Porous SnO2 nanospheres as sensitive gas sensors for volatile organic compounds detection†
Zhipeng
Li
,
Quanqin
Zhao
,
Weiliu
Fan
and
Jinhua
Zhan
*
Key Laboratory of Colloid and Interface Chemistry, Ministry of Education, Department of Chemistry, Shandong University, Jinan, 250100, Shandong, P.R. China. E-mail: jhzhan@sdu.edu.cn; Fax: +86-0531-8836-6280
First published on 31st January 2011
Abstract
Porous SnO2 nanospheres with high surface areas have been synthesized through a solvothermal method in the absence of any templates. The structure and morphology of the resultant products were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and nitrogen adsorption–desorption technique. The as-prepared SnO2 porous nanospheres with the diameters ranging from 90–150 nm are composed of small nanocrystals with average sizes of less than 10 nm. Results demonstrated that the formation of porous SnO2 nanospheres is ascribed to etching the center part of the nanospheres. It was found that hydrochloric acid and NaClO played important roles in determining the final morphologies of the porous SnO2 nanospheres. The gas sensing properties of the as-prepared porous SnO2 nanospheres were investigated. By the comparative gas sensing tests, the porous SnO2 nanospheres exhibited a superior gas sensing performance toward ppb level 2-chloroethanol and formaldehyde vapor, implying promising applications in detecting toxic volatile organic compounds (VOCs).
1. Introduction
Inorganic nanomaterials with hollow or porous structures have attracted considerable attention because of their promising applications such as gas sensors,1,2 efficient catalysts,3–5drug-delivery carriers,6,7 and photo-/electronic building blocks.8–10 For many applications, micro- and nanoscale materials display a strong correlation between their morphology and function. Porous and hollow materials are considered excellent candidates for many applications due to the fact that the catalytic, sensing, or photovoltaic activity often depends on the effective surface area and porosity.11 For some reactions occurring on the materials surface, the large specific surface area means that a significant fraction of atoms are located at the surface and can participate in surface reactions, providing more active sites and enhanced reactivity.Controlled synthesis strategies for hollow or porous nanostructures have generally involved the use of removable or sacrificial templates,12–14 soft template (micelles,15,16 emulsion,17,18 and gas bubbles19), and template-free methods. Recently, various one-pot template-free routes by using some physical phenomenon, such as the oriented attachment,20,21Ostwald ripening,22,23 the combination of oriented attachment and Ostwald ripening,24,25 Kirkendall effect,26,27 and etching-based inside-out evacuation,28,29 have been well developed to fabricate hollow or porous structures because of their simplicity, low cost and flexibility. Theoretically, the etching process involves the dissolution of the inner nanocrystallites with time or temperature evolution due to the high surface energies or poor crystalllinity, which provides facile options for the fabrication of hollow or porous structures. Examples include ZnO,30α-Fe2O3,31SnO2,32,33 and so on.
SnO2, an n-type semiconductor with a wide band gap (Eg = 3.6 eV), has been extensively studied and applied in the field of gas sensors,2,34–36lithium rechargeable batteries,29,37–39catalysts,40 and optoelectronic devices.41,42 In those applications, it must be noted that the performance is greatly influenced by the size, structure and morphology of SnO2 materials, especially for gas sensors typically depending on the surface area and porosity of semiconductor metal oxides. Herein, we report a simple template-free synthesis of porous SnO2 nanospheres with uniform structure and morphology. Results verify that the etching or dissolving of the inner crystals is responsible for the formation of highly porous SnO2 nanospheres. Furthermore, gas sensing properties of the as-prepared SnO2 products were investigated using 2-chloroethanol and formaldehyde as the representative target gases.
As we know, toxic or explosive volatile organic compounds (VOCs) are very dangerous for both the environment and human beings.43,44 For example, 2-chloroethanol is widely used in the manufacture of pesticides, plasticizers, plant-protection agents, and dye intermediates, leading to hypotension, metabolic acidosis, coma, and respiratory failure.45Formaldehyde, which can be released from buildings and decorative materials, is also seriously deleterious for human health. As a result, the admissible concentration of 2-chloroethanol and formaldehyde is strictly limited in the atmosphere. The OSHA (Occupational Safety and Health Administration, USA) sets the exposure limits of 5 ppm for 2-chloroethanol in workplace and 0.75 ppm for formaldehyde. Therefore, detection toward VOCs is of great significance for environmental safety and human health. In our study, the hierarchically porous SnO2 nanospheres show an excellent gas sensing performance for detecting VOCs. The effects of grain size and porosity of sensing materials on the gas sensing properties are also discussed.
