Me); MS (m/z): 232 (M+ + 1, 2.02), 231 (M+, 13.76), 159 (100); IR (KBr, cm−1): 3269, 3051, 2959, 2925, 2871, 2210, 1630, 1605, 1537, 1510, 1465, 1438, 1287, 1252, 1224, 1172, 1107, 1031; Anal. Calcd. for C14H17NO2: C 72.70, H 7.41, N 6.06; Found: C 72.69, H 7.41, N 6.07%.
N-(n-Butyl)-3-(4-fluorophenyl)propiolamide (1k).
Following the procedure for the preparation of 1g, the reaction of 0.9706 g (5.06 mmol) of ethyl 3-(4-fluorophenyl)propiolate, 2 mL of H2O and 2 mL of n-BuNH2 afforded 1k with some minor impurity after purification by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5/1). This product was further purified by recrystallization to afford 0.9623 g (87%) of 1k (n-hexane/CH2Cl2) as a solid, m.p.: 59.1–59.5 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.60–7.45 (m, 2H, Ar-H), 7.15–6.98 (m, 2H, Ar-H), [5.97 (bs), 5.85 (s), 1H, NH], [3.48 (q, J = 6.7 Hz), 3.35 (q, J = 6.6 Hz), 2H, N-CH2], 1.65–1.46 (m, 2H, CH2), 1.46–1.29 (m, 2H, MeCH2), 1.00–0.81 (m, 3H, Me); 19F NMR (282 MHz, CDCl3) δ −107.15, −107.61 (standard by frequency conversion of CDCl3); MS (m/z): 220 (M+ + 1, 1.17), 219 (M+, 7.79), 147 (100); IR (KBr, cm−1): 3304, 2964, 2935, 2868, 2229, 1624, 1598, 1538, 1505, 1471, 1400, 1375, 1351, 1307, 1231, 1157, 1095; Anal. Calcd. for C13H14FNO: C 71.21, H 6.44, N 6.39; Found: C 71.15, H 6.45, N 6.35%.
N-Benzyl-3-(3-thienyl)propiolamide (1l).
Following the procedure for the preparation of 1g, the reaction of 1.1863 g (6.59 mmol) of ethyl 3-(3-thienyl)propiolate, 2.5 mL of H2O and 2.5 mL of BnNH2 afforded 1l with some minor impurity after purification by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5/1–3/1). This product was further purified by recrystallization to afford 0.8678 g (54%) of 1l (n-hexane/CH2Cl2) as a solid, m.p.: 111.4–112.0 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.68–7.62 (m, 1H, Ar-H), 7.42–7.27 (m, 6H, Ar-H), 7.20–7.14 (m, 1H, Ar-H), 6.16 (bs, 1H, NH), [4.69 (d, J = 7.2 Hz), 4.54 (d, J = 5.7 Hz), 2H, N-CH2]; MS (m/z): 242 (M+ + 1, 12.00), 241 (M+, 64.86), 135 (100); IR (KBr, cm−1): 3215, 3105, 3036, 2847, 2218, 1625, 1558, 1494, 1452, 1420, 1359, 1294, 1225, 1172, 1089, 1030; Anal. Calcd. for C14H11NOS: C 69.68, H 4.59, N 5.80; Found: C 69.79, H 4.72, N 6.03%.
N-Benzyl-3-(2-thienyl)propiolamide (1m).
Following the procedure for the preparation of 1g, the reaction of 0.4535 g (2.52 mmol) of ethyl 3-(2-thienyl)propiolate, 1 mL of H2O and 1 mL of BnNH2 afforded 0.2171 g (36%) of 1m (eluent: petroleum ether/ethyl acetate = 5/1–3/1) as a solid, m.p.: 107.3–107.6 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.47–7.21 (m, 7H, Ar-H), 7.07–6.95 (m, 1H, Ar-H), [6.22 (bs), 6.05 (bs), 1H, NH], [4.67 (d, J = 6.3 Hz), 4.54 (d, J = 5.7 Hz), 2H, N-CH2]; MS (m/z): 242 (M+ + 1, 9.74), 241 (M+, 50.45), 135 (100); IR (KBr, cm−1): 3273, 3088, 2207, 1625, 1583, 1552, 1496, 1453, 1425, 1366, 1277, 1231, 1180, 1061, 1029, 1009; Anal. Calcd. for C14H11NOS: C 69.68, H 4.59, N 5.80; Found: C 69.74, H 4.60, N 6.12%.
