Facile synthesis of zirconium phosphonate-functionalized magnetic mesoporous silica microspheres designed for highly selective enrichment of phosphopeptides†
Jin
Lu
a,
Yan
Li
*b and
Chunhui
Deng
*a
aDepartment of Chemistry & Institutes of Biomedical Sciences, Fudan University, Shanghai, 200433, China. E-mail: chdeng@fudan.edu.cn; Fax: +86-21-65641740
bDepartment of Pharmaceutical Analysis, School of Pharmacy, Fudan University, Shanghai, 201203, China. E-mail: yanli@fudan.edu.cn; Tel: +86-21-51980057
First published on 24th January 2011
Abstract
In this work, we present a facile approach for the synthesis of zirconium phosphate (ZrP)-functionalized magnetic silica mesoporous microspheres for the selective enrichment of phosphopeptides. At first, magnetic mesoporous silica microspheres were prepared by directly coating mesoporous silica onto Fe3O4 magnetic microspheres, and then addition of phosphate onto the magnetic mesoporous silica microspheres was performed using 3-(trihydroxysilyl)propyl methylphosphate. The obtained phosphate-modified magnetic mesoporous microspheres were monodispersed with a mean diameter of 350 nm, and had an obvious mesoporous silica shell (∼65 nm). Finally, the resultant phosphate-functionalized magnetic mesoporous microspheres were incubated in ZrOCl2 solution with gentle stirring overnight for the loading of Zr4+ cations. The obtained Zr4+-functionalized materials were applied to the selective enrichment of phosphopeptides from both standard protein digestion and real samples. The enriched peptides were analyzed by MALDI–TOF MS and LC–ESI MS. Experimental results demonstrated that zirconium phosphonate-modified magnetic mesoporous silica microspheres show excellent potential for the selective enrichment of phosphopeptides.
1. Introduction
Protein phosphorylation is known to be a key event in intracellular signalling and other important cellular processes such as metabolism, transcription and apoptosis. Thus, the identification and localization of phosphorylation sites in proteins is very important to gain insight into the regulation of cellular signalling cascades and other biological networks.1Mass spectrometry (MS) is the technique of choice for proteome analysis due to its ability to not only identify a large number of proteins, mostly from their corresponding peptides, but also to identify, characterize and locate post-translational modifications.2 However, low abundances of phosphopeptides and the low degree of phosphopeptides typically necessitate isolation and concentration of phosphopeptides prior to MS analysis. The highly selective capture of phosphopeptides from proteolytic digests is a great challenge for the identification of phosphoproteins by mass spectrometry. A variety of approaches, including immunoprecipitation, chemical-modification strategies, strong cation exchange chromatography, immobilized metal affinity chromatography (IMAC) and metal oxide affinity chromatography (MOAC) have been used for this purpose.3 A novel zirconium phosphate (ZrP)-based approach used for trapping phosphopeptides was reported recently, and has been demonstrated to have higher selectivity and sensitivity for enriching phosphopeptides than the conventional Fe3+-IMAC beads do.4 The improved enrichment performance of the method is probably due to the coordination properties of the phosphate group, which lead to strong affinity interactions between the phosphate group and the ZrP. Due to the fact that nanomaterials have high surface areas, the designed synthesis of ZrP-modified nanomaterials, such as mesoporous silica, is very interesting and important for phosphoproteome research.5Magnetic materials have received a lot of attention due to their magnetism properties, and have been applied extensively in proteomics. Functionalized magnetic Fe3O4 materials have been widely applied to proteomics research, including protein fast digestion, phosphopeptide enrichment, and glycopeptide enrichment.6 Recently, mesoporous silica with a large surface area was widely studied for application in proteomics, including low-concentration peptide enrichment and selective capture of phosphopeptides.7 More recently, we synthesized magnetic mesoporous silica and applied them to the selective enrichment of endogenous peptides.8 The high surface area of the mesoporous silica shell benefits from further functionalization, and the magnetism properties allow convenient separation. Therefore, facile synthesis of ZrP functionalized magnetic mesoporous silica designed for selective enrichment of phosphopeptides is of great importance.