2. Results and discussion
Fig. 1a and 1b show the SEM images of as-prepared porous SnO2 nanospheres with diameters ranging from 90 to150 nm. From the high magnification SEM image in Fig. 1b the rough surface of SnO2 nanospheres can be observed, suggesting that the SnO2 nanospheres consist of small nanoparticles. The single SnO2 nanosphere is given in the inset of the Fig. 1b. The phase and purity of the products were determined by XRD. Fig. 2 illustrates the typical diffraction pattern, and all the peaks can be well indexed to the tetragonal rutile SnO2 structure (JCPDS card NO. 41-1445, ao = 4.738 Å, co = 3.187 Å). No characteristic peaks were observed for other impurities, revealing the high purity of the prepared SnO2 nanospheres. To further characterize the product, XPS analysis was conducted to investigate the surface compositions and chemical states of the as-prepared samples (Fig. S1 in ESI†). Fig. S1a shows that the Sn 3d spectrum of the sample, which can be assigned to a spin-orbit doublet at around 486.9 (3d5/2) and 495.3 eV (3d3/2), is in good agreement with the values given in the literature.33,46 In Fig. S2b, the peaks at 530.8 eV can be assigned to O 1s, indicating that oxygen atoms exist as O2− species. Both the XRD and XPS results verify that all the Sn2+ ions were oxidized to Sn4+ by strong oxidizing agents ClO−, and the final products were indeed SnO2. | ||
| Fig. 1 SEM images of as-prepared porous SnO2 nanospheres at (a) low and (b) high magnification. Inset in (b): the single porous SnO2 nanosphere. | ||
 | ||
| Fig. 2 XRD pattern of the porous SnO2 nanospheres. | ||
Detailed structural analysis of as-prepared porous SnO2 nanospheres was carried out by transmission electron microscopy (TEM), and electron diffraction (ED). From the TEM image (Fig. 3a), it is apparent that the products are uniform nanospheres with diameters of about 100 nm. The TEM image of individual porous SnO2 nanospheres (Fig. 3b) clearly reveals that the surface of SnO2 is rough and the samples were composed of numerous primary nanocrystallites with an average sizes of ca. 6 nm. The high-resolution TEM image of porous SnO2 nanospheres is given in Fig. 3c corresponding to the square part in Fig. 3b. Clear lattice fringes observed, and d-spacing are measured to 0.264 and 0.334 nm, which can be indexed to the SnO2 (101) and (110) crystal planes, respectively. An electron diffraction (ED) pattern is depicted in inset of Fig. 3b, suggesting the polycrystalline nature of the nanostructures. Although there is no direct proof, it can be seen from the SEM and TEM images that the inner part of the porous SnO2 nanospheres might be hollow as well.
 | ||
| Fig. 3 TEM images and SAED pattern of porous SnO2 nanospheres. (a) low-magnification TEM image of porous SnO2 nanospheres; (b) high-magnification TEM image of single SnO2 nanosphere; (c) HRTEM image from the interior space corresponding to the square part in (b). Inset in (c) is the SAED pattern of the SnO2 nanospheres. | ||
To further confirm the inner architectures of the porous SnO2 nanospheres, nitrogen adsorption/desorption analysis has been performed to estimate its textural properties. The representative nitrogen adsorption/desorption isotherm and the corresponding Barrett–Joyner–Halenda (BJH) pore size distribution plots (inset) of the porous SnO2 nanospheres are shown in Fig. 4. The N2 isotherm of the as-prepared porous SnO2 nanospheres is a type-V isotherm with a large type H3 hysteresis loop,47 which does not exhibit any limiting adsorption at high P/P0. The as-synthesized porous SnO2 nanospheres have a high BET surface area of 76.4 m2 g−1. As calculated by the BJH method from the desorption branch of the nitrogen isotherm, the pore size distribution (inset in Fig. 4) presents that the SnO2 nanospheres contain an average pore size of 32.8 nm, with the small mesopores in the order of 19 nm. The pores distributed in the SnO2 nanosphere are also observed in TEM image (Fig. 3b) between the adjacent nanoparticles, which probably arises from the imperfect attachment of the primary nanocrystallites.