N-(n-Butyl)-3-cyclohexenylpropiolamide (1n).
Following the procedure for the preparation of 1g, the reaction of 0.9073 g (5.10 mmol) of ethyl 3-cyclohexenylpropiolate, 2 mL of H2O and 2 mL of n-BuNH2 afforded 0.9200 g (88%) of 1n (eluent: petroleum ether/ethyl acetate = 3/1) as a liquid. 1H NMR (300 MHz, CDCl3) δ 6.41–6.27 (m, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.80 (bs, 1H, NH), [3.38 (q, J = 6.7 Hz), 3.29 (q, J = 6.7 Hz), 2H, N-CH2], 2.21–2.02 (m, 4H, CH2-CC-CH2), 1.69–1.45 (m, 6H, 3×CH2), 1.44–1.25 (m, 2H, MeCH2), 1.00–0.83 (m, 3H, Me); MS (m/z): 206 (M+ + 1, 1.98), 205 (M+, 8.93), 133 (100); IR (neat, cm−1): 3263, 3054, 2931, 2862, 2207, 1633, 1538, 1435, 1360, 1348, 1290, 1265, 1226, 1184, 1137, 1077; Anal. Calcd. for C13H19NO: C 76.06, H 9.33, N 6.82; Found: C 76.03, H 9.27, N 6.73%.
CH), 5.80 (bs, 1H, NH), [3.38 (q, J = 6.7 Hz), 3.29 (q, J = 6.7 Hz), 2H, N-CH2], 2.21–2.02 (m, 4H, CH2-CC-CH2), 1.69–1.45 (m, 6H, 3×CH2), 1.44–1.25 (m, 2H, MeCH2), 1.00–0.83 (m, 3H, Me); MS (m/z): 206 (M+ + 1, 1.98), 205 (M+, 8.93), 133 (100); IR (neat, cm−1): 3263, 3054, 2931, 2862, 2207, 1633, 1538, 1435, 1360, 1348, 1290, 1265, 1226, 1184, 1137, 1077; Anal. Calcd. for C13H19NO: C 76.06, H 9.33, N 6.82; Found: C 76.03, H 9.27, N 6.73%.
Reactions of 2-alkynamides with CO2
(Z)-3-Benzyl-5-benzylideneoxazolidine-2,4-dione (2a).
To the reaction vessel containing K2CO3 (54.5 mg, 0.39 mmol) were charged 1a (46.2 mg, 0.20 mmol) and 2 mL of DMSO sequentially under CO2 atmosphere. The CO2 gas from a CO2 balloon was dried by passing through a gas washing bottle with conc. H2SO4 and directed through a relief needle fixed with the rubber stopper to the reaction mixture. The resulting solution was heated at 30 °C with stirring. After 11 h, the reaction was quenched with 10 mL of H2O, extracted with ether (15 mL × 3), washed with brine, and dried over anhydrous Na2SO4. After filtration and evaporation, chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 20/1) of the crude product afforded 2a (34.0 mg, 62%) as a solid, m.p.: 158.2–159.2 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.79–7.70 (m, 2H, Ar-H), 7.50–7.29 (m, 8H, Ar-H), 6.78 (s, 1H, CH), 4.80 (s, 2H, N-CH2); 13C NMR (75 MHz, CDCl3): δ 162.0, 152.0, 137.4, 134.3, 131.1, 130.6, 130.5, 129.0, 128.9, 128.8, 128.5, 113.8, 43.8; MS (m/z): 280 (M+ + 1, 14.65), 279 (M+, 78.90), 118 (100); IR (KBr, cm−1): 1807, 1733, 1674, 1495, 1451, 1433, 1398, 1340, 1309, 1291, 1235, 1170, 1081, 1067; Anal. Calcd. for C17H13NO3: C 73.11, H 4.69, N 5.02; Found: C 73.20, H 4.72, N 4.96%.
