Sensing with fluorescent nanoparticles
Luca
Baù
a,
Paolo
Tecilla
b and
Fabrizio
Mancin
a
aDipartimento di Scienze Chimiche, Università di Padova, via Marzolo 1, 35131, Padova, Italy. E-mail: Fabrizio.mancin@unipd.it; Fax: +39 049 8275666; Tel: +39 049 8275239
bDipartimento di Chimica, Università di Trieste, via Giorgeri 1, 34127, Trieste, Italy. E-mail: ptecilla@units.it; Fax: +39 0405583903; Tel: +39 0405583925
First published on 21st October 2010
Abstract
Fluorescent chemosensors are chemical systems that can detect and signal the presence of selected analytes through variations in their fluorescence emission. Their peculiar properties make them arguably one of the most useful tools that chemistry has provided to biomedical research, enabling the intracellular monitoring of many different species for medical and biological purposes. In its simplest design, a fluorescent chemosensor is composed of a fluorescent dye and a receptor, with a built-in transduction mechanism that converts recognition events into variations of the emission properties of the fluorescent dye. As soon as fluorescent nanoparticles became available, several applications in the field of sensing were explored. Nanoparticles have been used not only as better-performing substitutes of traditional dyes but also as multivalent scaffolds for the realization of supramolecular assemblies, while their high surface to volume ratio allows for distinct spatial domains (bulk, external surface, pores and shells) to be functionalized to a comparable extent with different organic species. Over the last few years, nanoparticles proved to be versatile synthetic platforms for the implementation of new sensing schemes.
 Luca Baù | Luca Baù obtained his PhD in 2010 at the University of Padova, where he worked on the synthesis of organic molecules and nanosystems for use as intracellular fluorescent sensors, photosensitizers for photodynamic therapy and drug delivery systems. He is currently a post doctoral fellow at the same institution. His research interests lie in the area where organic chemistry and biology meet, his current focus being the synthesis of fluorescent probes for biomedical applications. |
 Paolo Tecilla | Paolo Tecilla received his PhD in chemistry from the University of Padova with Professor Tonellato in 1989. After postdoctoral work with Professor A. D. Hamilton at the University of Pittsburgh, he took a Lecturer position at the University of Padova. In 1998, he moved to the University of Trieste, where he is now a full Professor of Organic Chemistry. His scientific interests are in the field of supramolecular chemistry and focus on the metallocatalysis of hydrolytic reactions, fluorescent chemosensors and model membranes. |
 Fabrizio Mancin | Fabrizio Mancin received his PhD in Chemistry from the University of Padova with Professor Tonellato in 1999. On the same year he took a Lecturer position at the University of Padova, where he is now associate Professor. In 2002 he spent one year at the University of Toronto as post-doc researcher with Prof. Jik Chin. His research interest is focused on supramolecular chemistry, particularly on the development of artificial metallonucleases and on the design of fluorescence molecular chemosensors. Recently he has broadened his interest to include nanosystems for biomedical applications. |
1. Introduction
Fluorescent chemosensors are arguably one of the most useful tools that chemistry has provided to biomedical research.1 These molecular systems can detect and signal the presence of selected analytes through variations of their fluorescence emission properties.2 The unique role they play in biological studies stems from the combination of few fundamental properties. First, the possibility of integrating specific receptors into the molecular structure of the chemosensor provides a potentially exquisite selectivity. In addition, their molecular size minimizes physical perturbations to such delicate systems as living cells. Most importantly, the use of fluorescence emission as the analytical signal allows for highly sensitive measurements to be obtained with low-cost, even portable, instrumentation by exploiting different sensing modes, such as intensity, lifetime and polarization detection. Combined with the high spatial resolution of fluorescence microscopy, these features have enabled the intracellular monitoring of many different species for medical and biological purposes. Other important applications have also been pursued, from analytical tools for environmental and biochemical assays, to switches and logical gates for molecular information processing.3Since the seminal work of Tsien in the 1980s,4 the design of fluorescent chemosensors has been constantly evolving as more and more sophisticated sensing modes have been explored. In its simplest form, a fluorescent chemosensor is composed of a fluorescent dye and a receptor integrated into the same molecule.2 A built-in transduction mechanism converts recognition events into variations of the fluorescent dye emission properties. The design of a chemosensor may differ in the level of integration of the two units, in the transduction mechanism or in other subtler details. The receptor and dye units may be part of the same π system, or connected by electronically isolating spacer moieties.2 They may even be discrete entities, self-organized through non-covalent interactions.5 The sensing scheme may rely on emission quenching by the substrate, activation or deactivation of energy or electron transfer processes, alteration of internal charge transfer (ICT) excited states, or molecular movements resulting in a variation of the distance between two photoactive components.1 The latter effect may cause, in turn, emission quenching, formation of excimers, or switching of distance-dependent energy or electron transfer processes.1 More sophisticated designs use antenna groups or multi-dye systems, and may lead to yet other effects such as signal amplification or ratiometric signaling.6
As far as the output signal is concerned, intensity measurements are by far the most popular choice, mainly because they require relatively inexpensive instrumentation, in contrast with the more demanding lifetime and polarization measurements.1,2 Variations in the emission spectrum of a chemosensor may consist either in a general decrease of the intensity (quenching or on-off chemosensors), an increase of the emission (turn-on or off-on chemosensors) or a variation of the spectral shape (ratiometric chemosensors). While off-on behavior is usually preferred over the less sensitive on-off response, ratiometric signaling is by far the most useful. By using the ratio between the emission intensities at two different wavelengths as the analytical signal, the effect of such factors as photobleaching (the progressive decrease of emission due to photoinduced degradation), sensor concentration, polarity of the microenvironment, and fluctuations of the excitation source can be minimized.2
The signaling unit of a fluorescent chemosensor is a key component of the assembly, often limiting its practical application. A huge number of organic dyes, fluorescent proteins and luminescent metal complexes have been used for this purpose over the years. Even the most efficient among conventional fluorophores, however, suffer from some inherent limitations. Photobleaching, limited brightness (the product of extinction coefficient and fluorescence quantum yield) and short lifetimes are serious drawbacks in many applications.7 In the context of intracellular measurements, chemical interactions with biomolecules are another cause of concern.8 In the last decade, fluorescent nanoparticles have emerged as a new class of fluorophores with the potential to overcome these limitations.9 Some nanoparticles, such as quantum dots, are intrinsically fluorescent, while others can be made fluorescent by appropriate doping with fluorescent dyes or luminescent metals.