 | ||
| Fig. 4 Typical nitrogen adsorption–desorption isotherm and Barrett–Joyner–Halenda (BJH) pore size distribution plots (inset) of the porous SnO2 nanospheres. | ||
To understand the formation of porous SnO2 nanospheres, time-dependent experiments were conducted. As shown in Fig. 5, the products collected at different reaction times exhibit obvious discrepancy in morphology. At the early reaction stage, the products (Fig. 5a) were plenty of small aggregated nanocrystallites with size of about 30 nm, which have been confirmed by electron diffraction pattern (inset in Fig. 5a) suggesting its polycrystalline nature. When the reaction time was prolonged to 8 h, the SnO2 nanocrystallites grew to large spherical structures with dense cores, as shown in Fig. 5b. After 12 h, loosing took place at the interior part of nanospheres, which is indicated by the color contrast in TEM images (Fig. 5c). The porous SnO2 nanospheres with relatively porous structures can be observed at 18 h shown in Fig. 5d. On the basis of the time-dependent experiments, the possible formation process of SnO2 porous nanospheres was reasonably illustrated in Fig. 5e. In the initial stage of the reaction, primary nanocrystals with high energy aggregated to form small nanospheres with diameters of about 30 nm. Under the solvothermal conditions, these small nanospheres further agglomerated to construct large nanospheres to reduce the surface energies. As the reactions went on, the inner part of the large nanospheres began to dissolve gradually and became loose, which finally led to the porous structure. Because crystallization of the outer surfaces can be highly promoted by the copious surrounding solvent during solvothermal treatment,29 the primary nanocrystallites in the inner part of SnO2 nanospheres are poorly crystallized compared to those in the outer surface. As a result, the small nanocrystals in the inner region of nanospheres tend to dissolve when the reaction time was increased in this acid solution. Another important reason making the hollowing effect is that the nanocrystallites in the inner regions of the nanospheres have high surface energies compared to those in the outer surface, which in turn leads to a strong tendency to dissolve.22,33,48 So, the etching or dissolving of the inner part of SnO2 products resulted in the formation of porous nanospheres.
 | ||
| Fig. 5 TEM images of products obtained at 180 °C at different reaction times: (a) 2 h; (b) 8 h; (c) 12 h; and (d) 18 h. Inset in (a) shows the ED pattern of the SnO2 nanoparticles. (e) Schematic diagram of the formation of porous SnO2 nanospheres. | ||
It was also found that HCl played an important role in the formation of porous SnO2 nanospheres. As shown in Fig. 6a, the products synthesized in the absence of HCl were dense spheres with a diameter of about 2 µm. If excess HCl of 1 mL was added to original solution, the products are irregular particles (Fig. 6b). H2SO4 and HNO3 instead of HCl were used while the pH remained unchanged. As shown in Fig. 6c and 6d, the products were also irregular particles. Addition of HCl can inhibit hydrolysis of Sn4+ ions to a certain extent and slow down the growth of SnO2 nanocrystals, which can be verified by the diameters of porous SnO2 nanospheres and the dense spheres prepared in absence of HCl. This effect means that HCl slows down the growth of the SnO2 nanocrystals. It has been reported that Cl− ions can be adsorbed specifically providing the opportunity to kinetically control the shape of the SnO2 nanocrystals.49 In this work, NaClO acted as the oxidant to oxidize the Sn2+ to Sn4+ ions in the HCl solution. The effect of NaClO on the morphologies of SnO2 products was also studied by varyging the amount of the NaClO with the other synthetic conditions remaining unchanged. As is shown in Fig. 6e, only irregular nanoparticles were obtained when the experiments were performed without addding NaClO. Upon adding 1 mmol of NaClO (ClO−: Sn2+ = 1/2), the resultant products were composed of disordered nanoparticles and porous nanospheres (Fig. 6f). These results suggest that it is critical to oxidize Sn2+ to Sn4+ ions by NaClO before solvothermal treatment, which affects the hydrolysis of Sn4+ and the nucleation and growth of SnO2 nanocrystals. Therefore, HCl and NaClO were all crucial factors in determining the final morphologies of the porous SnO2 nanospheres.