In this work, we have developed a facile approach for the synthesis of ZrP-functionalized magnetic mesoporous silica microspheres, and demonstrated their ability for highly selective enrichment of phosphopeptides. The designed synthesis approach of ZrP-functionalized magnetic mesoporous silica microspheres is shown in Scheme 1. At first, mesoporous silica was directly coated on Fe3O4 microspheres, and then modification of phosphate onto the magnetic mesoporous silica microspheres was performed using 3-(trihydroxysilyl) propyl methylphosphate and Zr4+ ions which were located by chelating with phosphate groups. For the first time, we applied the ZrP-functionalized magnetic mesoporous silica microspheres for the highly selective enrichment of phosphopeptides. Because of the higher surface-to-volume ratio of the mesoporous silica microspheres, modification of the phosphate group is more efficient and beneficial for specific phosphorylated binding. Thus, a higher efficiency and an improved detection limit can be obtained. Finally, the synthesized ZrP-functionalized magnetic mesoporous silica microspheres were evaluated with tryptic digests of both standard protein and a rat brain lysate for the phosphoproteome analysis. All the experimental results demonstrated a good enrichment efficiency and selectivity of the method we report here.
 | ||
| Scheme 1 The synthesis strategy of zirconium phosphate-modified magnetic mesoporous microspheres. | ||
2. Experimental
2.1. Materials and chemicals
Bovine β-casein, bovine serum albumin, L-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK) treated trypsin (from bovine pancreas), ammonium bicarbonate (NH4HCO3), trifluoroacetic acid (TFA), 3-(trihydroxysilyl)propyl methylphosphate and 2,5-dihydroxybenzoic acid (DHB) were purchased from Sigma Chemical (St. Louis, MO). Acetonitrile was purchased from Merck (Darmstadt, Germany). All aqueous solutions were prepared using Milli-Q water by Milli-Q system (Millipore, Bedford, MA). All other chemicals and reagents were of the highest grade commercially available.2.2 Synthesis of ZrP-functionalized magnetic mesoporous silica microspheres
The Fe3O4 magnetic microspheres were synthesized through a hydrothermal reaction as described in our previous work. For mesoporous silica layer deposition, we introduce a one step hydrothermal reaction for direct coating of mesoporous silica instead of previous reported two step reactions.9 Compared to the previous work, the direct coating is very simple, fast and low-cost. 50 mg Fe3O4 microspheres and 500 mg CTAB were dispersed into 50 ml deionized water and dispersed by ultrasonic irradiation for half an hour. Then 50 ml 0.01 M NaOH solution and 400 ml deionized water was added into the mixture. The solution was ultrasonically dispersed for five minutes and incubated at 60 °C for 0.5 h. Finally, 2.5 ml tetraethylorthosilicate (TEOS)/ethanol (v/v: 1/4) solution was injected into the solution, and the obtained mixture was heated at 60 °C for 12 h. After the coating process, the product was washed at least 5 times with the help of a magnet and then dried in a vacuum at 50 °C for 12 h.For the modification of phosphate and Zr4+, 80 mg of the obtained Fe3O4@SiO2 magnetic mesoporous microspheres and 0.5 mL 3-(trihydroxysilyl)propyl methylphosphate were dispersed into 20 mL toluene and refluxed at 80 °C for 12 h. Then the microspheres were washed with anhydrous ethanol and deionized water five times. The resultant phosphate-functionalized magnetic mesoporous silica microspheres were incubated in a 0.2 M ZrOCl2 solution with gentle stirring overnight for the loading of Zr4+ cations. Finally, the prepared ZrP-functionalized magnetic mesoporous silica microspheres were rinsed with deionized water to remove the nonspecifically adsorbed Zr4+ cations, and dried in vacuum at 50 °C for 12 h.
2.3 Characterization of ZrP-functionalized magnetic mesoporous silica microspheres
The morphologies of the as-synthesized ZrP-functionalized magnetic mesoporous silica microspheres were studied under scanning electron microscopy (Philips XL30) and transmission electron microscopy (JEM-2100F). The energy-dispersive X-ray analysis was performed on the JEM-2100F. Fourier-transform infrared (FT-IR) spectra were collected on a Nicolet Fourier spectrophotometer, using KBr pellets (USA). Nitrogen adsorption and desorption isotherms were measured using a Micromeritics ASAP 2020. The samples were degassed in a vacuum at 200 °C for 8 h prior to measurement. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas (SBET) using adsorption data in a relative pressure range from 0.18 to 0.35. By using the Barrett–Joyner–Halenda (BJH) model, the pore volumes and pore size distributions were derived from the adsorption branches of the isotherms, and the total pore volumes (Vt) were estimated from the adsorbed amount at a relative pressure P/P0 of 0.992.2.4 Sample preparation
Bovine β-casein and bovine serum albumin were dissolved in 25 mM NH4HCO3 buffer at pH 8.3 and treated with trypsin (2%, w/w) for 16 h at 37 °C. The peptide mixture contained peptides that originated from a tryptic digestion of 0.02 pmol of β-casein and 2 pmol of bovine serum albumin.2.5 Preparation of the lysate of rat brain
Rats were sacrificed and the brains were promptly removed and placed in ice-cold homogenization buffer consisting of 7 M urea, 2 M thiourea and a mixture of protease inhibitor (1 mM phenylmethanesulfonylfluoride) and phosphatase inhibitors (0.2 mM Na3VO4, 1 mM NaF). After mincing with scissors and washing to remove blood, the brains were homogenized in a Pottter–Elvejhem homogenizer with a Teflon piston, using 5 mL of the homogenization buffer per 1 g of tissue. The suspension was homogenized for approximately 2 min, vortexed at 0 °C for 30 min, and then centrifuged at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 1.5 h. The supernatant contained the total brain proteins.
