Core-crosslinked compartmentalized cylinders
Felix H.
Schacher†
*,
Tobias
Rudolph
,
Markus
Drechsler
and
Axel H. E.
Müller
*
Makromolekulare Chemie II, Universität Bayreuth, Bayreuth, 95440, Germany. E-mail: axel.mueller@uni-bayreuth.de
First published on 9th November 2010
Abstract
We present a detailed study on the preparation of compartmentalized cylindrical nanoparticlesvia a templated approach: the polybutadiene part of a linear polybutadiene-block-poly(2-vinyl pyridine)-block-poly(tert-butyl methacrylate) block terpolymer, B420V280T790, having a bulk microstructure with PB cylinders covered by a P2VP double helix and embedded in a PtBMA matrix was selectively crosslinked. Subsequent sonication-assisted dissolution and chemical modifications such as quaternization (P2VP to P2VPq) and ester hydrolysis (PtBMA to poly(sodium methacrylate), PMANa) resulted in core-crosslinked cylinders soluble in organic and aqueous media. Different amounts of crosslinker and the influence of the sonication treatment on size and shape of the cylindrical aggregates were investigated. The cylinders always exhibit a compartmentalized corona. Under certain conditions, in particular quaternization of P2VP in mixtures of THF and MeOH, the helical arrangement of the P2VPq shell could be preserved even in solution, whereas in most other cases randomly distributed P2VP/P2VPq patches were observed. In aqueous solution at high pH, intramicellar interpolyelectrolyte complex (im-IPEC) formation occurred between the positively charged P2VPq shell and the negatively charged PMANa corona. We further show that different noble metal nanoparticles can be generated either selectively within the im-IPEC compartments (Pd) or randomly distributed among shell and corona of the cylinders (Au and Pt).
Introduction
The concept of self-assembly plays an important role for the design and the preparation of materials in areas where specific properties are required and is often linked to the field of block copolymers.1–5 Here, recent synthetic advances have distinctly broadened the accessible range of monomers, architectures, and morphologies.6–9 Further, not only different and more sophisticated building blocks are available but also a variety of preparation techniques10,11 and a multitude of substrates used resulted in more and more complex structures.12The self-assembly of ABC triblock terpolymers, both linear13 and miktoarm14 type, leads to more complex morphologies if compared to AB diblock copolymers. Multicompartmental morphologies have been reported, both in the bulk15,16 and in solution. For micellar aggregates, different examples have been reported so far: a compartmentalized core,14,17,18 shell,19,20 corona,21 or, more specifically, Janus micelles of various shapes.22
The formation of such multicompartment systems in solution has been a topic of on-going great interest over the past decade, one of the main reasons being the localization of different chemical or physical properties in close proximity. Several driving forces have been identified to facilitate this so far: the presence of hydrocarbon and fluorocarbon segments within the same material,23,24 non-conventional architectures like miktoarm-star polymers,14,18,25,26 non-covalent interactions between adjacent blocks,20 kinetic control,27 or the combination of building blocks with an extraordinarily high incompatibility.28 All these approaches essentially used pure block terpolymers with different sequences and architectures as the starting material, whereas other examples employ mixtures of block co- and terpolymers,29 partial crosslinking,30solvent mixtures,31 or even the removal of one of the blocks to induce compartmentalization.32
A totally different approach has been employed by Manners and co-workers: here, the epitaxial growth of block copolymer unimers onto previously formed seed micelles from semi-crystalline polyisoprene-block-polyferrocenylsilane (PI-b-PFS)33 has lead to a controlled block extension and to the formation of either core-34 or corona-compartmentalized35 cylindrical micellar structures.
Our approach for the preparation of compartmentalized cylindrical micelles uses the controlled crosslinking of the polybutadiene domain of a bulk sample, followed by sonication-assisted dissolution of the as-crosslinked film. The employed UV-photoinitiator, Lucirin-TPO®, has been shown earlier to selectively crosslink the PB compartments of polybutadiene-block-poly(2-vinylpyridine)-block-poly(tert-butyl methacrylate) (BVT)16 and polystyrene-block-polybutadiene-block-poly(tert-butyl methacrylate) (SBT)36 block terpolymers in the bulk as well as that of BV diblock copolymer micelles in solution.37 The sonication applied after crosslinking allows the fabrication of size-tunable, core-crosslinked solution structures with different shapes and geometries, depending on the bulk morphology used as the template. This has been shown for spherical,38 disc-like,39 and cylindrical particles.40 In our case, a multicompartmental bulk morphology has been used: B420V280T790, the subscripts denoting the corresponding degrees of polymerization, exhibits a cylindrical bulk morphology with PB cylinders, covered by a double-handed P2VP helix and embedded within a PtBMA matrix.16 Different amounts of crosslinker and sonication times have been used to elucidate the effect on length and shape of the resulting cylindrical particles and we present a detailed investigation of the solution structure at various stages with cryogenic transmission electron microscopy (cryo-TEM). Solubility and rigidity of the aggregates are further adjusted via quaternization of the shell, P2VP, and viaester hydrolysis of the corona, PtBMA, to yield poly(methacrylic acid) (PMAA). In aqueous solutions at high pH, these cylinders then can form intramicellar interpolyelectrolyte complexes (im-IPECs) as a result of the attraction between positively charged P2VPq and negatively charged, deprotonated PMANa compartments. This is in analogy to our recent work on spherical multicompartment micelles from polybutadiene-block-poly(1-methyl-2-vinylpyridinium)-block-poly(sodium methacrylate) BVqMANa triblock terpolymers.19 Finally, we demonstrate the use of such sophisticated solution structures as templates for the controlled formation of noble metal nanoparticles within selected domains and the generation of hybrid structures.