 | ||
| Fig. 6 TEM images of SnO2 samples obtained with the presence of different amounts of HCl and using H2SO4 and HNO3 instead of HCl: (a) without HCl; (b) with 1 mL of HCl; (c) with H2SO4; and (d) with HNO3. Inset in (a) gives the individual SnO2 dense sphere. (e) and (f) TEM images of the SnO2 products synthesized with addition of 0 and 1 mmol NaClO. | ||
The gas sensing properties of as-prepared porous SnO2 nanospheres for the detection of 2-chloroethanol and formaldehyde were investigated. It can be seen from Fig. S2 in ESI† that the porous nanostructures and morphologies of SnO2 nanospheres still remained after being aged for 3 days at 300 °C. In order to determine the optimum operating temperature, the response of the porous SnO2 nanospheres gas sensor to 10 ppm 2-chloroethanol and formaldehyde vapor in air were tested as a function of operating temperature, as shown in Fig. 7b. It is clear that the response of the gas sensor varied with operating temperature. The response first increased with temperature, up to 260 °C, and then gradually decreased. The maximum response for 2-chloroethanol and formaldehyde reached 9.3 and 7.6 at 260 °C. Therefore, the temperature of 260 °C was chosen for further examining the gas sensing characteristics of the as-prepared porous SnO2 nanospheres. Fig. 8a and 8b depicts the representative dynamic gas response of as-prepared porous SnO2 nanospheres sensing to 2-chloroethanol and formaldehyde with different concentrations when the sensor was working at 260 °C. A few cycles were successively recorded, corresponding to different 2-chloroethanol and formaldehyde concentrations ranging from 0.5 to 100 ppm. The conductance underwent a drastic rise upon the injection of both 2-chloroethanol and formaldehyde and dropped to its initial value after the sensor was exposed to air. When 2-chloroethnaol and HCHO with the same concentrations were re-injected into the glass chamber, the response had little change, indicating the good reversibility and repeatability. The lowest detection limit of the sensor was down to 0.5 ppm to 2-chloroethanol and formaldehyde, which is below the permissible exposure limits set by the OSHA (Occupational Safety and Health Administration, USA). Response and recovery times are also important parameters in a gas sensor. For 1 ppm 2-chloroethanol, the response and recovery times of the as-prepared SnO2 nanospheres were 15 s and 17 s, respectively. For 1 ppm HCHO, the response and recovery times were 13 s and 14 s, respectively.
 | ||
| Fig. 7 (a) Schematic illustration of the sensor element and (b) sensor response versus operating temperature of porous SnO2 nanospheres to 10 ppm 2-chloroethanol and formaldehyde. | ||
 | ||
| Fig. 8 Dynamic response-recovery curves of the gas sensor based on prepared porous SnO2 nanospheres toward volatile organic compounds at a series of concentrations: (a) 2-chloroethanol; (b) formaldehyde. | ||
The gas sensing mechanism for semiconducting metal oxides can be ascribed to the change in electrical conductivity resulting from the chemical interaction of gas molecules with the surface of semiconductor metal oxides.50 This mechanism mainly involves the gas adsorption, charge transfer, and desorption process. Once the SnO2 sensing materials are exposed to air at a high temperature (here 260 °C), oxygen molecules are adsorbed on the SnO2 surface and ionized oxygen species (O2−, O2−, or O−) are formed through trapping electrons from the conductance band of SnO2. Then, SnO2 shows a high resistance state in air due to the formation of a space-charge region. When reductive 2-chloroethanol and formaldehyde gas molecules approach the SnO2 surface, they will react with the oxygen species, which results in electrons being released to the surface layer of SnO2. This effect eventually increases the conductivity of the SnO2 nanospheres.