(Z)-5-Benzylidene-3-(n-butyl)oxazolidine-2,4-dione (2b).
Following the procedure for the preparation of 2a, the reaction of 40.1 mg (0.20 mmol) of 1b and 55.8 mg (0.40 mmol) of K2CO3 in DMSO (2 mL) afforded 39.0 mg (80%) of 2b (eluent: petroleum ether/ethyl acetate = 25/1) as a solid, m.p.: 82.7–83.4 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.80–7.71 (m, 2H, Ar-H), 7.50–7.36 (m, 3H, Ar-H), 6.76 (s, 1H, CH), 3.65 (t, J = 7.4 Hz, 2H, N-CH2), 1.78–1.62 (m, 2H, CH2), 1.46–1.30 (m, 2H, MeCH2), 0.96 (t, J = 7.4 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3): δ 162.4, 152.3, 137.5, 131.0, 130.7, 130.4, 129.0, 113.3, 40.0, 29.6, 19.8, 13.5; MS (m/z): 246 (M+ + 1, 4.12), 245 (M+, 25.65), 118 (100); IR (KBr, cm−1): 1816, 1721, 1666, 1495, 1449, 1413, 1354, 1315, 1264, 1236, 1187, 1082, 1006; Anal. Calcd. for C14H15NO3: C 68.56, H 6.16, N 5.71; Found: C 68.88, H 6.40, N 5.72%.
(Z)-5-Benzylidene-3-ethyloxazolidine-2,4-dione (2c).
Following the procedure for the preparation of 2a, the reaction of 34.2 mg (0.20 mmol) of 1c and 55.4 mg (0.40 mmol) of K2CO3 in DMSO (2 mL) afforded 28.4 mg (66%) of 2c (eluent: petroleum ether/ethyl acetate = 25/1) as a solid, m.p.: 123.6–124.3 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.85–7.68 (m, 2H, Ar-H), 7.52–7.35 (m, 3H, Ar-H), 6.75 (s, 1H, CH), 3.71 (q, J = 7.2 Hz, 2H, N-CH2), 1.32 (t, J = 7.2 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3): δ 162.1, 152.0, 137.5, 131.0, 130.7, 130.4, 129.0, 113.2, 35.3, 13.0; MS (m/z): 218 (M+ + 1, 5.78), 217 (M+, 43.70), 118 (100); IR (KBr, cm−1): 1813, 1735, 1682, 1496, 1452, 1441, 1415, 1372, 1347, 1320, 1246, 1211, 1169, 1115, 1080, 1045; Anal. Calcd. for C12H11NO3: C 66.35, H 5.10, N 6.45; Found: C 66.31, H 5.33, N 6.46%.
(Z)-3-Allyl-5-benzylideneoxazolidine-2,4-dione (2d).
Following the procedure for the preparation of 2a, the reaction of 36.7 mg (0.20 mmol) of 1d and 56.2 mg (0.41 mmol) of K2CO3 in DMSO (2 mL) afforded 32.6 mg (72%) of 2d (eluent: petroleum ether/ethyl acetate = 15/1) as a solid, m.p.: 79.7–80.5 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.84–7.68 (m, 2H, Ar-H), 7.50–7.36 (m, 3H, Ar-H), 6.78 (s, 1H, CH-Ar), 6.00–5.78 (m, 1H, CH-CH2), 5.45–5.25 (m, 2H, CH2), 4.25 (d, J = 5.7 Hz, 2H, N-CH2); 13C NMR (75 MHz, CDCl3): δ 161.8, 151.8, 137.4, 131.1, 130.6, 130.5, 129.5, 129.0, 119.6, 113.6, 42.2; MS (m/z): 230 (M+ + 1, 6.03), 229 (M+, 41.47), 118 (100); IR (KBr, cm−1): 1816, 1740, 1682, 1452, 1433, 1403, 1351, 1238, 1176, 1101; Anal. Calcd. for C13H11NO3: C 68.11, H 4.84, N 6.11; Found: C 68.47, H 5.01, N 5.99%.