Experimental
Materials
All solvents and reagents (Sigma-Aldrich, p.a. grade) were used without further purification. Lucirin-TPO (2,4,6-trimethylbenzoyldiphenylphosphine oxide), the UV-photoinitiator we used, was kindly provided by BASF and used as received.Polymer synthesis
The used block terpolymer, B420V280T790 (or B14V18T68165, where the subscripts denote the weight fractions of the corresponding blocks and the superscript is the overall molecular weight in kg mol−1), was synthesized via sequential living anionic polymerization in THF. Details about polymerization and characterization can be found elsewhere.16Bulk film casting
100 mg of the block copolymer and the respective amount of crosslinker (calculated according to the weight fraction of polybutadiene) were weighed into a small glass vial and dissolved in approximately 10 mL of THF. The vials were placed in a desiccator and the solvent was allowed to evaporate slowly (usually this takes around a week). Further, the whole process was carried out in the dark to prevent the UV-photoinitiator from decomposing. Afterwards, the polymer films were annealed in a vacuum oven at 50 °C for 48 hours.Crosslinking
Crosslinking of the as-cast and dried polymer films with additional UV-photoinitiator was carried out with an UV lamp (Hoehnle UVA-Hand 250) equipped with a quartz glass filter for 1 hour. Crosslinking efficiencies were estimated viaSoxhlet extraction of the crosslinked specimens with THF for 48 hours and were typically around 75%.Sonication
The crosslinked polymer films were swollen in a non-selective solvent (THF, dioxane, or different solvent mixtures where noted, the concentration was roughly 1 g/L) for at least 1 hour prior to sonication treatment. Sonication was carried out using a Branson W-450 sonifier equipped with a tungsten tip at 10% amplitude for 1 second, followed by a 2 seconds break. The sonication time will be denoted in minutes, where this is the summarized exposure time without taking into account the breaks in between. During the sonication the vials containing the micellar solutions were water-cooled.Quaternization of the P2VP block
Unless stated otherwise, quaternization of the P2VP compartment was carried out by adding a 10-fold excess of methyl iodide (compared to P2VP) to the micellar solution in THF. After 24 hours, the unreacted quaternizing agent was removed viadialysis against THF. After quaternization, the solutions usually exhibited a slight yellowish kink. We have shown earlier that the quaternization efficiency of P2VP to P2VPq under such conditions is around 80%.20Hydrolysis of PtBMA to PMANa
Directly after quaternization of the P2VP block, a 10-fold excess of HCl (32%) compared to the PtBMA was added and the solution was heated to 95 °C under reflux for 24 hours. Afterwards, dialysis was first carried out against a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF and deionized water, followed by another dialysis against a pH 10 buffer solution (Fluka, H3BO3/KCl/NaOH). To achieve a stable dispersion of the particles in water under these conditions, a short additional sonication treatment (10–30 seconds) was necessary.
:1 mixture of THF and deionized water, followed by another dialysis against a pH 10 buffer solution (Fluka, H3BO3/KCl/NaOH). To achieve a stable dispersion of the particles in water under these conditions, a short additional sonication treatment (10–30 seconds) was necessary.
Nanoparticle synthesis
The metal precursor salts (HAuCl4, K2PtBr6, and Na2PdCl4) were dissolved in the same pH 10 buffer solution used for the cylindrical particles (Fluka, H3BO3/KCl/NaOH). Then, 0.1 to 0.5 equivalent of the metal precursor (calculated according to the amount of P2VP present in solution) were added to the micellar solution. The solutions were kept in the dark during the whole process to avoid light-induced reduction of the metal salts. After 24 hours mixing, any non-complexed metal precursor was removed viadialysis against pH 10 buffer solution, and the remainder was reduced via either UV (UVA-Hand 250, Hoehnle) for 60 minutes or via the addition of 2 eq. (according to the metal precursors) of NaBH4. In the latter case, any excess of reducing agent was again removed via further dialysis.Dialysis
Dialysis membrane tubes (MWCO 12–14000 g/mol, Regenerated Celluloseester) were purchased from Spectra/Por. Prior to use, the tubes were immersed into de-ionized water for 1 h to open the pores.
Characterization
Dynamic light scattering (DLS)
DLS measurements were performed in sealed cylindrical scattering cells (d = 10 mm) at an angle of 90° on an ALV DLS/SLS-SP 5022F equipment consisting of an ALV-SP 125 laser goniometer with an ALV 5000/E correlator and a He–Ne laser with the wavelength λ = 632.8 nm. The CONTIN algorithm was applied to analyze the obtained correlation functions. Apparent hydrodynamic radii were calculated according to the Stokes–Einstein equation. Prior to the light scattering measurements the sample solutions were filtered using Millipore PTFE filters with a pore size of 5 µm.Transmission electron microscopy (TEM)
TEM images were taken with a Zeiss CEM902 EFTEM electron microscope operated at 80 kV or a Zeiss EM922 OMEGA EFTEM electron microscope operated at 200 kV. Both machines are equipped with an in-column energy filter. Samples were prepared through deposition of a drop of micellar solution (concentration typically 0.1 g/L) onto the TEM grid (Cu, 200 mesh). Afterwards the remaining solvent was removed with a filter paper.Cryogenic transmission electron microscopy (cryo-TEM)