The comparative sensing studies among the various SnO2 materials, including porous SnO2 nanospheres, small nanoparticles with the average diameters of 6 nm (TEM image is given in Fig. S3 of ESI†), commercial powders (see Fig. S4 in ESI†), and dense spheres, were further performed to depict the excellent gas sensing performance of porous SnO2 nanospheres. In order to examine the influence of the samples morphology on the gas sensing properties to 2-chloroethanol and formaldehyde, several SnO2 samples including SnO2 nanoparticles, commercial powders, and dense spheres, were used as references for comparison, and their gas sensing properties were also studied under the same test conditions. The relationship between sensor response and gas concentrations of 2-chloroethanol and formaldehyde vapor is depicted in Fig. 9a and 9b. Apparently, the sensor based porous SnO2 nanospheres exhibit much higher response than those of other three kinds of SnO2 materials. For example, at a determined HCHO concentration of 1 ppm, the response value of porous SnO2 nanospheres is 3.14, whereas it is 1.27, 0.22, and 0.13 for SnO2 nanoparticles, commercial powders, and dense spheres, respectively. Similarly, for 2-chloroethanol detection, the as-prepared SnO2 nanospheres also demonstrate enhanced performance. These results suggest that the morphology and structure of SnO2 materials have a great influence on the gas sensing performance.
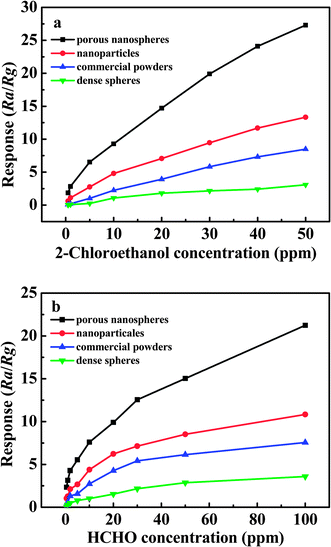 | ||
| Fig. 9 The relationship between response (Ra/Rg) of various SnO2 samples and different gas concentrations: (a) 2-chloroethanol; (b) formaldehyde. | ||
It has been reported that the response (Ra/Rg) of metal-oxide gas sensors highly depends on the grain size and porosity of sensing material.51–55 For instance, Xu et al. reported that the increase of gas sensitivity was remarkable when the average grain size (D) of SnO2 elements was below about 10 nm.53 Yamazoe et al. have confirmed that the critical grain sizes of SnO2 for sensing H2 or CO were in the region of 5–15 nm.56 According to the model of the grain size effect in n-type semiconducting metal oxide gas sensors developed by Avner Rothschild,52 when D<2L (L = thickness of the depletion layer) the depletion region extends throughout the whole grain and the conductance is grain-controlled. The previous reported results have demonstrated that the thickness of the depletion layer (L) of SnO2 in air is about 3 nm.57 In our work, the size of primary nanocrystallites of porous SnO2 nanospheres is about 6–7 nm, which is nearly equal to the 2L, implying the small SnO2 crystallites are fully depleted. On the contrary, for the dense spheres and commercial powders, whose diameters are about 2 µm and 60 nm (D≫2L), their conductance is controlled by grain boundaries, in which the interior parts of the grains do not contribute to the gas response. This small size effect means that a significant fraction of the tin atoms are surface atoms that can participate in surface reactions, providing more active sites to adsorb more oxygen species and target molecules. In addition, the sensing tests conducted with dispersive SnO2 nanoparticles (diameters of 6 nm) show that their response is lower than the porous SnO2 nanospheres, suggesting that porosity is also an important factor for gas sensing properties. The high porosity and 3D morphology of porous SnO2 nanospheres can also significantly facilitate gas diffusion and mass transportation in sensing material.11,51,58
We also measured the response of the porous SnO2 nanospheres to other volatile organic compounds with a concentration of 1 ppm, such as ethanol, acetone, chlorophenol, toluene, acetonitrile, and chloroform at the operating temperature of 260 °C. The sensor based on porous SnO2 nanospheres exhibited high response to these gases, as shown in Fig. 10 and Table 1 in ESI†. So it is believed that the as-prepared porous SnO2 nanospheres maybe have potential applications in the detection of volatile organic compounds.