Quantum dots (QDs) are crystals of semiconductor materials a few nanometres in size, with a size-dependent electronic structure which is responsible for some peculiar photophysical properties.7,10 Their broad absorption and narrow emission spectrum can be finely tuned by varying the size and composition of the particles, which are also remarkably bright and stable against photobleaching. These features make QDs especially useful both in energy transfer applications and in simultaneous assays exploiting multiple excitation and emission wavelengths.7,10 Organic dyes and luminescent transition metal complexes can be also embedded into silica or polymer nanoparticles.9,11 The inclusion of dyes in a polymer matrix results in decreased emission quenching and increased photostability due to oxygen and solvent exclusion.11 Many of the optical properties of quantum dots can thus be obtained with biocompatible materials and easy, low-cost preparation procedures.
As soon as fluorescent nanoparticles became available, several applications in the field of sensing were explored (Fig. 1). Nanoparticles were only used, at first, as better-performing substitutes of traditional dyes in biolabeling applications and fluorescence-based assays,12 but it soon became clear that these unique objects, combining the properties of extended solids and molecular species, had much more to offer than that. The high amount of reactive sites found on their surface make them ideal multivalent scaffolds for the realization of supramolecular assemblies, while their high surface to volume ratio allows for distinct spatial domains (bulk, external surface, pores and shells) to be functionalized to a comparable extent with different organic species. Over the last few years, nanoparticles proved to be versatile synthetic platforms for the implementation of new chemosensing schemes.
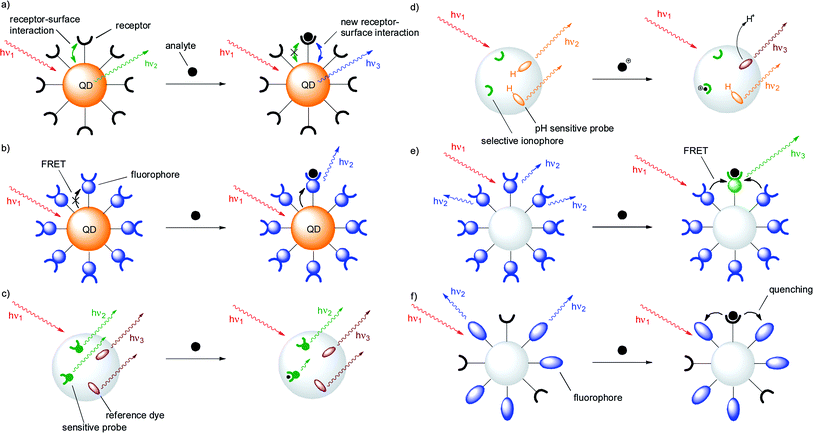 | ||
| Fig. 1 Representative sensing schemes in fluorescent nanoparticles: (a) Analyte binding to a surface receptor causes spectral changes in a quantum dot by altering its surface electronic structure; (b) analyte binding to a surface receptor affects an energy transfer process from a quantum dot to an organic fluorophore; (c) analyte binding to a nanoparticle embedded chemosensor in the presence of a co-embedded reference dye triggers a ratiometric response in a PEBBLE sensor; (d) ion binding to a nanoparticle embedded ionophore is reported by a co-embedded pH-sensitive dye in a PEBBLE sensor; (e) analyte binding to one surface-grafted chemosensor affects several surrounding units, resulting in signal amplification; (f) receptors and fluorophores are individually grafted to the surface of a nanoparticle in a self-organized sensor. | ||
2. Sensing quantum dots
In the most naïve approach, a chemosensor would be built by simply sticking a fluorescent dye and a receptor together in the most synthetically straightforward way. Unfortunately, such a strategy is rarely effective without a transduction mechanism in place. The recognition of an analyte by the receptor does not necessarily affect the emission of the fluorophore, unless the analyte itself is a fluorescence quencher. Even so, only the less sensitive on-off response can be obtained. The peculiar physical properties of quantum dots, however, seem to offer a way to exploit this attractively simple approach. The emission of quantum dots originates from the recombination of photogenerated electron-hole pairs, which takes place predominantly on the surface of the nanocrystals. As a consequence, the binding of charged species, such as metal cations, close to the surface of quantum dots may affect and even increase their emission. This concept provides the unique possibility to create fluorescent chemosensors by simply grafting ligands to the particles (Fig. 1a). The first demonstration of this concept was described