(Z)-5-Benzylidene-3-isopropyloxazolidine-2,4-dione (2e).
Following the procedure for the preparation of 2a, the reaction of 37.6 mg (0.20 mmol) of 1e and 82.9 mg (0.60 mmol) of K2CO3 in DMSO (2 mL) afforded 31.5 mg (68%) of 2e (eluent: petroleum ether/ethyl acetate = 25/1) as a solid, m.p.: 123.8–125.0 °C (n-hexane/CH2Cl2). The reaction of 37.0 mg (0.20 mmol) of 1e and 132.7 mg (0.41 mmol) of Cs2CO3 in DMSO (2 mL) afforded 20.8 mg (46%) of 2e. 1H NMR (300 MHz, CDCl3) δ 7.80–7.69 (m, 2H, Ar-H), 7.50–7.36 (m, 3H, Ar-H), 6.72 (s, 1H, CH), 4.44 (heptet, J = 7.0 Hz, 1H, N-CH), 1.49 (d, J = 7.0 Hz, 6H, 2×Me); 13C NMR (75 MHz, CDCl3): δ 162.2, 151.5, 137.2, 131.0, 130.8, 130.3, 129.0, 112.9, 45.3, 19.4; MS (m/z): 232 (M+ + 1, 5.15), 231 (M+, 34.78), 118 (100); IR (KBr, cm−1): 1815, 1738, 1686, 1666, 1496, 1452, 1403, 1388, 1371, 1347, 1249, 1207, 1181, 1071, 1021, 1002; Anal. Calcd. for C13H13NO3: C 67.52, H 5.67, N 6.06; Found: C 67.48, H 5.73, N 6.11%.
(Z)-3-(n-Butyl)-5-(4-n-propylbenzylidene)oxazolidine-2,4-dione (2g).
Following the procedure for the preparation of 2a, the reaction of 48.2 mg (0.20 mmol) of 1g and 56.0 mg (0.41 mmol) of K2CO3 in DMSO (2 mL) afforded 39.3 mg (69%) of 2g (eluent: petroleum ether/ethyl acetate = 25/1) as a liquid. 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.4 Hz, 2H, Ar-H), 7.23 (d, J = 8.4 Hz, 2H, Ar-H), 6.74 (s, 1H, CH), 3.64 (t, J = 7.4 Hz, 2H, N-CH2), 2.61 (t, J = 7.7 Hz, 2H, Ar-CH2), 1.78–1.57 (m, 4H, 2×MeCH2), 1.46–1.30 (m, 2H, CH2), 1.02–0.89 (m, 6H, 2×Me); 13C NMR (75 MHz, CDCl3): δ 162.4, 152.3, 145.8, 136.9, 131.1, 129.1, 128.2, 113.5, 40.0, 37.9, 29.6, 24.2, 19.8, 13.7, 13.5; MS (m/z): 288 (M+ + 1, 6.70), 287 (M+, 34.43), 160 (100); IR (neat, cm−1): 2960, 2925, 2873, 1822, 1738, 1674, 1608, 1511, 1442, 1404, 1371, 1345, 1301, 1246, 1192, 1162, 1092, 1042; HRMS Calcd for C17H21NO3 (M+): 287.1521, Found: 287.1521.
(Z)-3-(n-Butyl)-5-(4-methylbenzylidene)oxazolidine-2,4-dione (2h).