 | ||
| Fig. 10 Response values of the porous SnO2 nanospheres sensor to different gases of 1 ppm. | ||
3. Conclusions
In summary, a template-free solvothermal method was demonstrated for the synthesis of hierarchically porous SnO2 nanospheres with high surface area and porosity, which were composed of interconnected SnO2 nanocrystallites with a diameter of 5–10 nm. On the basis of the morphology study on the evolution of porous SnO2 nanospheres by TEM, the etching-induced inside-out dissolution of the inner region is proposed to account for the formation of these porous nanostructures. HCl acted as not only the inhibitor for Sn4+ hydrolysis but also the adsorbent, which played an important role in the formation of porous SnO2 nanospheres. The obtained SnO2 nanospheres exhibited excellent gas sensing performance towards 2-chloroethanol and formaldehyde vapor due to the high surface area and porous nanosturctures, suggesting the SnO2 nanomaterial is a promising candidate for detecting toxic volatile organic compounds (VOCs).4. Experimental
Tin dichloride (SnCl2·2H2O, ≥98.0%), sodium hypochlorite (NaClO), and tin dioxide powders (SnO2, ≥99.5%, 40–70 nm) were purchased from Aladdin Reagent Co. Ltd. Hydrochloric (HCl, 36%), sulfuric acid (H2SO4, 98%), and nitric acid (HNO3, 65%) were obtained from Shanghai Chemical Reagent Company and used as received. In a typical procedure, 0.6 mL of HCl and 2 mmol of SnCl2·2H2O were dissolved in 30 mL anhydrous ethanol under vigorous magnetic stirring. 2 mmol of NaClO was then added into the ethanol solution. The obtained white slurry was transferred into a Teflon-lined steel autoclave and heated for 24 h at 180 °C in an electric oven. After the autoclave was cooled to room temperature, white products were collected and thoroughly washed with distilled water and absolute ethanol and dried in a vacuum for 12 h at 60 °C.The as-prepared samples were characterized by XRD on a Bruker D8 advanced X-ray diffractometer equipped with graphite monochromatized Cu Kα radiation (λ = 1.5418 Å). The morphology and structure of the as-synthesized SnO2 nanospheres were studied by FE-SEM (JSM-6700F), TEM (JEOL 6300, 100 kV), and high-resolution transmission electron microscopy (HRTEM, JEM-2100, 200 kV). XPS spectra were recorded by a PHI 5300 X-ray photoelectron spectrometer with Al Kα radiation. The BET surface area and Barrett–Joyner–Halenda pore size and pore distribution were determined by N2adsorption/desorption at 77 K using a QuadraSorb SI surface area analyzer after degassing the samples at 100 °C for 10 h. Gas sensing properties of the sample were measured using a China HW-30A gas sensitivity instrument. The schematic illustration of the sensor element is shown in Fig 7a.
The gas sensor was fabricated as follows: SnO2 powders were mixed with water, and then coated onto an Al2O3 tube on which two platinum wires had been installed at each end. The gas sensing element was aged at 300 °C for 3 days in order to improve its stability and repeatability. A schematic diagram of electrical circuit of the instrument is shown in Fig. S5 in ESI†. The operating temperature was controlled by adjusting the heating voltage (VH), using a Ni-Cr alloy coil placed through the tube for heating the sensing material. In this test, a load resistor was serialized and a constant working voltage (VW) at 5 V was applied to this system. By monitoring the load resistor voltage (VO), the response of the SnO2 sensor in air or in the test gases can be measured. The resistance or conductance of the sensor can be calculated from Ohm's law: Rs = (5/VO-1)RL. The sensor response was defined as the ratio S= Ra/Rg, where Ra and Rg were the resistance measured in air and in target gas atmosphere, respectively. The response and recovery times were defined as the time to reach 90% of the final signal. The measurement was processed by a static process: a given amount of the tested gas was injected into a glass chamber and mixed with air. When the response reached a constant value, the sensor was taken to recover in air.
Acknowledgements
We thank the helpful discussion with Prof. Yitai Qian and financial support from National Natural Science Foundation of China (NSFC 50972083 and 21075077), Independent Innovation Foundation of Shandong University (IIFSDU-2009JQ011), the Key Project of Chinese Ministry of Education (No. 109098), National Basic Research Program of China (973 Program 2007CB936602). Weiliu Fan thanks financial support from NSFC (50802056).References
- H. Zhang, Q. Zhu, Y. Zhang, Y. Wang, L. Zhao and B. Yu, Adv. Funct. Mater., 2007, 17, 2766–2771 CrossRef CAS.