by Rosenzweig and his co-workers, who used CdS quantum dots coated with polyphosphate, L-cysteine, or thioglycerol, as water-soluble metal ion sensors.13 The nature of the ligands had a dramatic effect on the type and selectivity of luminescence response. Polyphosphate-capped CdS QDs showed no selectivity and displayed an on-off behavior. Thioglycerol-capped QDs were selective towards copper and iron ions over several other transition metal ions with an on-off response. Finally, L-cysteine-capped QDs responded selectively to zinc ions with an off-on behavior and detection limits below 1 μM.Similar sensing schemes have been later exploited for several others analytes including metal cations (Fig. 2), anions and organic molecules, even if in some cases a simple quenching of the QD emission by the substrate is likely responsible for the observed behaviour.14 Sensitivity towards organic molecules was achieved by decorating the QD surface with more sophisticated recognition units: calixarenes were used to bind and signal acetylcholine (Fig. 3)14c while cyclodextrins allowed the discrimination of enantiomeric amino acids.14a
 | ||
| Fig. 2 Cu(I) sensing system based on PEGylated CdS clusters: (a) cluster structure; (b) photoluminescence increase upon Cu(I) addition in water, as a consequence of the perturbation of the surface electronic structure of the quantum dots (adapted from ref. 14b). | ||
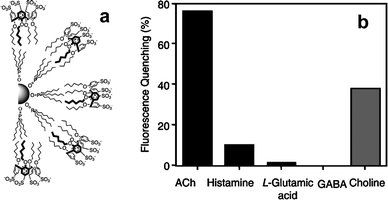 | ||
| Fig. 3 CdSe/ZnS QDs for acetyl choline sensing: (a) Schematic representation of calixarenes coated QDs; (b) fluorescence quenching of QDs in the presence of neurotransmitter compounds and choline (1.6 mM, PBS buffer pH 7.4; adapted from ref. 14c). | ||
Besides the modification of surface properties, other mechanisms typical of molecular chemosensors can also be exploited. For instance, a photoinduced electron transfer (PET) from the coating molecules to the valence band of an excited QD results in emission quenching. If the electron donor is bound to a substrate, this process may be interrupted and the emission restored. Several groups have recently explored this mechanism. Singaram and co-workers (Fig. 4) showed that the addition of a boronic acid substituted viologen to commercially available CdSe/ZnS QDs with carboxylic groups on the surface results in a strong quenching of the emission from the QDs. Likely, electrostatic interactions cause the absorption of the viologen derivative on the anionic surface of the QDs and the achieved proximity allows the viologen to quench the QD emission trough a PET mechanism.15 Subsequent addition of glucose converts the boronic groups into the corresponding glucose boronate esters, which are worse electron donors, at the same time decreasing the affinity of the quencher molecule for the QD surface. As a result, the QD emission increases. Glucose concentrations in the 2–20 millimolar range can be detected in water at pH 7.4. Amine functionalized QDs can also be used in the same way: in this case, however, the quenching efficiency of the viologen derivative is lower, probably because of weaker interactions with the positively charged QD surface.
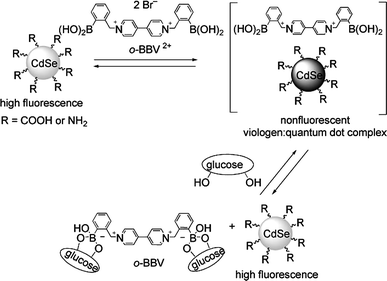 | ||
| Fig. 4 Working scheme the turn-on glucose sensing system of Singaram et al.: a viologen quencher detaches from the surface of quantum dots upon esterification of pendant boronic acids with glucose (reproduced from ref. 15). | ||
Mareque-Rivas and co-workers (Fig. 5) used a similar approach to develop a sensing system for cyanide anions.16 A 2,2′-bipyridine copper(II) complex was adsorbed on the trioctylphosphine oxide coating layer of CdSe QD, resulting in the quenching of the QD emission by an electron transfer from the excited QD to the metal complex. Addition of cyanide ions caused the displacement of the copper(II) ions from the bipy complex, thus restoring the QD emission.