Following the procedure for the preparation of 2a, the reaction of 43.3 mg (0.20 mmol) of 1h and 56.6 mg (0.41 mmol) of K2CO3 in DMSO (2 mL) afforded 35.8 mg (69%) of 2h (eluent: petroleum ether/ethyl acetate = 25/1) as a solid, m.p.: 87.0–88.4 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 8.1 Hz, 2H, Ar-H), 7.23 (d, J = 8.1 Hz, 2H, Ar-H), 6.73 (s, 1H, CH), 3.64 (t, J = 7.4 Hz, 2H, N-CH2), 2.38 (s, 3H, Ar-Me), 1.77–1.61 (m, 2H, CH2), 1.46–1.25 (m, 2H, MeCH2), 0.95 (t, J = 7.2 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3): δ 162.4, 152.3, 141.0, 136.9, 131.0, 129.7, 127.9, 113.4, 39.9, 29.6, 21.5, 19.8, 13.5; MS (m/z): 260 (M+ + 1, 4.24), 259 (M+, 25.13), 132 (100); IR (KBr, cm−1): 2959, 2877, 1817, 1735, 1675, 1608, 1514, 1440, 1404, 1362, 1345, 1318, 1288, 1239, 1161, 1094, 1065, 1042; Anal. Calcd. for C15H17NO3: C 69.48, H 6.61, N 5.40; Found: C 69.43, H 6.66, N 5.35%.
(Z)-3-(n-Butyl)-5-(4-methoxybenzylidene)oxazolidine-2,4-dione (2i).
Following the procedure for the preparation of 2a, the reaction of 45.7 mg (0.20 mmol) of 1i and 55.1 mg (0.40 mmol) of K2CO3 in DMSO (2 mL) afforded 36.7 mg (67%) of 2i (eluent: petroleum ether/ethyl acetate = 25/1–15/1) as a solid, m.p.: 106.0–106.6 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.73–7.65 (m, 2H, Ar-H), 6.98–6.88 (m, 2H, Ar-H), 6.70 (s, 1H, CH), 3.84 (s, 3H, OMe), 3.63 (t, J = 7.4 Hz, 2H, N-CH2), 1.75–1.61 (m, 2H, CH2), 1.45–1.27 (m, 2H, MeCH2), 0.95 (t, J = 7.5 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3): δ 162.5, 161.3, 152.4, 136.0, 132.9, 123.4, 114.4, 113.3, 55.3, 39.9, 29.6, 19.8, 13.5; MS (m/z): 276 (M+ + 1, 6.32), 275 (M+, 36.31), 148 (100); IR (KBr, cm−1): 2979, 2952, 2867, 2835, 1814, 1740, 1678, 1603, 1514, 1447, 1408, 1345, 1307, 1257, 1163, 1095, 1064, 1027; Anal. Calcd. for C15H17NO4: C 65.44, H 6.22, N 5.09; Found: C 65.38, H 6.30, N 5.03%.
(Z)-3-Benzyl-5-(4-methoxybenzylidene)oxazolidine-2,4-dione (2j).
Following the procedure for the preparation of 2a, the reaction of 53.3 mg (0.20 mmol) of 1j and 55.6 mg (0.41 mmol) of K2CO3 in DMSO (2 mL) afforded 42.2 mg (68%) of 2j (eluent: petroleum ether/ethyl acetate = 20/1–10/1) as a solid, m.p.: 117.8–118.7 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.75–7.65 (m, 2H, Ar-H), 7.50–7.40 (m, 2H, Ar-H), 7.40–7.27 (m, 3H, Ar-H), 6.98–6.88 (m, 2H, Ar-H), 6.73 (s, 1H, CH), 4.78 (s, 2H, N-CH2), 3.84 (s, 3H, OMe); 13C NMR (75 MHz, CDCl3): δ 162.1, 161.3, 152.1, 135.9, 134.5, 133.0, 128.81, 128.77, 128.4, 123.3, 114.5, 113.8, 55.3, 43.6; MS (m/z): 310 (M++1, 13.88), 309 (M+, 68.64), 148 (100); IR (KBr, cm−1): 1805, 1735, 1666, 1601, 1572, 1512, 1455, 1443, 1430, 1401, 1348, 1312, 1259, 1174, 1091, 1070, 1025; Anal. Calcd. for C18H15NO4: C 68.89, H 4.89, N 4.53; Found: C 69.75, H 5.01, N 4.64%.