A drop of the sample solution (c ≈ 0.5 g/L, solvent being either THF or water) was placed on a lacey carbon-coated copper TEM grid (200 mesh, Science Services, München, Germany), where most of the liquid was removed with blotting paper, leaving a thin film stretched over the grid holes. The specimens were shock vitrified by rapid immersion into liquid nitrogen (liquid ethane in the case of water as the solvent) in a temperature controlled freezing unit (Zeiss Cryobox, Zeiss NTS GmbH, Oberkochen, Germany) and cooled to approximately 90 K. The temperature was monitored and kept constant in the chamber during all the preparation steps. After freezing the specimens, they were inserted into a cryo-transfer holder (CT3500, Gatan, München, Germany) and transferred to a Zeiss EM922 OMEGA EFTEM instrument. Examinations were carried out at temperatures around 90 K. The transmission electron microscope was operated at an acceleration voltage of 200 kV. Zero-loss filtered images (ΔE = 0 eV) were taken under reduced dose conditions (100–1000 e/nm2). All images were registered digitally by a bottom mounted CCD camera system (Ultrascan 1000, Gatan), combined and processed with a digital imaging processing system (Gatan Digital Micrograph 3.9 for GMS 1.4).Scanning electron microscopy (SEM)
SEM was carried out on a Leo Gemini 1530. The specimens were dried under vacuum overnight and coated with approximately 2 nm Pd.Fourier transform infrared spectroscopy (FTIR)
FTIR measurements were performed using a Perkin Elmer Spectrum One FTIR spectrometer. The samples were introduced as freeze-dried powders.Results and discussion
Block terpolymer synthesis
The synthesis and characterization of the block terpolymer employed here, B420V280T790 (or B14V18T68165, where the subscripts denote the weight fractions of the corresponding blocks and the superscript is the overall molecular weight in kg/mol), as well as of several BVT block terpolymers of different compositions, were performed via sequential living anionic polymerization in THF at low temperatures and were described recently.16 For simplicity, the polymer will be denoted as BVT throughout the manuscript. The polymer structure and the microphase-separated bulk morphology are shown in Fig. 1.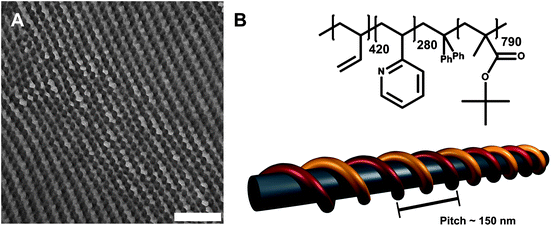 | ||
| Fig. 1 TEM micrograph showing the bulk morphology of B420V280T790 (A), the scale bar corresponds to 500 nm; structure, composition, and schematic bulk morphology, depicting a P2VP double helix with a pitch of roughly 150 nm (B).16 | ||
Fig. 1A shows a TEM micrograph of a microtome cut specimen after crosslinking in the bulk. To enhance the contrast, the P2VP phase was stained with iodine. Grey polybutadiene cylinders can be seen, covered with a P2VP double helix (black parts), both embedded in a PtBMA matrix (light grey, deteriorating through the incident electron beam). The helical pitch according to TEM measurements in the bulk is around 150 nm. The reason for the formation of a non-continuous shell around the PB core is most probably the high incompatibility between PB and P2VP.41 This has already been shown for multicompartment micelles prepared via direct dissolution of other BVT block terpolymers in acetone as a selective solvent.28 In that way, an additional interface is created in-between the first (PB) and the third block (PtBMA). The terpolymer composition, structure, and the proposed bulk morphology are shown in Fig. 1B.
Bulk crosslinking and sonication-assisted dissolution
To achieve a controlled and selective crosslinking of the PB domains, BVT bulk films with additional Lucirin-TPO® as UV-photoinitiator (5 to 20 wt% according to the PB content of the used block terpolymer) were cast from THF and afterwards crosslinked via UV irradiation for 60 minutes. The whole process is shown in Scheme 1. This particular photoinitiator has already been used to crosslink PB in the bulk16 and in solution.37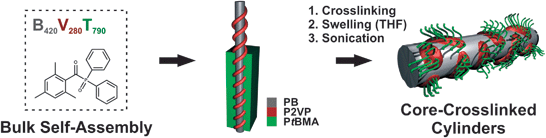 | ||
| Scheme 1 Bulk film casting and crosslinking of BVT, followed by sonication-assisted dissolution of the core-crosslinked BVT domains. | ||
After crosslinking, the polymer films are first swollen in a non-selective solvent, typically THF. Subsequently, sonication-assisted dissolution leads to size-tunable core-crosslinked cylindrical particles. Apparent hydrodynamic radii obtained viadynamic light scattering (DLS) in THF dependent on the applied sonication time are shown in Fig. 2A.
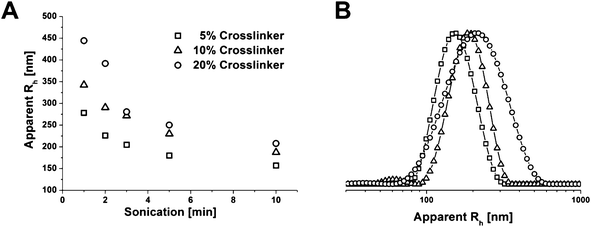 | ||
| Fig. 2 Apparent hydrodynamic radii obtained viaDLS for different amounts of crosslinker and sonication times (A); DLS CONTIN plots for cylinders after 5 minutes sonication and with 5 (-□-), 10 (-△-), and 20% (-○-) crosslinking agent (B). | ||
The apparent size of the crosslinked particles in THF decreases with longer sonication times, although, this seems to level out after 10 minutes. This can be explained in a way that larger structures provide more surface and are thus stronger affected by the sonication treatment. Similar observations have been made for disc-like39 and cylindrical Janus structures,42 and fiber-like cylindrical micelles of polyisoprene-block-polyferrocenylsilane (PI-b-PFS).43 In the latter case the micelles exhibited a semi-crystalline PFS core and, therefore, were shown to be rod-like and rigid whereas the aggregates obtained by crosslinking of the PB core of BVT block terpolymers are supposed to behave more worm-like. The amount of crosslinker incorporated also seems to play a role, at least for the particle sizes after short sonication times. For 20% of TPO, 〈Rh〉z,app = 440 nm was obtained after 1 minute sonication whereas the corresponding values are 〈Rh〉z,app = 340 nm for 10% and 〈Rh〉z,app = 280 nm for 5% of added crosslinking agent. Of course, one has to keep in mind that these values are based on the evaluation according to the CONTIN algorithm, assuming a spherical particle shape and are, therefore, only partially applicable. Fig. 2B shows representative DLS CONTIN plots for BVT core-crosslinked cylinders after 5 minutes sonication. At a closer look, one can also see a second distribution of smaller fragments, at least for 10% of added TPO (-△-), with 〈Rh〉z,app ≈ 60 to 70 nm. A tentative explanation might be that this occurs due to loose aggregates being present where the crosslinking has not or only partially been successful. If the overall crosslinking efficiency is determined viaSoxhlet extraction with hot THF for 48 hours, around 75% of insoluble material remains for both 10 and 20% of crosslinker added. In that respect, and for simplicity reasons, for the rest of the manuscript samples with 10% crosslinker and 5 minutes sonication time will be used unless otherwise mentioned.