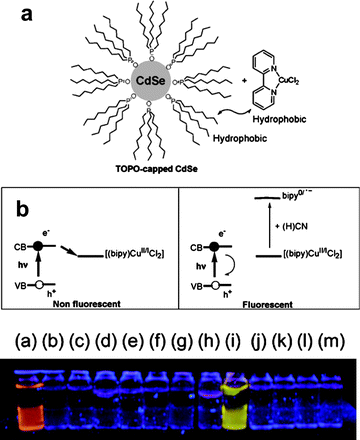 | ||
| Fig. 5 Cyanide sensing QD studied by Mareque-Rivas et al.: (a) QD schematic structure; (b) sensing scheme and naked-eye fluorescent detection of cyanide: the analyte displaces a quenching ion from a surface-grafted ligand, and the emission is restored as a result (vial (i) contains different potentially interfering anions than in the other vials; no Cu(I) complex in vial (a); adapted from ref. 16). | ||
Santra and co-workers coated the surface of CdS:Mn/ZnS QDs with azacrown derivatives to obtain a cadmium(II) sensor. In this case, however, the sensitivity was in the millimolar range and several transition metal ions interfered with the cadmium ion detection.17 Finally, QDs coated with a urea-bearing ferrocene derivative were used by Callan and co-workers to selectively detect fluoride ions in the sub-millimolar range (detection limit 0.074 mM in chloroform).18 Apparently, the binding of fluoride to the urea receptors alters the reduction potential of the ferrocenyl groups causing the interruption of the PET process.
A more sophisticated PET-based strategy was devised by Benson and coworkers (Fig. 6), who designed a single-molecule biosensor for maltose in the micromolar range.19 The authors prepared a chimeric protein composed by a maltose-binding (MBP) domain and a metallothionein (MT) domain. The MBP-MT protein was selectively conjugated with a ruthenium(II) complex and grafted to a CdSe QD through the cysteine residues of the MT domain. The ruthenium(II) complex quenches the QD emission through a PET process. Maltose binding to the MBP domain causes the protein to undergo a severe conformational change that increases the distance between the Ru(II) complex and the QD, thus interrupting the PET process and restoring the emission.
 | ||
| Fig. 6 Working scheme of the PET-based maltose sensing system studied by Benson et al.: maltose binding to a surface-grafted MBP protein conjugated with a quencher triggers a conformational change in the protein and restores the emission (reproduced from ref. 19). | ||
The most largely exploited sensing mechanism, however, is the activation/deactivation of Förster resonance energy transfer (FRET) processes (Fig. 1b).7 Very recently, P.-T. Chou and coworkers developed a ratiometric sensor for potassium ions based on a mixture of two 15-crown-5 functionalized CdSe/ZnS QDs of different sizes.20 The different size of the QDs makes them suitable as a FRET pair. Potassium ions, when present, are bound in a sandwich-like manner by the crown ether units grafted to different nanoparticles. This results in the activation of an energy transfer process between different particles and in the consequent modification of the emission spectrum of the mixture.
Several other sensing schemes involving FRET processes have been designed. The general idea is to use the analyte recognition to modulate the energy transfer process between the QD and a dye appended to its surface. As in the previous example, such modulation will produce a ratiometric response of the sensing system to the analyte.
Molecular beacons (fluorescent DNA sensors based on hybridization) are the most explored application. In a typical probe, a QD is functionalized with a DNA sequence complementary to the target sequence, while the sample to be analyzed is conjugated, using standard labeling procedures, with a fluorophore whose absorption spectrum overlaps with the emission of the QD. If the target DNA is present, successful hybridization results in the activation of a FRET process where the QD emission decreases and the fluorophore emission becomes stronger.7 This strategy has also been exploited in DNA-functionalized gold nanoparticles, with the particles acting as quenchers.21
Analyte detection may also be achieved by pursuing the reverse process, i.e. interruption or modulation of FRET due to analyte-induced displacement of a fluorophore from a QD-bound receptor. Such an approach was first explored by Mattoussi and co-workers in 2003 using CdSe QDs conjugated with a maltose binding protein (MBP).22 A sugar derivative functionalized with a non-emissive dye was bound to the protein resulting in FRET quenching of the QD emission. Upon maltose addition, the quencher is displaced by the protein and the QD emission restored. A 2,4,6-trinitrotoluene (TNT) sensor was also realized in a subsequent work (Fig. 7) using a CdSe/ZnS QD, an anti-TNT antibody and a quencher-conjugated TNT analogue.23
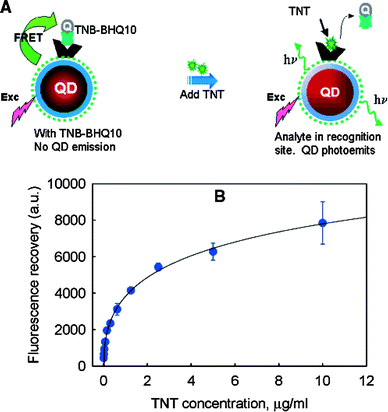 | ||
| Fig. 7 TNT sensing QDs based on the displacement of a quencher conjugate (TNT-BHQ10) from a QD-bound anti-TNT antibody. (a) Sensing scheme; (b) fluorescence recovery (reproduced from ref. 23). | ||
A similar approach was recently explored by Willner and coworkers (Fig. 8) using a boronic acid derivative instead of MBP.24 Displacement of dye-labeled galactose or dopamine derivatives from the boronic acid functionalized CdSe/ZnS QDs resulted in the interruption of FRET and gave rise to a ratiometric response. Submillimolar concentrations of galactose and glucose and micromolar concentrations of dopamine could be detected in water at pH 7.4.