(Z)-3-(n-Butyl)-5-(4-fluorobenzylidene)oxazolidine-2,4-dione (2k).
Following the procedure for the preparation of 2a, the reaction of 43.2 mg (0.20 mmol) of 1k and 56.2 mg (0.41 mmol) of K2CO3 in DMSO (2 mL) afforded 37.2 mg (72%) of 2k (eluent: petroleum ether/ethyl acetate = 25/1) as a solid, m.p.: 102.6–103.8 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.81–7.69 (m, 2H, Ar-H), 7.18–7.05 (m, 2H, Ar-H), 6.71 (s, 1H, CH), 3.65 (t, J = 7.4 Hz, 2H, N-CH2), 1.78–1.60 (m, 2H, CH2), 1.46–1.28 (m, 2H, MeCH2), 0.95 (t, J = 7.4 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3): δ 163.6 (d, J = 251.4 Hz), 162.3, 152.2, 137.2 (d, J = 2.7 Hz), 133.1 (d, J = 8.4 Hz), 127.0 (d, J = 3.2 Hz), 116.2 (d, J = 22.7 Hz), 112.0, 40.1, 29.6, 19.8, 13.5; 19F NMR (282 MHz, CDCl3): δ −108.0 (standard by frequency conversion of CDCl3); MS (m/z): 264 (M+ + 1, 3.56), 263 (M+, 21.57), 136 (100); IR (KBr, cm−1): 2959, 2873, 1819, 1728, 1674, 1602, 1511, 1447, 1416, 1373, 1356, 1310, 1292, 1239, 1186, 1163, 1085, 1058, 1010; Anal. Calcd. for C14H14FNO3: C 63.87, H 5.36, N 5.32; Found: C 63.98, H 5.41, N 5.23%.
(Z)-3-Benzyl-5-(thiophen-3-ylmethylene)oxazolidine-2,4-dione (2l).
Following the procedure for the preparation of 2a, the reaction of 47.9 mg (0.20 mmol) of 1l and 55.1 mg (0.40 mmol) of K2CO3 in DMSO (2 mL) afforded 34.8 mg (61%) of 2l (eluent: petroleum ether/ethyl acetate = 15/1) as a solid, m.p.: 155.9–156.4 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.83–7.75 (m, 1H, Ar-H), 7.52–7.41 (m, 3H, Ar-H), 7.40–7.28 (m, 4H, Ar-H), 6.84 (s, 1H, CH), 4.78 (s, 2H, N-CH2); 13C NMR (75 MHz, CDCl3): δ 161.9, 151.9, 136.6, 134.4, 132.0, 130.7, 128.9, 128.8, 128.6, 128.5, 126.7, 107.8, 43.8; MS (m/z): 286 (M+ + 1, 16.21), 285 (M+, 100); IR (KBr, cm−1): 3102, 3031, 2947, 1808, 1735, 1674, 1518, 1495, 1456, 1438, 1406, 1354, 1343, 1322, 1249, 1212, 1156, 1090, 1069; Anal. Calcd. for C15H11NO3S: C 63.14, H 3.89, N 4.91; Found: C 63.18, H 4.09, N 4.99%.
(Z)-3-Benzyl-5-(thiophen-2-ylmethylene)oxazolidine-2,4-dione (2m).