Characterization of core-crosslinked cylinders
The structure of the core-crosslinked BVT cylinders in THF was investigated using transmission electron microscopy (TEM). Therefore, samples were taken both directly after crosslinking and swelling in THF and after sonication, and deposited onto carbon-coated TEM grids. The resulting micrographs are shown in Fig. 3. Fig. 3A depicts the particles directly after swelling and without further sonication treatment. Cylindrical objects with lengths of several µm can be seen. As expected, they appear rather like worms instead of rigid rods. This can be attributed to the low glass transition temperature of 1,2-polybutadiene. Even after crosslinking, the PB core can still swell to a certain extent in good solvents like THF. Similar effects have been shown for spherical micelles with a PB core in acetone and THF/dioxane after crosslinking with S2Cl2.28 During sonication, the cylinders break into smaller fragments which are found in TEM. This is shown in Fig. 3B after 5 minutes of sonication treatment. The resulting length distribution seems rather broad. 300 cylinders were measured using the ImageJ software, yielding a number-averaged length of Ln = 850 nm and a polydispersity index, Lw/Ln = 1.27. If compared, the z-averaged length from TEM measurements (Lz = 1350 nm) exceeds the value obtained by DLS (〈Rh〉z = 230 nm) by a factor of ∼6. A possible explanation for this discrepancy is that the cylinders may adopt a coiled conformation in solution due to the flexible PB core, whereas deposition onto a TEM grid leads to stretching. It is obvious that the crosslinked PB forms the core of these cylindrical aggregates. However, the expected helix around the PB core, formed by P2VP as shown in the bulk for this material, cannot be directly seen from the TEM micrographs. Instead, dark spots appear on both sides of the PB cylinder. To elucidate this, the P2VP compartment was stained by iodine, which is known to form charge-transfer complexes with P2VP and enhance contrast in TEM.28 This is shown in Fig. 3C. More or less randomly distributed dark spots can be observed on the cylindrical PB core. Obviously, the P2VP helix is rearranged through the swelling of the terpolymer in THF. The presence of THF induces changes in the interfacial balance of the system and so the situation reported for the bulk is altered. To exclude drying artifacts during the sample preparation, cryo-TEM measurements in THF were performed as well. This is shown in Fig. 3D.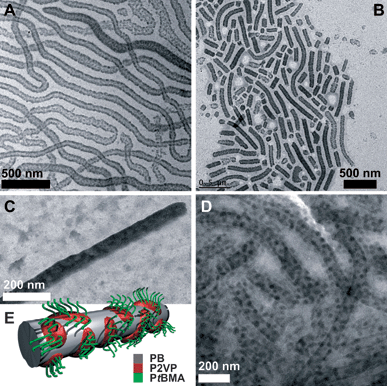 | ||
| Fig. 3 TEM micrographs of BVT cylinders after crosslinking (A), sonication for 5 minutes (B), staining with iodide (C); cryo-TEM micrograph of BVT in THF after crosslinking and sonication for 5 minutes (D), proposed solution structure of the block terpolymer particles (E). | ||
Cryo-TEM is an almost in-situ technique which allows the unperturbed visualization of self-assembled structures directly in solution after flash vitrification.27 In the cryo-TEM image in Fig. 3D cylindrical objects with an approximate core width of 70–80 nm can be seen, the crosslinked PB core of the particles. They bear randomly distributed, spherical dark patches with a size of 15–20 nm. These, to our opinion, are the discontinuous P2VP corona parts after a rearrangement from the original helix bulk structure in THF as a good solvent. Surrounding this compartmentalized cylindrical core is the corona, PtBMA, not visible here. This is summarized in the proposed solution structure depicted in Fig. 3E. According to the block sequence BVT, the PtBMA corona is emerging from the P2VP patches present on the PB core of the cylinders, generating a surface of PB in-between, which is directly exposed to the solvent/the solvent-swollen corona. To further evaluate the effect of the sonication treatment on the length distribution of the cylinders, the length of approx. 300 particles was measured from TEM micrographs for 10% crosslinker and sonication times of 1, 5, and 10 minutes. The resulting distributions are shown in Fig. 4A and the obtained number- and weight-average lengths are listed in Fig. 4B together with the corresponding apparent hydrodynamic radii from DLS measurements. After 1 minute of sonication (black distribution), cylinder lengths up to 4 µm were found, the average Ln was 950 nm and Lw/Ln = 2.19. After 5 minutes (blue distribution), smaller values with Ln = 850 nm and Lw/Ln = 1.27 were obtained. A few cylinders with lengths up to 2.5 µm were found but this was rather exceptional. Finally, after 10 minutes (red distribution), the length decreased drastically, giving Ln = 390 and Lw/Ln = 1.32.