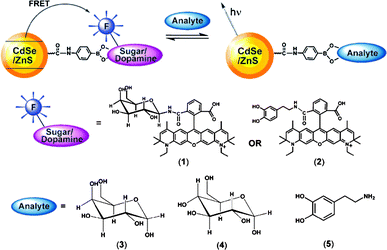 | ||
| Fig. 8 Competitive analysis of monosaccharides or dopamine using fluorophore-labeled galactose or dopamine and boronic acid functionalized QDs (reproduced from ref. 24). | ||
Conformational changes of MBP upon maltose binding were also exploited by the Mattoussi group to modulate FRET.25 In a work published simultaneously with the previously discussed PET system of Benson et al.,19 an engineered MBP was functionalized with a Cy3 dye and bound to CdSe trough a C-terminal pentahystidine fragment. In the resulting nanobiosensor, the FRET process is highly efficient and QD excitation at 300 nm results in an emission spectrum containing mainly the Cy3 features. Maltose addition increases the donor–acceptor distance resulting in a decrease of the Cy3 emission.
As an alternative to donor–acceptor distance variations, FRET can also be modulated if the absorption spectrum of the acceptor is modified by analyte recognition. Such changes may decrease or increase the overlap between the QD emission and the acceptor absorption thus modulating the efficiency of FRET. This approach has been explored by conjugating pH-sensitive dyes to QDs. In particular, Nocera and co-workers functionalized polymer-coated CdSe/ZnS QDs with a pH-sensitive squaraine dye.26 At pH values below 7.5, the overlap between squaraine absorption and QD emission is high and the emission spectrum is dominated by the features of the organic dye. When the pH is raised, squaraine absorption is blue shifted and FRET becomes less efficient: the emission originates predominantly from quantum dots. This results in a remarkable ratiometric behavior that allows the determination of pH with better accuracy than the dye alone in highly scattering environments.
3. Sensing dye-doped particles: PEBBLEs
The unusual optical properties of QDs have attracted great interest and stimulated the development of many ingenious nanosensors, which were discussed in the previous paragraph. However, QDs have only been used in most cases as “better” organic dyes, and the same sensing schemes could be, and often have been, exploited with conventional dyes. More complex sensing schemes, exploiting the inherent multivalency of nanoparticles to trigger collective and cooperative processes, have been explored using dye-doped nanoparticles.The first approach to the realization of dye-doped sensing nanoparticles was proposed in the late 1990s by Kopelman and co-workers for studies in live cells and dubbed PEBBLE (Probes Encapsulated By Biologically Located Embedding or, more recently, Photonic Explorers for Bioanalysis with Biologically Localized Embedding).8,27 In its simplest embodiment, the PEBBLE approach involves the encapsulation of classical fluorescent chemosensors into water-soluble polymeric nanoparticles (Fig. 1c, d). The semi-permeable and transparent nature of the matrix allows the analyte to interact with a dye that reports the interaction via a change in the emitted fluorescence. This sensing scheme may look the same as that of classical chemosensors, but a closer inspection reveals several distinct advantages. First, the inclusion of chemosensors into nanoparticles minimizes the interaction with cell components such as proteins or membranes. Multifunctional systems implementing complex sensing schemes can also be easily realized. For instance, reference dyes can be added to enable ratiometric sensing, or ionophore/chromoionophore combinations can be used in order to take advantage of highly selective but non-fluorescent ionophores.
As an example, Kopelman and co-workers28 reported on a ratiometric sensor for intracellular oxygen obtained by including a ruthenium complex [Ru(dpp)3]2+ and the dye Oregon Green 488 in silica PEBBLEs with diameters ranging from 100 to 400 nm. In the presence of oxygen, the fluorescence emission of [Ru(dpp)3]2+ is strongly quenched while the emission of Oregon Green is not affected, thus allowing the ratiometric determination of oxygen levels. These PEBBLEs were introduced into living cells using gene gun delivery techniques and used for the monitoring of variations of the oxygen level in the cytosol. Similar results were reported by the same research group29 using 120 nm diameter ormosil PEBBLEs doped with a platinum porphyrin (platinum(II) octaethylporphine ketone), an oxygen-sensitive dye, and octaethylporphine as a reference dye. Compared to the previous example, this ormosil PEBBLE sensor shows higher sensitivity, a wider linear range and longer excitation and emission wavelengths. This last feature is especially important in intracellular measurements, where the background noise from fluorescent biomolecules (“autofluorescence”) is often a source of confusion. Moreover, these PEBBLE sensors display excellent reversibility and stability with respect to leaching and long-term storage. Other examples of oxygen sensing PEBBLEs have been reported by Kopelman et al. and others using organic polymeric nanoparticles.30
The versatility of PEBBLEs was proved by targeting several different analytes. pH is probably one of the most obvious targets, not least because of the large number of pH-sensitive dyes available. Core-shell silica nanoparticles were used by Wiesner and co-workers for ratiometric pH sensing in vitro (Fig. 9).31 Tetramethylrhodamine, which is pH-insensitive, was covalently included into 50 nm silica particles that were subsequently coated with a 10 nm thick shell doped with the pH-responsive dye fluorescein. The core-shell structure of the particles, whereby sensitive units are excluded from the solvent-inaccessible core and thus prevented from contributing to the background signal, results in increased sensitivity.