000 g for 1.5 h. The supernatant contained the total brain proteins.
Appropriate volumes of protein sample were precipitated as above, lyophilized to dryness, and redissolved in a reducing solution (6 M guanidine hydrochloride, 100 mM NH4HCO3, pH 8.3) with the protein concentration adjusted to 2 μg μL−1. Then, 200 μg of this protein sample (100 μL volume) were mixed with 10 μL of 0.5 M DTT. The mixture was incubated at 37 °C for 1 h, and then 20 μL of 0.5 M 2-iodoacetamide were added and incubated for an additional 30 min at 37 °C in the dark. The protein mixtures were exchanged into 50 mM NH4HCO3 buffer, pH 8.5, and incubated with trypsin (40:1) at 37 °C overnight. All animal administration procedures were performed in strict accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals (China).
2.6 Selective enrichment of phosphopeptides from tryptic digestion of standard proteins and rat brain
First, the peptide mixtures originating from tryptic digestions were diluted by 50% acetonitrile and 0.1% TFA aqueous solution (v/v), and a suspension of ZrP-functionalized magnetic mesoporous silica microspheres (400 μg) was added into 200 μL of diluted peptide mixture. Then the mixed solutions were vibrated at 25 °C for 30 min. After that, with the help of magnet, the pellucid solutions were taken out. After that, the ZrP-functionalized magnetic mesoporous silica microspheres were washed with 50% acetonitrile and 0.1% TFA aqueous solution (v/v) three times. Finally, the peptides captured by ZrP-functionalized magnetic mesoporous silica microspheres were eluted with 5 μL of 0.4 M ammonia aqueous solution for 10 min, and the eluate was analyzed by MALDI–TOF MS.The tryptic digests of rat brain were lyophilized and then dissolved in loading buffer. 2 μg of ZrP-functionalized magnetic mesoporous silica microspheres was added into 400 μL of diluted rat brain digests. Then the mixed solutions were vibrated at 25 °C for 30 min. After that, with the help of a magnet, the phosphopeptides adsorbed to the magnetic mesoporous silica microspheres were collected and washed with 50% (v/v) acetonitrile and 0.1% (v/v) TFA aqueous solution three times. Then the ZrP-functionalized magnetic mesoporous silica microspheres were eluted with 50 μL of 0.4 M aqueous ammonia solution for 30 min. The eluate was lyophilized and then dissolved in 35 μL loading phase. Finally, the solution was submitted for LC–ESI–MS analysis.
2.7 MALDI-TOF MS Analysis
The above phosphopeptides eluted from ZrP-functionalized magnetic mesoporous silica microspheres were deposited on the MALDI target using the dried droplet method. 0.5 μL of the washing buffer was deposited on the plate and then another 0.5 μL of a mixture of 20 mg mL−12,5-dihydroxybenzoic acid in 50% acetonitrile and 1% H3PO4 aqueous solution (v/v) was introduced as a matrix. MALDI–TOF MS experiments were performed in positive ion mode on a 4700 Proteomics Analyzer (Applied Biosystems, USA) with the Nd-YAG laser at 355 nm, a repetition rate of 200 Hz and an acceleration voltage of 20 kV.2.8 Nano-flow LC–ESI–MS
A Nano-LC MS/MS experiment was performed on an HPLC system composed by 2 LC-20AD nano-flow LC pumps and 1 LC-20AB micro-flow LC pump (all from Shimadzu Corporation, Tokyo, Japan) connected to an LTQ mass spectrometer (Thermo Electron Corporation, San Jose, CA). Sample injection was done via an SIL-20 AC auto-sampler (Shimadzu Corporation, Tokyo, Japan) and loaded onto a CAPTRAP column (0.5 × 2 mm, MICHROM Bioresources Inc., Auburn, CA) for 5 min at a flow rate of 60 μL min−1. The sample was subsequently separated by a PICOFRIT C18 reverse-phase column (0.075 × 100 mm, New Objective Inc., Woburn, MA) at a flow rate of 300 nL min−1. The mobile phases consisted of 5% acetonitrile with 0.1% formic acid (phase A and the loading phase) and 95% acetonitrile with 0.1% formic acid (phase B). To achieve proper separation, a 120-min linear gradient from 5 to 45% phase B was employed. The separated sample was introduced into the mass spectrometervia a 15 μm silica tip (New Objective Inc., Woburn, MA) adapted to a DYNAMIC nano-electrospray source (Thermo Electron Corporation, San Jose, CA). The spray voltage was set at 2.0 kV and the heated capillary at 210 °C. The mass spectrometer was operated in data-dependent mode and each cycle of duty consisted one MS survey scan at the mass range 400–2000 Da, followed by MS2 experiments for the 8 strongest peaks and MS3 experiments for the peak corresponding to the neutral loss of 97.97, 48.99, 32.67 Da among the three most intense fragment ions using the LTQ section. Peptides were fragmented in the LTQ section using collision-induced dissociation with helium with the normalized collision energy value set at 35%. Previously fragmented peptides were excluded for 30 s.2.9 Peptide sequencing and data interpretation