Following the procedure for the preparation of 2a, the reaction of 47.1 mg (0.20 mmol) of 1m and 81.6 mg (0.59 mmol) of K2CO3 in DMSO (2 mL) afforded 30.7 mg (55%) of 2m (eluent: petroleum ether/ethyl acetate = 15/1) as a solid, m.p.: 177.2–178.0 °C (n-hexane/CH2Cl2). 1H NMR (300 MHz, CDCl3) δ 7.59 (d, J = 4.8 Hz, 1H, Ar-H), 7.53–7.40 (m, 3H, Ar-H), 7.40–7.28 (m, 3H, Ar-H), 7.12 (t, J = 4.4 Hz, 1H, Ar-H), 7.01 (s, 1H, CH), 4.78 (s, 2H, N-CH2); 13C NMR (75 MHz, CDCl3): δ 161.5, 151.6, 135.6, 134.4, 133.5, 133.1, 132.0, 128.9, 128.8, 128.5, 128.1, 107.4, 43.8; MS (m/z): 286 (M+ + 1, 13.07), 285 (M+, 74.61), 124 (100); IR (KBr, cm−1): 3104, 1810, 1728, 1668, 1495, 1457, 1435, 1398, 1344, 1318, 1247, 1231, 1167, 1071, 1051; Anal. Calcd. for C15H11NO3S: C 63.14, H 3.89, N 4.91; Found: C 63.11, H 3.97, N 5.01%.
Acknowledgements
Financial support from the Major State Basic Research and Development Program (2009CB825300) and Project for Basic Research in Natural Science Issued by Shanghai Municipal Committee of Science (No. 08dj1400100). Shengming Ma is a Qiu Shi Adjunct Professor at Zhejiang University. We thank Mr. Suhua Li in this group for reproducing the results presented in entries 3, 9, and 13 in Table 2.
Notes and references
- For a most recent review, see: T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 Search PubMed.
- For recent reports, see:
(a) M. Yoshida and M. Ihara, Angew. Chem., Int. Ed., 2001, 40, 616 CrossRef CAS;
(b) M. Shi and Y.-M. Shen, J. Org. Chem., 2002, 67, 16 CrossRef CAS;
(c) M. Yoshida, M. Fujita, T. Ishii and M. Ihara, J. Am. Chem. Soc., 2003, 125, 4874 CrossRef CAS;
(d) M. Yoshida, M. Fujita and M. Ihara, Org. Lett., 2003, 5, 3325 CrossRef CAS;
(e) Y. Gu, F. Shi and Y. Deng, J. Org. Chem., 2004, 69, 391 CrossRef CAS;
(f) Y. Kayaki, M. Yamamoto, T. Suzuki and T. Ikariya, Green Chem., 2006, 8, 1019 RSC;
(g) W. Yamada, Y. Sugawara, H. M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604 CrossRef CAS;
(h) M. Yoshida, T. Murao, K. Sugimoto and M. Ihara, Synlett, 2007, 575 CrossRef CAS;
(i) Y. Kayaki, M. Yamamoto and T. Ikariya, J. Org. Chem., 2007, 72, 647 CrossRef CAS;
(j) M. Yoshida, Y. Komatsuzaki and M. Ihara, Org. Lett., 2008, 10, 2083 CrossRef CAS;
(k) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS;
(l) S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072 CrossRef CAS.
- Using electrochemical method, see:
(a) L. Rossi, M. Feroci, M. Verdecchia and A. Inesi, Lett. Org. Chem., 2005, 2, 731 CrossRef CAS;
(b) M. A. Casadei, F. M. Moracci, G. Zappia, A. Inesi and L. Rossi, J. Org. Chem., 1997, 62, 6754 CrossRef CAS;
(c) M. A. Casadei, S. Cesa, F. M. Moracci, A. Inesi and M. Feroci, J. Org. Chem., 1996, 61, 380 CrossRef CAS;
(d) M. A. Casadei, S. Cesa and A. Inesi, Tetrahedron, 1995, 51, 5891 CrossRef CAS;
(e) Using strong bases LDA or n-BuLi, see: G. M. Coppola and R. E. Damon, J. Heterocycl. Chem., 1995, 32, 1133 Search PubMed;
(f) A. R. Katritzky, W.-Q. Fan, A. E. Koziol and G. J. Palenik, Tetrahedron, 1987, 43, 2343 CrossRef CAS;
(g) A. R. Katritzky and W.-Q. Fan, Org. Prep. Proced. Int., 1987, 19, 263 CrossRef CAS;
(h) G. M. Coppola and R. E. Damon, J. Heterocycl. Chem., 1995, 32, 1133 CrossRef CAS.