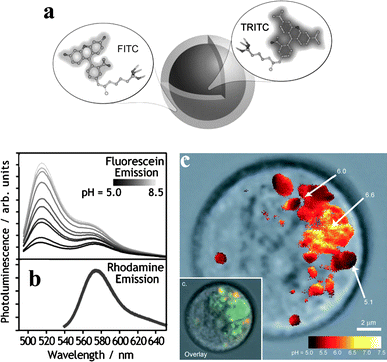 | ||
| Fig. 9 Core-shell silica nanoparticles doped with rhodamine and fluorescein for ratiometric pH sensing: (a) nanoparticle scheme; (b) ratiometric response; (c) false-color ratiometric imaging of pH in various intracellular compartments (inset: overlaid image; adapted from ref. 31). | ||
A different sensing scheme was demonstrated by Wolfbeis and co-workers.32 The emission intensity of a fluorescent nanoparticle is modulated by the pH-dependent absorption of a polyaniline coating layer. The organic conducting polymer acts as a screen by limiting the amount of exciting light reaching the fluorophores in a variable fashion, depending on the amount of protonated residues. By using fluorescent dyes with absorption centred on either side of the isosbestic point of polyaniline, it was possible to obtain an increase or a decrease of fluorescence with increasing pH depending on the excitation wavelength. With a pKa around 7, polyaniline is especially suitable for pH sensing in a physiological environment.
The PEBBLE approach has also been extended to more challenging analytes. Anions are notoriously hard for chemosensors to detect in aqueous environments, as most anion-selective receptors rely on hydrogen bonding. Other systems are based on less sensitive transduction mechanisms, such as collisional quenching. Nanoparticles may offer a way out of this conundrum by providing a platform for sensing schemes that would not be feasible at the single-molecule level. One of the first examples of anion-responsive nanosensors was devised by Kopelman and co-workers33 by exploiting the same principles used in the realization of ion-selective electrodes and fiber optic probes.34 In this approach, chloride ions are extracted into a solid matrix by a selective ionophore (Fig. 1d). The simultaneous co-extraction of H+ gives rise to a local pH change, which is monitored by a pH-sensitive fluorescent dye and used for the indirect determination of chloride. The authors encapsulated a commonly used chloride-selective ionophore and a pH-sensitive fluorescent dye into hydrophobic poly(decyl methacrylate) nanoparticles. An ionic additive was added in order to maintain the ionic strength. At physiological pH these PEBBLEs have a limit of detection of 0.2 mM, with a linear range of 0.4–190 mM. These PEBBLEs were also tested in C6 glioma cells after insertion with a gene gun.
In a recent and related work, Mohr and co-workers prepared a ratiometric sensor by doping polyacrylamide nanoparticles with the chloride-sensitive fluorescent probe lucigenin and a sulforhodamine derivative as a reference dye.35 The nanoparticles were successfully delivered to Chinese hamster ovary cells and mouse fibroblasts with the help of the transfection reagent Lipofectamine. Even though the efficiency of encapsulated lucigenin (Stern–Volmer constant 53 M−1) is remarkably lower than that of the free dye (Stern–Volmer constant 250 M−1), the co-encapsulation of a reference dye makes these sensors much more reliable, as they are not affected by changes in sensor concentration.
The versatility of the PEBBLE approach is also demonstrated by the possibility of using multifunctional nanoparticles with a magnetic core in order to recover and recycle the sensors36 or to achieve an enhanced sensitivity.37
4. Signal amplification in dye-doped particles
The confinement of a relatively large number of dye molecules in the small volume of the nanoparticles may trigger collective phenomena which would not be otherwise observed (Fig. 1e). These effects were first described by McBranch, Whitten and their co-workers, who showed in 2001 that the absorption of a cationic cyanine on the surface of silica nanoparticles leads to the formation of extended J-aggregates.38 Such “self-assembled polymers” detect the presence of the negatively charged anthraquinone disulfonate through fluorescence quenching. The measured quenching constant is four orders of magnitude greater than that of the free dye in solution. Such an increment is due to both the enhanced binding of the substrate to the polycationic cyanine assembly on the particle and the amplified quenching of an exciton delocalized over many dye units.A second example of collective processes made possible by the spatial organization of dyes in PEBBLE-like nanoparticles has been reported by Montalti and coworkers.39 The authors prepared silica nanoparticles (30 nm in diameter) in which a dansylated polyamine, obtained by reaction of dansyl chloride with 3-[2-(2-aminoethylamino)ethylamino]propyltrimethoxy-silane, is covalently included in the silica matrix by co-polymerization with tetraethoxysilane (TEOS) in basic conditions. Due to the presence of the polyamine receptor, the nanoparticles bind metal ions with relatively high affinity and the addition of copper(II), cobalt(II) and nickel(II) results in quenching of the fluorescence emission of the dansyl unit. Interestingly the observed quenching is much stronger than expected, allowing detection of nanomolar concentration of analyte. This effect has been explained with the occurrence of multiple simultaneous quenching processes favoured by the dense packing of the fluorescent dyes in the nanoparticles. As a consequence of this tight packing, a single quenching ion bound to one receptor can interact with several nearby fluorophores. The results obtained allowed to estimate that a single copper ion is able to quench up to 13 dansyl units, leading to strong signal amplification.