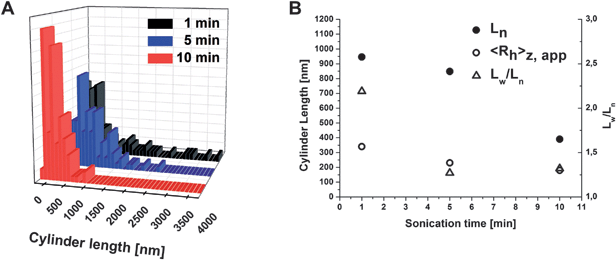 | ||
| Fig. 4 Cylinder length distributions for BVT cylinders after crosslinking with 10% TPO and 1 (black), 5 (blue), and 10 minutes (red) sonication in THF (A); obtained values for number-average cylinder length, apparent 〈Rh〉z from DLS experiments, and Lw/Ln (B). | ||
To conclude, the PB part of BVT can be crosslinked in the bulk in a controlled fashion. Above 10 weight%, relative to the PB compartment, the amount of added crosslinker seems to have no influence on the final degree of crosslinking or, respectively, the amount of insoluble material generated according to Soxhlet extraction. The length of the resulting cylindrical particles and, to a certain extent, the cylinder length distribution, can be controlled viasonication in THF as a non-selective solvent. The helical arrangement of the P2VP middle block in the bulk is changed to a patchy, compartmentalized corona in solution, most probably due to the swelling and, hence, volume increase of the P2VP through THF.
Cylinder modification
Both the solubility and the surface characteristics of the particles can be influenced by polymer analogous reactions after self-assembly. This is desirable as in that way those structures can be preserved in different environments, e.g. non-selective solvents. Such modifications include crosslinking of the micellar core28 or shell,44 quaternization reactions, or corona transformations such as ester hydrolysis.20For BVT, as the core is already crosslinked, two particular modifications can be further pursued: first, the quaternization of the middle block, P2VP, and, second, the hydrolysis of the PtBMA compartment to poly(sodium methacrylate) (PMANa). Quaternization would lead to P2VPq, a strong polyelectrolyte, and to the generation of charges along the cylinders whereas a PMANa corona would render the cylinders water-soluble at high pH and, further, would introduce hydrogen bonding with P2VP or interpolyelectrolyte complex formation (IPECs) with P2VPq. This has been shown recently for spherical micelles of BVT,28BVqT,20 and BVqMAA block terpolymers.19 If not mentioned otherwise, all modification were performed on cylinders priorly crosslinked with 10% TPO in the bulk and sonicated at a concentration of roughly 1 g/L for 5 minutes, and thus having lengths in the range of 1 µm (cf.Fig. 2 and 3).
Quaternization and crosslinking of the P2VP shell
Quaternization of the P2VP shell was carried out in THF using MeI as methylation agent during 24 hours at RT. Earlier work has shown that an approximate quaternization efficiency of ca. 80% is reached via this method due to the localization of the nitrogen close to the backbone in P2VP.19 One reason for this modification is that P2VPq should be less soluble in THF and, therefore, the helical structure of the discontinuous shell around the PB core of the cylinders might be preserved even in solution. However, after quaternization in pure THF the same, patchy structures were obtained as those shown in Fig. 3. If, on the contrary, 20 volume% of MeOH were added to the THF solution of the BVT cylinders (the bulk film of the terpolymer was swollen and sonicated for 5 minutes in that mixture), and the quaternization reaction was carried out in that solvent mixture, the outcome was different (Fig. 5). Fig. 5A shows a TEM micrograph of BVqT cylinders deposited directly after quaternization in THF:MeOH 80:20. A dark double helix wrapped around the PB core can be seen. To definitely exclude any drying artifacts here, the same sample solution was subjected to cryo-TEM: this is shown in Fig. 5B. Also here, the double helix can be seen; the cylinders again have a thickness of about 70 nm, the helix itself has a diameter of roughly 25 nm, and the helical pitch, i.e. the distance necessary for a full rotation of one of the strands, was found to be approximately 170 nm.
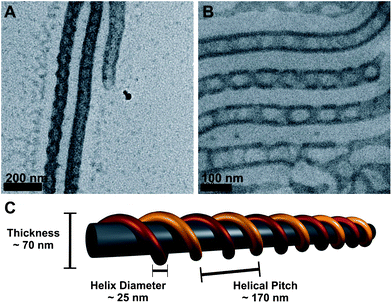 | ||
| Fig. 5
TEM micrograph of core-crosslinked and quaternized BVqT cylinders cast from THF:MeOH 80:20 solution (A); cryo-TEM image of the same sample (B); proposed solution structure highlighting important distances (C), the two strands of the P2VPq double helix are shown in yellow and red for easier visualization. | ||
Presumably, the used solvent mixture prevents the rearrangement of the non-continuous P2VPq shell around the PB cylindrical core and the reported bulk morphology for BVT is preserved. In the bulk, values of 50 nm for the cylinder thickness and 150 nm for the helical pitch were reported,16 the slightly increased values in solution can be explained by a slight swelling in THF:MeOH 80:20. To our knowledge, this is the first example for such helical micellar morphology in solution. Other reported solution-based examples used multivalent counterions to direct the structure of cylindrical polymer brushes45 or multi-amines to trigger the coiling of cylindrical, charged block terpolymer micelles.46 Liu and co-workers observed the rearrangement of spherical micelles into double and triple helices for poly(butyl methacrylate)-block-poly(cinnamoyl ethyl methacrylate)-block-poly(tert-butyl acrylate) over several months.47 Several other studies were carried out on bulk structures of polystyrene-block-polybutadiene-block-poly(methyl methacrylate) (SBM) block terpolymers.48–50 Here, also a double-stranded helical arrangement of the middle block, PB, was found.