- Y. E. Chang, D. Y. Youn, G. Ankonina, D. J. Yang, H. G. Kim, A. Rothschild and I. D. Kim, Chem. Commun., 2009, 4019–4021 RSC.
- W. W. Wang, Y. J. Zhu and L. X. Yang, Adv. Funct. Mater., 2007, 17, 59–64 CrossRef CAS.
- H. P. Liang, H. M. Zhang, J. S. Hu, Y. G. Guo, L. J. Wan and C. L. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540–1543 CrossRef CAS.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642–7643 CrossRef CAS.
- Y. Zhu, T. Ikoma, N. Hanagata and S. Kaskel, Small, 6, 471–478 Search PubMed.
- W. Wei, G. H. Ma, G. Hu, D. Yu, T. McLeish, Z. G. Su and Z. Y. Shen, J. Am. Chem. Soc., 2008, 130, 15808–15810 CrossRef CAS.
- D. Grosso, C. Boissiere and C. Sanchez, Nat. Mater., 2007, 6, 572–575 CrossRef CAS.
- S. C. Yang, D. J. Yang, J. Kim, J. M. Hong, H. G. Kim, I. D. Kim and H. Lee, Adv. Mater., 2008, 20, 1059–1064 CrossRef CAS.
- A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391–4395 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- M. Yang, J. Ma, C. L. Zhang, Z. Z. Yang and Y. F. Lu, Angew. Chem., Int. Ed., 2005, 44, 6727–6730 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- Y. Zeng, X. Wang, H. Wang, Y. Dong, Y. Ma and J. N. Yao, Chem. Commun., 2010, 46, 4312–4314 RSC.
- D. Liu and K. Nakashima, Inorg. Chem., 2009, 48, 3898–3900 CrossRef CAS.
- A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS.
- C. I. Zoldesi and A. Imhof, Adv. Mater., 2005, 17, 924–928 CrossRef CAS.
- G. R. Bourret and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 6657–6659 CrossRef CAS.
- Q. Peng, Y. J. Dong and Y. D. Li, Angew. Chem., Int. Ed., 2003, 42, 3027–3030 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS.
- D. B. Yu, X. Q. Sun, J. W. Zou, Z. R. Wang, F. Wang and K. Tang, J. Phys. Chem. B, 2006, 110, 21667–21671 CrossRef CAS.
- B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS.
- Y. Chang, J. J. Teo and H. C. Zeng, Langmuir, 2005, 21, 1074–1079 CrossRef CAS.
- G. Z. Chen, C. X. Xu, X. Y. Song, S. L. Xu, Y. Ding and S. X. Sun, Cryst. Growth Des., 2008, 8, 4449–4453 CrossRef CAS.
- B. P. Jia and L. Gao, J. Phys. Chem. C, 2008, 112, 666–671 CrossRef CAS.
- X. Liang, X. Wang, Y. Zhuang, B. Xu, S. Kuang and Y. Li, J. Am. Chem. Soc., 2008, 130, 2736–2737 CrossRef CAS.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS.
- Y. J. Xiong, B. Wiley, J. Y. Chen, Z. Y. Li, Y. D. Yin and Y. N. Xia, Angew. Chem., Int. Ed., 2005, 44, 7913–7917 CrossRef CAS.
- X. W. Lou, Y. Wang, C. L. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325–2329 CrossRef CAS.
- F. Li, Y. Ding, P. Gao, X. Xin and Z. L. Wang, Angew. Chem., Int. Ed., 2004, 43, 5238–5242 CrossRef CAS.
- C. J. Jia, L. D. Sun, Z. G. Yan, L. P. You, F. Luo, X. D. Han, Y. C. Pang, Z. Zhang and C. H. Yan, Angew. Chem., Int. Ed., 2005, 44, 4328–4333 CrossRef CAS.
- Z. J. Miao, Y. Y. Wu, X. R. Zhang, Z. M. Liu, B. X. Han, K. L. Ding and G. M. An, J. Mater. Chem., 2007, 17, 1791–1796 RSC.