-
(a) J. W. Clark-Lewis, Chem. Rev., 1958, 58, 63 CrossRef CAS;
(b) P. C. Unangst, D. T. Connor, W. A. Cetenko, R. J. Sorenson, C. R. Kostlan, J. C. Sircar, C. D. Wright, D. J. Schrier and R. D. Dyer, J. Med. Chem., 1994, 37, 322 CrossRef CAS;
(c) D. A Heerding, L. T. Christmann, T. J. Clark, D. J. Holmes, S. F. Rittenhouse, D. T. Takata and J. W. Venslavsky, Bioorg. Med. Chem. Lett., 2003, 13, 3771 CrossRef CAS;
(d) R. L. Dow, B. M. Bechle, T. T. Chou, D. A. Clark, B. Hulin and R. W. Stevenson, J. Med. Chem., 1991, 34, 1538 CrossRef CAS;
(e) Y. Momose, T. Maekawa, T. Yamano, M. Kawada, H. Odaka, H. Ikeda and T. Sohda, J. Med. Chem., 2002, 45, 1518 CrossRef CAS;
(f) Y.-L. Li, Y.-X. Song and T.-J. Guo, Cont. Chem. Indu., 2003, 32, 18 Search PubMed;
(g) Y.-L. Li, W.-R. Miao and Y.-H. Zhou, Chin. J. Pest., 2005, 44, 503 Search PubMed.
-
(a) T. L. Patton, J. Org. Chem., 1967, 32, 383 CrossRef CAS;
(b) A. Oku, S. Nakaoji, T. Kadono and H. Imai, Bull. Chem. Soc. Jpn., 1979, 52, 2966 CrossRef CAS;
(c) A. Zask, J. Org. Chem., 1992, 57, 4558 CrossRef CAS;
(d) A. Benavides, R. Martínez, H. A. Jiménez-Vázquez, F. Delgado and J. Tamariz, Heterocycles, 2001, 55, 469 CrossRef CAS.
- G. Chen, C. Fu and S. Ma, Org. Lett., 2009, 11, 2900 CrossRef CAS.
- Crystal data for compound 2a: C17H13NO3, MW = 279.28, monoclinic, space group P2(1)/c, final R indices [I > 2σ(I)], R1 = 0.0411, wR2 = 0.0979, R indices (all data) R1 = 0.0487, wR2 = 0.1027, a = 6.4285(4) Å, b = 31.4664(18) Å, c = 7.0702(4) Å, α = 90°, β = 106.517(2)°, γ = 90°, V = 1371.16(14) Å3, T = 173(2) K, Z = 4, reflections collected/unique 15862/2426 (Rint = 0.0299), number of observations [I > 2σ(I)] 2086, parameters: 190. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Center. CCDC 779363.
-
(a) S. A. Jr. Lang and E. Cohen, J. Med. Chem., 1975, 18, 441 CrossRef;
(b) Y. Iwanami, Nippon Kagaku Zasshi, 1962, 83, 600 CAS;
(c) A. Yokoyama, K. Ashida and H. Tanaka, Chem. Pharm. Bull., 1964, 12, 690 CAS;
(d) H. Jiang, S. Ma, G. Zhu and X. Lu, Tetrahedron, 1996, 52, 10945 CrossRef CAS;
(e) H. Imase, K. Noguchi, M. Hirano and K. Tanaka, Org. Lett., 2008, 10, 3563 CrossRef CAS;
(f) G. T. Crisp and M. J. Millan, Tetrahedron, 1998, 54, 637 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.