The peak lists for the MS2 and MS3 spectra were generated from the raw data by Bioworks version 3.3 (Thermo Electron) with the following parameters: mass range 600–3500, intensity threshold 1400, and minimum ion count 10. The generated peak lists were searched by the Sequest program included in Bioworks against the International Protein Index (IPI) database (IPI rat v3.45 fasta with 40189 entries). The MS/MS spectra were searched with a precursor ion mass tolerance of 2 Da, a fragment ion mass tolerance of 1 Da, full tryptic specificity was applied, two missed cleavages were allowed, and with fixed modification, carboxyamidomethylation (C); variable modifications, phosphorylation (S, T, Y) and oxidation (M). For the searching with MS/MS/MS data, besides the above settings, dynamic modifications were also set for water loss on Ser and Thr (−18 Da). For the identification of phosphopeptides based only on MS/MS or MS/MS/MS spectra, the following criteria were used for filtering the database searching results: cross-correlation value (Xcorr) > 2.0, 2.9, and 3.73 for singly, doubly, and triply charged ions, respectively; delta Cn value (ΔCn) > 0.1. For the phosphopeptide identifications derived from MS/MS/MS spectra used to validate the MS/MS identifications, the following criteria were used: Xcorr > 1.5, 2.4, and 2.96; ΔCn > 0.1. Manual validation was further carried out for peptides passing the above criteria. Criteria used for manual validation included the following: (a) The phosphoric acid neutral loss peak to phosphoserine and phosphothreonine must be the dominant peak. (b) The spectrum must be of good quality with fragment ion clearly above the base-line noise. (c) Sequential members of the b- or y-ion series were observable in the mass spectra. (d) For multiply phosphorylated peptides, the peptides derived from MS2 must be confirmed by MS3 spectra in the same MS cycle.
3. Results and discussion
3.1 Synthesis and characterization of ZrP-functionalized magnetic mesoporous silica microspheres
The designed synthesis strategy of ZrP-functionalized magnetic mesoporous silica microspheres is shown in Scheme 1. Firstly, mesoporous silica was directly coated on Fe3O4 microspheres, and then, modification of phosphate onto the magnetic mesoporous silica microspheres was performed using 3-(trihydroxysilyl)propyl methylphosphate, and Zr4+ ions were located by chelating with phosphate groups. The morphology of prepared Fe3O4@SiO2 magnetic mesoporous microspheres was obtained by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1a, the obtained magnetic mesoporous silica microspheres are monodispersed and have nearly uniform size. From Fig. 1b we can see that the mean diameter of the obtained magnetic mesoporous silica microspheres is about 350 nm, and an obvious mesoporous silica shell (∼65 nm) was observed. Therefore, we can conclude that the direct coating of mesoporous silica on Fe3O4 microspheres has been successfully performed using the proposed approach. Fourier transform infrared spectroscopy (FT-IR) was employed to investigate the functional groups modified on the Fe3O4@SiO2 magnetic mesoporous microspheres. The magnetic mesoporous microspheres show absorption bands ∼800 and ∼1100 cm−1 for Si–O–Si vibrations (Fig. 2a). After phosphate modification, the FT-IR spectrum of phosphate-functionalized magnetic mesoporous microspheres (Fig. 2b) shows obvious absorption peaks at 1066 and 1160 cm−1, which can be attributed to the P–O–Si and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations respectively. It indicates that the phosphate groups have been successfully modified on the surface of the magnetic mesoporous silica microspheres. Then the obtained phosphate-functionalized magnetic mesoporous silica microspheres were dispersed in ZrOCl2 solution and Zr4+ cations were immobilized on the surface. The energy-dispersive X-ray analysis (EDXA) (Fig. 1c) of the illuminating electron beams on the obtained ZrP-functionalized magnetic mesoporous silica microspheres reveals the existence of Fe, Zr, Si, P and O elements (the Cu peaks in the spectrum originate from the Cu TEM grid), further confirming the successful modification of phosphate and immobilization of Zr4+. Furthermore, compared to the previous work,9 the amount of Zr is significantly improved, which demonstrated that the mesoporous silica microspheres with larger surface area can facilitate better modification of ZrP. These results show that ZrP-modified magnetic mesoporous silica microspheres were successfully synthesized using the proposed facile approach. Additionally, the porosity of the magnetic mesoporous silica microspheres was characterized by nitrogen sorption at 77 K. Fig. 3 displays the N2 adsorption–desorption isotherm and the corresponding pore size distribution curve for the magnetic mesoporous silica microspheres. The BET surface area and total pore volume were calculated to be 223.15 m2 g−1 and 0.25 cm3 g−1 respectively, indicating that the sample has a relatively high surface-to-volume ratio.