In a subsequent study, again Montalti and co-workers reported the first example of signal amplification for a turn-on system.40 They coated the surface of 20 nm commercial silica nanoparticles with a trialcoxysilane derivative of the fluoroionophore TSQ (Fig. 10). In the absence of zinc ions the excited states localized on the TSQ molecules deactivate mostly in a nonradiative way because the radiative path is quenched by an intramolecular proton and electron transfer.41 When Zn(II) is added in a small amount, it is complexed by the TSQ units (with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand to metal stoichiometry) interrupting the quenching process. Fluorescence switch-on is accompanied by a red-shift of the absorption of the complexed units. Such shift allows an energy-transfer process that funnels the excitation energy absorbed by the surrounding uncomplexed TSQ molecules towards the fluorescent excited states. As a result, the observed emission increase is greater than that achieved by individual TSQ units in solution. The effect is small, but interestingly it is not observed if the TSQ units are dispersed inside the silica matrix.41 This suggests that such cooperative effects require a high density of dyes, which is more easily obtained by surface functionalization.
:1 ligand to metal stoichiometry) interrupting the quenching process. Fluorescence switch-on is accompanied by a red-shift of the absorption of the complexed units. Such shift allows an energy-transfer process that funnels the excitation energy absorbed by the surrounding uncomplexed TSQ molecules towards the fluorescent excited states. As a result, the observed emission increase is greater than that achieved by individual TSQ units in solution. The effect is small, but interestingly it is not observed if the TSQ units are dispersed inside the silica matrix.41 This suggests that such cooperative effects require a high density of dyes, which is more easily obtained by surface functionalization.
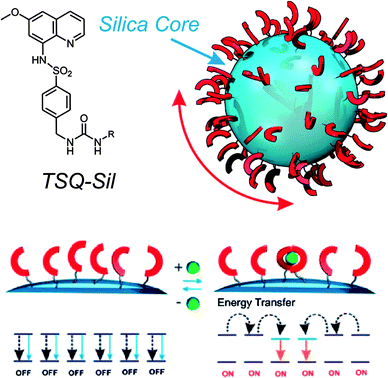 | ||
| Fig. 10 Amplified turn-on response described by Montalti et al. for TSQ-coated silica nanoparticles (adapted from ref. 40). | ||
An energy transfer process was also exploited by Prasad and co-workers (Fig. 11) to obtain signal amplification in an ORMOSIL pH sensor.42 The authors prepared a triethoxysilyl derivative of naphthalenylvinylpyridine (NVP) by reaction with (3-isocyanatopropyl)triethoxysilane (ICTES). The sol–gel polymerization in aqueous micellar solution of the NVP-ICTES derivative with vinyltriethoxysilane (VTES) affords sphere-shaped silica nanoparticles, about 30 nm in size, in which the dye is covalently linked to the silica matrix and uniformly distributed throughout the nanoparticle volume. NVP responds ratiometrically to pH, with a fluorescence red-shift from a high-energy neutral form (blue) to a low-energy protonated form (yellow). The red-shifted absorption of the protonated form partially overlaps with the tail of the blue emission from the neutral form. In the NVP-loaded PEBBLEs, the two protonation states exist in two distinct spatial compartments, the acid form being confined to the surface layer, and an energy transfer from the inner neutral dye to the protonated form on the surface is observed. This process, which is made possible by the organization of dye molecules in the nanospace inside the particles, amplifies the emitted signal and increases the sensitivity of the sensor with respect to the free dye in solution, where FRET is not observed. Moreover, the energy transfer process increases the apparent pKa of the dye to 6.4, a suitable value for biological applications. A similar behaviour was also reported with two-photon excitation.