- S. Y. Ho, A. S. W. Wong and G. W. Ho, Cryst. Growth Des., 2009, 9, 732–736 CrossRef CAS.
- H. R. Kim, K. I. Choi, K. M. Kim, I. D. Kim, G. Z. Cao and J. H. Lee, Chem. Commun., 2010, 46, 5061–5063 RSC.
- C. H. Wang, X. F. Chu and M. M. Wu, Sens. Actuators, B, 2007, 120, 508–513 CrossRef.
- X. M. Yin, C. C. Li, M. Zhang, Q. Y. Hao, S. Liu, Q. H. Li, L. B. Chen and T. H. Wang, Nanotechnology, 2009, 130, 455503 CrossRef.
- J. F. Ye, H. J. Zhang, R. Yang, X. G. Li and L. M. Qi, Small, 2010, 6, 296–306 CrossRef CAS.
- J. S. Chen, C. M. Li, W. W. Zhou, Q. Y. Yan, L. A. Archer and X. W. Lou, Nanoscale, 2009, 1, 280–285 RSC.
- R. Demir-Cakan, Y. S. Hu, M. Antonietti, J. Maier and M. M. Titirici, Chem. Mater., 2008, 20, 1227–1229 CrossRef.
- G. Wang, W. Lu, J. H. Li, J. Choi, Y. S. Jeong, S. Y. Choi, J. B. Park, M. K. Ryu and K. Lee, Small, 2006, 2, 1436–1439 CrossRef CAS.
- J. Y. Liu, T. Luo, T. S. Mouli, F. L. Meng, B. Sun, M. Q. Li and J. H. Liu, Chem. Commun., 2010, 46, 472–474 RSC.
- H. J. Snaith, H. J. Snaith and C. Ducati, Nano Lett., 2010, 10, 1259–1265 CrossRef CAS.
- K. Mitsubayashi, K. Yokoyama, T. Takeuchi and I. Karube, Anal. Chem., 1994, 66, 3297–3302 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Jr, Science, 1997, 276, 1045–1051 CrossRef CAS.
- Y. T. Chen, D. Z. Hung, C. C. Chou, J. J. Kang, Y. W. Cheng, C. M. Hu and J. W. Liao, J. Health Sci., 2009, 55, 525–531 CrossRef CAS.
- H. J. Ahn, H. C. Choi, K. W. Park, S. B. Kim and Y. E. Sung, J. Phys. Chem. B, 2004, 108, 9815–9820 CrossRef CAS.
- Z. H. Jing and J. H. Zhan, Adv. Mater., 2008, 20, 4547–4551 CrossRef CAS.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492–3495 CrossRef CAS.
- X. G. Han, M. S. Jin, S. F. Xie, Q. Kuang, Z. Y. Jiang, Y. Q. Jiang, Z. X. Xie and L. S. Zheng, Angew. Chem., Int. Ed., 2009, 48, 9180–9183 CrossRef CAS.
- T. Seiyama, A. Kato, K. Fujiishi and M. Nagatani, Anal. Chem., 1962, 34, 1502–1503 CrossRef CAS.
- M. Tiemann, Chem.–Eur. J., 2007, 13, 8376–8388 CrossRef CAS.
- A. Rothschild and Y. Komem, J. Appl. Phys., 2004, 95, 6374–6380 CrossRef CAS.
- C. Xu, J. Tamaki, N. Miura and N. Yamazoe, Sens. Actuators, B, 1991, 3, 147–155 CrossRef.
- F. Gyger, M. Hubner, C. Feldmann, N. Barsan and U. Weimar, Chem. Mater., 2010, 22, 4821–4827 CrossRef CAS.
- T. Kida, T. Doi and K. Shimanoe, Chem. Mater., 2010, 22, 2662–2667 CrossRef CAS.
- N. Yamazoe, Sens. Actuators, B, 1991, 5, 7–19 CrossRef.
- H. Ogawa, M. Nishikawa and A. Abe, J. Appl. Phys., 1982, 53, 4448 CrossRef CAS.
- J. H. Lee, Sens. Actuators, B, 2009, 140, 319–336 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and Table 1. See DOI: 10.1039/c0nr00728e |
| This journal is © The Royal Society of Chemistry 2011 |