O vibrations respectively. It indicates that the phosphate groups have been successfully modified on the surface of the magnetic mesoporous silica microspheres. Then the obtained phosphate-functionalized magnetic mesoporous silica microspheres were dispersed in ZrOCl2 solution and Zr4+ cations were immobilized on the surface. The energy-dispersive X-ray analysis (EDXA) (Fig. 1c) of the illuminating electron beams on the obtained ZrP-functionalized magnetic mesoporous silica microspheres reveals the existence of Fe, Zr, Si, P and O elements (the Cu peaks in the spectrum originate from the Cu TEM grid), further confirming the successful modification of phosphate and immobilization of Zr4+. Furthermore, compared to the previous work,9 the amount of Zr is significantly improved, which demonstrated that the mesoporous silica microspheres with larger surface area can facilitate better modification of ZrP. These results show that ZrP-modified magnetic mesoporous silica microspheres were successfully synthesized using the proposed facile approach. Additionally, the porosity of the magnetic mesoporous silica microspheres was characterized by nitrogen sorption at 77 K. Fig. 3 displays the N2 adsorption–desorption isotherm and the corresponding pore size distribution curve for the magnetic mesoporous silica microspheres. The BET surface area and total pore volume were calculated to be 223.15 m2 g−1 and 0.25 cm3 g−1 respectively, indicating that the sample has a relatively high surface-to-volume ratio.
 | ||
| Fig. 1 (a) SEM images of the magnetic mesoporous silica microspheres; (b) TEM images of the magnetic mesoporous silica microspheres; (c) The energy dispersive X-ray (EDX) spectrum data of the ZrP-functionalized magnetic mesoporous silica microspheres. | ||
 | ||
| Fig. 2 The FTIR spectra of (a) magnetic mesoporous silica microspheres and (b) phosphate-modified magnetic mesoporous silica microspheres. | ||
 | ||
| Fig. 3 Nitrogen adsorption and desorption isotherms of magnetic mesoporous silica microspheres at 77 K, and the corresponding pore-size distribution (inset) calculated by the BJH method from the adsorption branch. | ||
3.2 Selective enrichment of phosphopeptides using ZrP-functionalized magnetic mesoporous silica microspheres
The ZrP-functionalized magnetic mesoporous silica microspheres were first used to isolate phosphopeptides from tryptic digests of β-casein, which is commonly contaminated with a trace of α-casein. A direct analysis of 4 × 10−8 M β-casein by MALDI–TOF MS indicates that almost no phosphopeptides were detected and the non-phosphorylated peptides dominate the spectrum (Fig. 4a). However, after enrichment by ZrP-functionalized magnetic mesoporous silica microspheres, the signals for the β-casein phosphopeptides (marked with a black asterisk) significantly increase and dominate the spectrum (Fig. 4b). Furthermore, two phosphopeptides derived from α-casein (marked with a red asterisk) were also observed and no non-phosphorylated peptides were observed, indicating a high enrichment efficiency of the magnetic mesoporous silica microspheres. The signals marked with a circumflex could be assigned to a dephosphorylated fragment of phosphopeptide through loss of H3PO4. | ||
| Fig. 4 MALDI mass spectrum of peptides derived from β-casein, (a) without enrichment and (b) enriched by ZrP-functionalized magnetic mesoporous silica microspheres, where the black * indicates the phosphopeptides of β-casein, the red * indicates phosphopeptides of α-casein, and # indicates the metastable losses of phosphoric acid. | ||
To evaluate the specificity of enrichment of phosphopeptides with ZrP-functionalized magnetic mesoporous silica microspheres, a relatively complex sample consisting of a peptide mixture originating from a tryptic digest of β-casein (4 × 10−8 M) and bovine serum albumin (2 × 10−6 M) with a ratio of 1:50, was used as the test sample. The spectrum obtained for MALDI–TOF MS of the peptide mixture is shown in Fig. 5a. The MS spectrum for the analysis of phosphopeptides enriched with ZrP-functionalized magnetic mesoporous silica microspheres is shown in Fig. 5b. It is obvious that no non-phosphopetides were described in Fig. 5b. The differences suggest that ZrP-functionalized magnetic mesoporous silica microspheres could specifically isolate and enrich the phosphopeptides.