A further possible modification of the BVT cylinders is the hydrolysis of the PtBMA corona, generating poly(sodium methacrylate) (PMANa) and rendering the particles water-soluble at pH-values higher than 5.51 If this step is performed after prior quaternization of the P2VP compartment (BVqT), intramicellar interpolyelectrolyte complexes (im-IPECs) can be formed between the positively charged P2VPq and the negatively charged PMANa. This has been recently shown for spherical micelles of polybutadiene-block-poly(1-methyl-2-vinylpyridinium)-block-poly(methacrylic acid) (BVqMAA) triblock terpolymers, where the resulting im-IPECs formed a patchy, discontinuous shell.19,20Hydrolysis of the PtBMA was performed directly after quaternization of the P2VP (and removal of any excess of quaternizing agent, MeI, viadialysis) by adding HCl (10 eq. compared to PtBMA) to the BVqT cylinders in THF and heating to 95 °C for 24 hours. This is also shown in Scheme 2. Directly after hydrolysis, the reaction mixture was dialyzed against an aqueous pH 10 buffer solution. Assuming that im-IPEC formation occurs during the dialysis process and taking into account the chain lengths of the participating blocks (280 for P2VPq and 790 for PMANa), an average excess chain length of 510 units of PMANa should form the corona of the resulting BVqMANa cylinders.
 | ||
| Scheme 2 Quaternization of P2VP to P2VPq and ester hydrolysis from PtBMA to PMANa. | ||
The aqueous dispersions were almost opaque, compared to slightly opalescent solutions beforehand. This indicates that some aggregation occurs. One possible explanation is that the ester hydrolysis did not proceed to full completion, as indicated by FTIR-measurements (not shown here). Residual PtBMA could be identified through characteristic bands at 1720 cm−1 (–COOR), 1394 cm−1 and 1368 cm−1 (both t-Bu).20 Nevertheless, a few additional seconds of sonication were enough to break up the formed aggregates. This leads to partial clarification and, supposably, deaggregation, resulting in stable dispersions of the BVqMANa cylinders. Fig. 6A shows a SEM image of such particles after deposition onto previously cleaned silicon wafers. The width of the PB core is still around 70–80 nm and the collapsed shell/corona can be seen by the uneven appearance of the cylinder surface. The same sample was used for TEM analysis, as shown in Fig. 6B. Despite the additional sonication, the cylinders seem to connect, forming network-like structures. The P2VPq compartments can be visualized as dark, randomly distributed spots on the PB core surface, further surrounded by a rather fuzzy grey haze, most probably the PMANa corona.
 | ||
| Fig. 6 SEM (A) and TEM (B) images of BVqMANa particles deposited from aqueous solution at pH 10; cryo-TEM micrographs of BVqMANa cylinders in aqueous solution at pH 10 (C and D). | ||
We also performed cryo-TEM on the BVqMANa particles: this is shown in Fig. 6C and D (at higher magnification). As observed in Fig. 6B, a pronounced tendency for the cylinders to connect or form networks was observed also in the cryo-TEM (Fig. 6C) throughout the whole sample. At higher magnification, Fig. 6D shows darker patches/compartments distributed on the PB core of the cylinders. Those are, most presumably, the result of the im-IPEC formation between P2VPq and PMANa under these conditions. The excess PMANa, forming the corona, cannot be distinguished here due to low contrast.
The conclusions that can be drawn from this part are that basically the same post-self-assembly modifications could be performed on BVT cylinders as shown earlier for spherical analogues with different compositions.20 This results in positively charged BVqT or water-soluble and (overall) negatively charged BVqMANa particles. In the latter case, the overall charge can be estimated by comparing the block lengths of the positively charged (P2VPq280) and negatively charged (PMANa790) block. Further, the presence of both PMANa and P2VPq within the same particle leads to the formation of im-IPECs, creating a (for that system) new type of discontinuous shell, as shown by cryo-TEM measurements. Most remarkably, however, is that the observed swelling and rearrangement of the P2VP helix around the PB core in THF (Fig. 3) can be suppressed by the addition of 20 vol% MeOH during the quaternization (Fig. 5).
Formation of polymer/nanoparticle hybrids
The ability of certain polymers to attract, stabilize, or selectively bind metal nanoparticles (or the corresponding salt precursors) has led to the preparation of a variety of different composite or hybrid structures. Referring to the solution state, mostly spherical52,53 and cylindrical block copolymer brushes have been employed as templates.40,54–57 Among the most commonly reported metal nanoparticles incorporated are Au,40,52Ag,40,58Pt,52,55 and Pd,52,54,59 but also metal oxides and sulfides have been reported.60–62 Interest in these structures is mainly driven by combining the properties of both polymers (processability, flexibility, and the huge variety of accessible building blocks with today's synthetic methods) and metal nanoparticles (optical properties, catalytic activity,63 chemical sensing64). Suitable polymeric “stabilizers” or compartments are both positively and negatively charged polyelectrolytes like poly(vinylpyrrolidone) (PVP),65 poly(vinylpyridines) (P2VP and P4VP),40poly(ethylene imine) (PEI),66poly(N-isopropylacrylamide) (PNiPAAm),52 poly(2-dimethylaminoethyl methacrylate) (PDMAEMA),54,55 and their quaternized analogues,55poly(acrylic acid) (PAA)58 or PMAA.67 In the case of PNiPAAm, the resulting hybrid particles showed switchable “on–off” catalytic activity of the embedded metal-NPs due to the LCST and the resulting volume transition of the smart PNiPAAm shell upon heating.52 We have recently shown that in the case of spherical multicompartment micelles with a discontinuous im-IPEC shell (P2VPq and PMANa) and a PMANa corona the formation of Au–NP takes place predominantly at the IPEC/corona interface and within the PMANa corona.19We generated Au, Pt, and Pd nanoparticles within BVqMANa particles, using HAuCl4, K2PtBr6, and Na2PdCl4 as metal precursors. The loading was calculated according to the amount of P2VPq present and was typically between 10 and 50 mol% metal precursor. Reduction of the precursors (and NP formation) was carried out using UV irradiation for 60 minutes (Au) or a two-fold excess of NaBH4 (Pt, Pd) in solution and, in the latter case, subsequent dialysis to remove any unreacted NaBH4. We had two main intentions: first, the transfer of the already established protocol to more complex systems and a variety of metal nanoparticles and, second, to compare different metal precursors concerning their preferential deposition. In the ideal case, a metal precursor could be found that selectively forms NPs within the P2VPq/im-IPEC compartments. TEM and cryo-TEM images of BVqMANa cylinders/metal nanoparticle hybrids are shown in Fig. 7. We will denote the hybrids as NP@BVqMANa.