 | ||
| Fig. 5
MALDI mass spectrum of (a) the peptide mixture originated from β-casein and BSA with a ratio of 1:50 and (b) phosphopeptiedes enriched form peptide mixture using ZrP-functionalized magnetic mesoporous silica microspheres, where * indicates the phosphopeptides and # indicates the metastable losses of phosphoric acid. | ||
To study the detection limit of the ZrP-functionalized magnetic mesoporous silica microspheres used for enrichment of phosphopeptides, β-casein digest solutions with different concentrations were applied. The mass spectra are shown in Fig. 6. It can be seen that when the concentration of β-casein digest is 2 × 10−9 M, after enrichment by the ZrP-functionalized magnetic mesoporous silica microspheres, the ion signals from the phosphopeptides can still be detected, which indicates the high detection sensitivity of the microspheres. The results suggest that the ZrP-functionalized magnetic mesoporous silica microspheres we report here are promising materials for the enrichment of phosphopeptides.
 | ||
| Fig. 6 MALDI mass spectra of phosphopeptides enriched from β-casein with different concentrations using ZrP-functionalized magnetic mesoporous silica microspheres, where * indicates phosphopeptides and # indicates the metastable losses of phosphoric acid. | ||
To investigate whether the mesoporous microspheres can be recycled and reused for phosphopeptides enrichment, the materials were regenerated by washing with the buffer solution three times. The regenerated ZrP-functionalized magnetic mesoporous silica microspheres were reused to enrich phosphopeptides from 2 × 10−7 M β-casein digest five times. As shown in Fig. 7, the MS spectrum of phosphopeptides after enrichment five times is similar to that for the first time, confirming that our new synthesized materials can be recycled and reused for phosphopeptides enrichment.
 | ||
| Fig. 7 MALDI mass spectrum of phosphopeptides enriched from β-casein using ZrP-functionalized magnetic mesoporous silica microspheres, (a) for the first time and (b) for the fifth time. | ||
To further evaluate the performance of ZrP-functionalized magnetic mesoporous silica microspheres for phosphopeptide enrichment, the microspheres were employed to enrich phosphopeptides from tryptic digest of rat brain. 2 mg ZrP-functionalized magnetic mesoporous silica microspheres were used to enrich the phosphopeptides derived from 100 μg of rat brain protein digest. The peptides eluted from ZrP-functionalized magnetic mesoporous silica microspheres were analyzed by LC–ESI–MS. Four technical replicate runs were conducted. The acquired MS/MS and MS/MS/MS spectra were searched separately by the Sequest program as the Experimental section described. The searching results were filtered with Xcorr values obtained by statistical calculation of reverse database searching results. For p < 0.05, the Xcorr > 2.0, 2.9, and 3.73 for singly, doubly, and triply charged ions, respectively and ΔCn > 0.1 were used to filter the MS/MS database searching results. Due to the poor quality of the spectra for the phosphopeptides, the MS/MS/MS spectra were used to validate the phosphopeptides identified from the MS/MS. For the MS/MS/MS