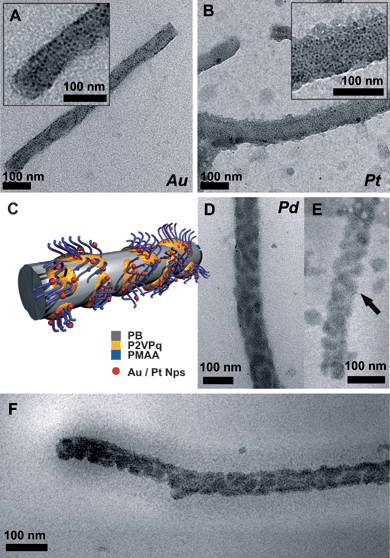 | ||
| Fig. 7 TEM micrographs of BVqMANa cylinders/metal nanoparticle hybrids deposited from aqueous solution; Au (A), Pt (B), the insets showing a higher magnification, and Pd (D), the proposed solution structure (C); cryo-TEM images from Pd@BVqMANa (E and F). | ||
Fig. 7A shows Au@BVqMANa at 10% loading. The Au–NPs (round black dots, approx. 5–10 nm in size, see inset in Fig. 7A) are randomly distributed all over the cylinders, indicating a location within both the corona, PMANa, and the im-IPEC/P2VPq domains. If preferential localization of the NPs within the P2VPq domains would occur, the patches would be visible. This is in accordance with previous results obtained for spherical BVqMANa multicompartment micelles.19 The same accounts for Pt@BVqMANa, shown in Fig. 7B, the inset shows a higher magnification. However, the situation is different for Pd@BVqMANa, depicted in Fig. 7D: here, the Pd-NPs are located in patches, distributed along the PB core of the cylinders. This indicates that a preferential loading of the P2VPq/im-IPEC domains took place. Pd–NPs have been generated both in P2VP68 and PAA69 domains of block copolymers, but in both cases no “competing” polyelectrolyte has been present. We therefore propose that in the case of BVqMANa NP generation and stabilization are favored within the im-IPEC domains containing P2VPq. To exclude drying artifacts, we also performed cryo-TEM on the aqueous solution containing Pd@BVqMANa (Fig. 7E and F). Fig. 7F depicts a single cylindrical particle; the PB core is covered with dark im-IPEC patches also containing the generated Pd-NPs. Further, the PMANa corona can be visualized as a grey shade around the core with a thickness of approximately 50–60 nm. Fig. 7E shows an enlargement of a different BVqMANa micelle. In this case, even the double-helical structure of the im-IPEC shell is preserved, indicated by the arrow.
Conclusions
We have successfully transferred a complex, compartmentalized morphology of a BVT triblock terpolymer from the bulk to the solution state via selective, UV-induced crosslinking of the polybutadiene block. In that way, size-tunable cylindrical particles with a PB core, a discontinuous P2VP shell, and a PtBMA corona could be prepared. Both the appearance and the solubility of these cylindrical particles can be adjusted viapolymer-analogous modifications such as quaternization, crosslinking (both P2VP), ester hydrolysis (PtBMA), or the generation of hybrid structures via the controlled deposition of metal nanoparticles within selected compartments. This methodology represents a facile toolbox to control the functionality of soft, polymer-based particles with a size of 100 nm up to several µm. We further showed that by selecting a solvent mixture of appropriate polarity, the swelling of the P2VP domains can be suppressed and cylinders covered by a P2VP double helix can be generated in organic solvents.In combination with our recent work on BVT block terpolymers in the bulk,16 in solution,28 and on the corresponding terpolymers after quaternization and/or ester hydrolysis,19,20 we think that we can convincingly show that the BVT system provides a straightforward and versatile platform for the generation of sophisticated and multifaceted nanostructures both in the bulk and in solution. One driving-force for that has already been identified: the high incompatibility of PB and P2VP, resulting in the formation of discontinuous morphologies over a broader composition range than expected16 and, also, within environments of unforeseen selectivity.28 Concerning future work, the emphasis will be put on utilizing both hydrophilic and hydrophobic compartments present in such self-assembled structures for the selective deposition of NPs or other guest molecules.
Acknowledgements
The VolkswagenStiftung is gratefully acknowledged for financial support within the framework “Complex Materials”. We would like to thank Melanie Förtsch for some of the TEM micrographs. The authors thank Antti Nykänen for help with the drawings in Figure 5.References
- Z. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290 CrossRef CAS.
- S. I. Stupp, V. LeBonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384–389 CrossRef.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- A.-V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082–5104 CrossRef CAS.
- N. Hadjichristidis and S. Pispas, Adv. Polym. Sci., 2006, 200, 37–55 CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- D. Baskaran and A. H. E. Müller, Prog. Polym. Sci., 2007, 32, 173–219 CrossRef CAS.
- J. Zhu and R. C. Hayward, Angew. Chem., Int. Ed., 2008, 47, 2113–2116 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS.
- C.-A. Fustin, V. Abetz and J.-F. Gohy, Eur. Phys. J. E, 2005, 16, 291–302 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS.
- U. Breiner, U. Krappe, V. Abetz and R. Stadler, Macromol. Chem. Phys., 1997, 198, 1051–1083 CrossRef CAS.
- F. Schacher, J. Yuan, H. G. Schoberth and A. H. E. Müller, Polymer, 2010, 51, 2021–2032 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245–1249 CrossRef CAS.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS.
- F. Schacher, E. Betthausen, A. Walther, H. Schmalz, D. V. Pergushov and A. H. E. Müller, ACS Nano, 2009, 3, 2095–2102 CrossRef CAS.
- F. Schacher, A. Walther and A. H. E. Müller, Langmuir, 2009, 25, 10962–10969 CrossRef CAS.
- H. Schmalz, J. Schmelz, M. Drechsler, J. Yuan, A. Walther, K. Schweimer and A. M. Mihut, Macromolecules, 2008, 41, 3235–3242 CrossRef CAS.