spectra, Xcorr > 1.5, 2.4, and 2.96 and ΔCn > 0.1 were used as criteria. Comparing the phosphopeptides identified from MS/MS and MS/MS/MS spectra, it was found that the sequences of 54 phosphopeptides were the same in both cases. Fig. 8 is an example of identification of doubly charged phosphopeptide TAKDSpDDDDDVTVTVDR. From the spectra it can be seen that b- and y-ion series are consistent with the theoretically predicted peaks in both MS/MS and MS/MS/MS spectra, and in MS/MS spectrum the peak at m/z 924.3 represents the doubly charged form of the selected precursor ion at m/z 1946.7 by losing an H3PO4 group. Besides the above 54 phosphopeptides, more phosphopeptides, including multiply phosphorylated peptides, have been identified by the MS/MS spectra, which could not be validated by MS/MS/MS spectrum. However, until now, strictly universal validation criteria have not been established and defined; it has too many subjective factors for interpretation of the spectra of multiply phosphorylated peptides. Therefore, only singly phosphorylated peptide identifications were validated manually in this work. After manual validation, an additional 99 singly phosphorylated peptides were finally identified from MS/MS spectra. In total, 218 phosphorylation sites—175 on serine (80.27%), 38 on threonine (17.43%) and 5 on tyrosine (2.29%)—were identified. The detailed information of the phosphopeptides enriched from tryptic digests of rat brain by using ZrP-functionalized magnetic mesoporous silica microspheres are listed in the ESI (see Table S1).†
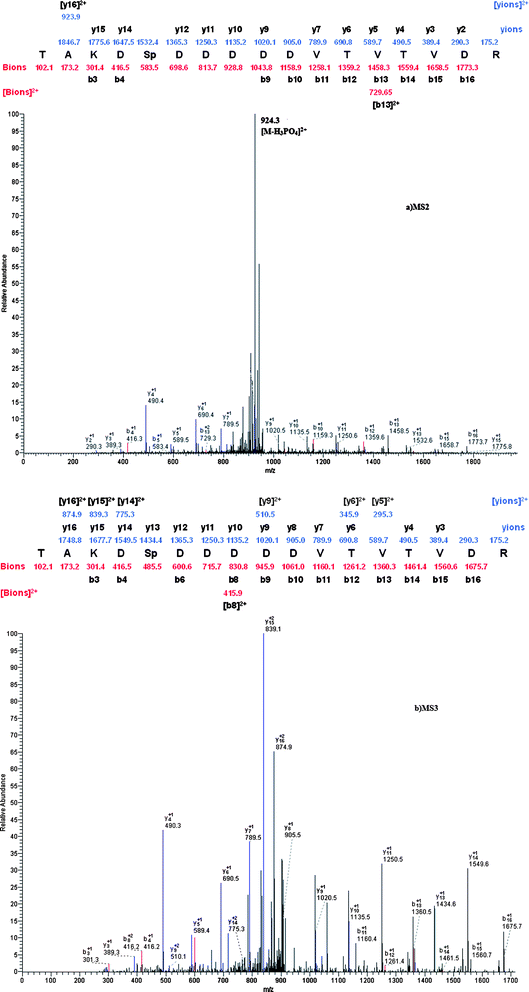 | ||
| Fig. 8 MS2 spectrum of the doubly charged form of a singly phosphorylated peptide identified from the tryptic digest of rat brain. The identified phosphopeptide was TAKDSpDDDDDVTVTVDR. b) MS3 spectrum of the doubly charged neutral loss peak at m/z 924.3. | ||
4. Conclusions
In this work, we successfully synthesized a novel functionalized magnetic material, ZrP-functionalized magnetic mesoporous silica microspheres, and demonstrated their potential for efficient and selective enrichment of phosphopeptides. In the study, ZrP-functionalized magnetic mesoporous silica microspheres were demonstrated to have the ability for selective enrichment of phosphopeptides by using standard protein digestion. Furthermore, we applied ZrP-functionalized magnetic mesoporous silica microspheres for selective enrichment of phosphopeptides from tryptic digestion of rat brain, and the experimental results showed that the approach based on ZrP-functionalized magnetic mesoporous silica microspheres would be effective for phosphopeptides enrichment.Acknowledgements
The work was supported by the National Natural Science Foundation of China (Project: 20875017, 21075022 and 30873132), Technological Innovation Program of Shanghai (09JC1401100), the National Basic Research Priorities Program (Project: 2007CB914100/3), and Shanghai Leading Academic Discipline Project (B109).References
- (a) M. J. Hubbard and P. Cohen, Trends Biochem. Sci., 1993, 18, 172 CrossRef CAS; (b) T. Hunter, Philos. Trans. R. Soc. London, Ser. B, 1998, 353, 583 CrossRef CAS; (c) J. D. Graves and E. G. Krebs, Pharmacol. Ther, 1999, 82, 11.