- A. Walther and A. H. E. Müller, Soft Matter, 2008, 4, 663–668 RSC.
- K. Skrabania, H. von Berlepsch, C. Böttcher and A. Laschewsky, Macromolecules, 2010, 43, 271–281 CrossRef CAS.
- S. Kubowicz, J. F. Baussard, J. F. Lutz, A. F. Thünemann, H. von Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262–5265 CrossRef CAS.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2009, 25, 13718–13725 CrossRef CAS.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2008, 41, 8815–8822 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS.
- F. Schacher, A. Walther, M. Ruppel, M. Drechsler and A. H. E. Müller, Macromolecules, 2009, 42, 3540–3548 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2005, 39, 765–771.
- R. H. Zheng, G. J. Liu and X. H. Yan, J. Am. Chem. Soc., 2005, 127, 15358–15359 CrossRef CAS.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2008, 24, 12001–12009 CrossRef CAS.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, ACS Nano, 2010, 4, 1907–1912 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef.
- H. Wang, W. Lin, K. P. Fritz, G. D. Scholes, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2007, 129, 12924–12925 CrossRef CAS.
- A. Walther, A. Göldel and A. H. E. Müller, Polymer, 2008, 49, 3217–3227 CrossRef CAS.
- A. Walther, A. S. Goldmann, R. S. Yelamanchili, M. Drechsler, H. Schmalz, A. Eisenberg and A. H. E. Müller, Macromolecules, 2008, 41, 3254–3260 CrossRef CAS.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS.
- A. Walther, X. Andrè, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187–6198 CrossRef CAS.
- A. Walther, J. Yuan, V. Abetz and A. H. E. Müller, Nano Lett., 2009, 9, 2026–2030 CrossRef CAS.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, John Wiley & Sons, New York, 4th edn, 1999 Search PubMed.
- A. Walther, M. Drechsler, S. Rosenfeldt, L. Harnau, M. Ballauff, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2009, 131, 4720–4728 CrossRef CAS.
- G. Guèrin, H. Wang, i. Manners and M. A. Winnik, J. Am. Chem. Soc., 2008, 130, 14763–14771 CrossRef CAS.
- G. Sun, N. S. Lee, W. L. Neumann, J. N. Freskos, J. J. Shieh, R. B. Dorshow and K. L. Wooley, Soft Matter, 2009, 5, 3422–3429 RSC.
- Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Yuan, L. Harnau, M. Ballauff and A. H. E. Müller, Soft Matter, 2009, 5, 379–384 RSC.
- S. Zhong, H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2008, 4, 90–93 RSC.
- J. Dupont, G. Liu, K.-I. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS.
- U. Breiner, U. Krappe, V. Abetz and R. Stadler, Macromol. Chem. Phys., 1997, 198, 1051 CrossRef CAS.
- U. Krappe, R. Stadler and I. Voigt-Martin, Macromolecules, 1995, 28, 4558–4561 CrossRef CAS.
- H. Jinnai, T. Kaneko, K. Matsunaga, C. Abetz and V. Abetz, Soft Matter, 2009, 5, 2042–2046 RSC.
- J. Leyte and M. Mandel, J. Polym. Sci., Part A: Polym. Chem., 1964, 2, 1879–1891 Search PubMed.
- M. Ballauff and Y. Lu, Polymer, 2007, 48, 1815–1823 CrossRef CAS.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617–620 CrossRef CAS.
- Q. Ye, X. Wang, H. Hu, D. Wang, S. Li and F. Zhou, J. Phys. Chem. C, 2009, 113, 7677–7683 CrossRef CAS.
- J. Yuan, F. Schacher, M. Drechsler, A. Hanisch, Y. Lu, M. Ballauff and A. H. E. Müller, Chem. Mater., 2010, 22, 2626–2634 CrossRef CAS.
- J. Yuan and A. H. E. Müller, Polymer, 2010, 51, 4015–4036 CrossRef CAS.
- R. S. Yelamanchili, A. Walther, A. H. E. Müller and J. Breu, Chem. Commun., 2008, 489–491 RSC.
- Y. Lu, Y. Mei, M. Schrinner, M. Ballauff, M. W. Möller and J. Breu, J. Phys. Chem. C, 2007, 111, 7676–7681 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- N. Duxin, F. Liu, H. Vali and A. Eisenberg, J. Am. Chem. Soc., 2005, 127, 10063–10069 CrossRef CAS.
- F. Polzer, D. A. Kunz, J. Breu and M. Ballauff, Chem. Mater., 2010, 22, 2916–2922 CrossRef CAS.
- X. Yan, G. Liu, F. Liu, B. Z. Tang, H. Peng and A. B. Pakhomov, Angew. Chem., Int. Ed., 2001, 40, 3593–3596 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CrossRef CAS.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- D. K. Park, S. J. Lee, J.-H. Lee, M. Y. Choi and S. W. Han, Chem. Phys. Lett., 2010, 484, 254–257 CrossRef CAS.
- L. Bai, H. Zhu, J. S. Thrasher and S. C. Street, ACS Appl. Mater. Interfaces, 2009, 1, 2304–2311 CrossRef CAS.
- V. Kozlovskaya, E. Kharlampieva, S. Chang, R. Muhlbauer and V. V. Tsukruk, Chem. Mater., 2009, 21, 2158–2167 CrossRef CAS.
- S. Akasaka, H. Mori, T. Osaka, V. H. Mareau and H. Hasegawa, Macromolecules, 2009, 42, 1194–1202 CrossRef CAS.
- D. Li, J. R. Dunlap and B. Zhao, Langmuir, 2008, 24, 5911–5918 CrossRef CAS.
Footnote |
| † Present address: Institut für Organische und Makromolekulare Chemie, Friedrich-Schiller-Universität Jena, Humboldtstraße 10, D-07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de |
| This journal is © The Royal Society of Chemistry 2011 |