- (a) D. E. Kalume, H. Molina and A. Pandey, Curr. Opin. Chem. Biol., 2003, 7, 64 CrossRef CAS; (b) S. A. Beausoleil, M. Jedrychowski, D. Schwartz, J. E. Elias, J. Villén, J. Li, M. A. Cohn, L. C. Cantley and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12130 CrossRef CAS; (c) M. R. Larsen, M. B. Trelle, T. E. Thingholm and O. N. Jensen, BioTechniques, 2006, 40, 790 CAS; (d) O. N. Jensen, Nat. Rev. Mol. Cell Biol., 2006, 7, 391 CrossRef CAS; (e) E. S. Witze, W. M. Old, K. A. Resing and N. G. Ahn, Nat. Methods, 2007, 4, 798 CrossRef CAS.
- (a) S. Ficarro, O. Chertihin and V. A. Westbrook, J. Biol. Chem., 2003, 278, 11579 CrossRef CAS; (b) T. S. Nuhse, A. Stensballe, O. N. Jensen and S. C. Peck, Mol. Cell. Proteomics, 2003, 2, 1234 CrossRef; (c) A. Gruhler, J. V. Olsen, S. Mohammed and P. Mortensen, Mol. Cell. Proteomics, 2005, 4, 310 CrossRef CAS; (d) Y. Oda, T. Nagasu and B. T. Chait, Nat. Biotechnol., 2001, 19, 379 CrossRef CAS; (e) H. Zhou, J. D. Watts and R. Aebersold, Nat. Biotechnol., 2001, 19, 375 CrossRef CAS; (f) M. R. Larsen, T. E. Thingholm and O. N. Jensen, Mol. Cell. Proteomics, 2005, 4, 873 CrossRef CAS; (g) H. K. Kweon and K. Hakansson, Anal. Chem., 2006, 78, 1743 CrossRef CAS.
- (a) H. J. Zhou, S. Y. Xu, M. L. Ye, S. Feng, C. S. Pan, X. G. Jiang, X. Li, G. H. Han, Y. Fu and H. F. Zou, J. Proteome Res., 2006, 5, 2431 CrossRef; (b) S. Feng, M. L. Ye, H. J. Zhou, X. G. Jiang, X. N. Jiang, H. F. Zou and B. L. Gong, Mol. Cell. Proteomics, 2007, 6, 1656 CrossRef CAS; (c) J. Y. Wei, Y. J. Zhang, J. L. Wang, F. Tan, J. F. Liu, Y. Cai and X. H. Qian, Rapid Commun. Mass Spectrom., 2008, 22, 1069 CrossRef; (d) S. Y. Xu, J. C. Whitin, T. T. S. Yu, H. J. Zhou, D. Z. Sun, H. J. Sue, H. F. Zou, H. J. Cohen and R. N. Zare, Anal. Chem., 2008, 80, 5542 CrossRef CAS.
- (a) G. Nonglaton, I. O. Benitez, I. Guisle, M. Pipelier, J. Leger, D. Dubreuil, C. Tellier, D. R. Talham and B. J. Bujoli, J. Am. Chem. Soc., 2004, 126, 1497 CrossRef CAS; (b) D. W. Qi, Y. Mao, J. Lu, C. H. Deng and X. M. Zhang, J. Chromatogr., A, 2010, 1217, 2606 CrossRef CAS.
- (a) Y. Li, D. W. Qi, C. H. Deng, P. Y. Yang and X. M. Zhang, J. Proteome Res., 2008, 7, 1767 CrossRef CAS; (b) X. Q. Xu, C. H. Deng, M. X. Gao, W. J. Yu, P. Y. Yang and X. M. Zhang, Adv. Mater., 2006, 18, 3289 CrossRef CAS; (c) H. M. Chen, X. Q. Xu, N. Yao, C. H. Deng, P. Y. Yang and X. M. Zhang, Proteomics, 2008, 8, 2778 CrossRef CAS; (d) H. M. Chen, D. W. Qi, C. H. Deng, P. Y. Yang and X. M. Zhang, Proteomics, 2009, 9, 380 CrossRef CAS.
- (a) Y. Zhang, C. Chen, H. Q. Qin, R. A. Wu and H. F. Zou, Chem. Commun., 2010, 46, 2271 RSC; (b) L. H. Hu, H. J. Zhou, Y. H. Li, S. T. Sun, L. H. Guo, M. L. Ye, X. F. Tian, J. R. Gu, S. L. Yang and H. F. Zou, Anal. Chem., 2009, 81, 94 CrossRef CAS.
- H. M. Chen, S. S. Liu, H. L. Yang, Y. Mao, C. H. Deng, X. M. Zhang and P. Y. Yang, Proteomics, 2010, 10, 930 CAS.
- Y. H. Deng, D. W. Qi and C. H. Deng, J. Am. Chem. Soc., 2007, 130, 28.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0nr00896f |
| This journal is © The Royal Society of Chemistry 2011 |