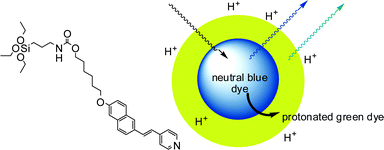 | ||
| Fig. 11 Core-shell silica nanoparticles doped with a two-photon pH sensitive dye.42 Protonation of the dye in the outer shell result in energy transfer from the neutral dyes in the core to the protonated ones in the shell, with consequent signal amplification. | ||
A further evolution in the use of energy transfer to obtain signal amplification has been recently reported by McNeill and co-workers (Fig. 12) for oxygen sensing.43 The authors embedded the oxygen-sensitive dye Pt(II) octaethylporphine (PtOEP) inside conjugated polymers nanoparticles made of polyfluorene derivatives poly(9,9-dihexylfluorene) (PDHF) and poly(9,9-dioctylfluorene) (PFO). In this case, the polymer matrix of the nanoparticles is not a mere scaffold but an active component of the sensing scheme. The conjugated polymer absorbs the excitation light and efficiently transfers it to the PtOEP dye (the energy transfer efficiency is 87% for the PFO nanoparticles). This results in a strong emission of the dye at 650 nm, with a phosphorescence brightness more than 1000 times higher than that of conventional oxygen sensing dyes, and 5–10 times higher than that of PtOEP-doped silica particles. This achievement allowed the authors to push oxygen detection down to the single-particle level. The cellular uptake of these particles was also demonstrated.
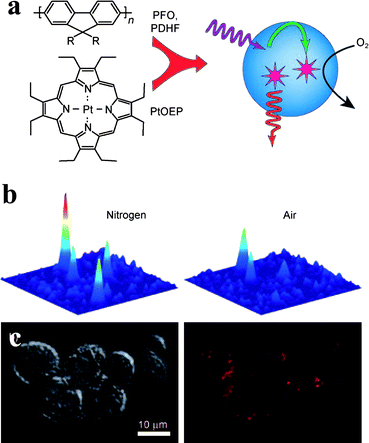 | ||
| Fig. 12 Conjugated polymer nanoparticle for molecular oxygen monitoring: (a) nanoparticle and sensing scheme: the incident light is efficiently absorbed by the polymeric matrix and transferred to an oxygen chemosensor, thus enhancing the effective brightness of the assembly; (b) single-particle intracellular oxygen sensing (adapted from ref. 43). | ||
5. Self-organized sensors on nanoparticles
The examples of the previous section show how the spatial confinement of several photoactive units within the small volume of a nanoparticle can be exploited in order to achieve cooperative or collective effects, such as signal amplification, which significantly improve the performance of the sensing agent. The degree of cooperation between different components can be further increased by taking advantage of the dense packing of active units that is achievable on the surface of nanoparticles and of the possibility of organizing those units in distinct spatial domains. In the sensing quantum dots described earlier, a similar process is likely to occur when QDs coated with weakly-binding molecules, such as cysteine or thioglycerol, bind metal cations at micromolar concentration.13 Several functional groups from different molecules are probably cooperating to bind the analyte. As we will show in this paragraph, many examples of the employment of such effects are based on polymeric nanoparticles.A first demonstration of this approach was reported by Raymo and Cejas.44 The authors functionalized the surface of commercially available silica nanoparticles (20 nm diameter) with 2,7-diazapyrenium dications and used them for the detection of dopamine in water solutions. Addition of dopamine or catechol results in emission quenching as consequence of the formation of donor/acceptor complexes between the dye and the substrate. A tighter surface coverage of the nanoparticles results into an increased sensitivity, suggesting that a cooperative effect is at work: the spatially close 2,7-diazapyrenium units on the surface cooperate in binding the substrate, favouring the formation of acceptor/donor/acceptor sandwich complexes.44a
In this example the cooperation between the active units ensures increased affinity and sensitivity. Still, whole chemosensors are linked to the particles and the working scheme of the assembly is not fundamentally different from that of the individual units. A different strategy has also been explored, in which fluorescent dyes and receptors are separate elements that spontaneously self-assemble on a proper template (Fig. 1f).5a,b In 2003, we functionalized commercially available silica particles (20 nm in diameter) with triethoxysilyl derivatives of picolinamide, a Cu(II)-selective ligand, and of the fluorescent dye dansylamide (Fig. 13).45a Grafting these components to the surface of nanoparticles ensures the reciprocal spatial proximity required for Cu(II) bound to the ligand units to quench the emission from the fluorophores. In a 9:1 DMSO–water solution, the coated silica nanoparticles selectively detect copper ions down to micromolar concentrations and the operative range of the sensor can be tuned by simply changing the ratio between the two components. Most importantly, it was possible to obtain clear evidence of the cooperation of the ligand subunits in creating binding sites with an increased affinity for the substrate.45a The preparation of a small library of coated silica nanoparticles with the same ligand and different fluorophores proved the versatility of this approach.45b The emission spectra of the library span a wide range of wavelengths, from 300 to 600 nm (correspondingly, excitation wavelengths are in the range 285–466 nm), allowing the choice of the most suitable sensor for any desired application. Using a stronger ligand for the Cu(II) ion, we were able to exploit collective processes, similar to those reported by Montalti et al. and discussed in the previous section,39 in which a single metal ion is capable of quenching the emission of about 10 surrounding dye molecules, thus extending the detection limit of the sensor down to the nanomolar range.
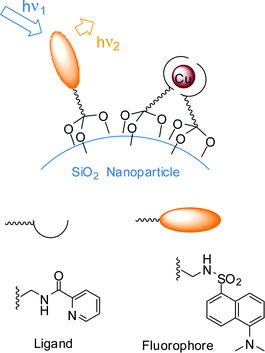 | ||
| Fig. 13 Working scheme of Cu(II) sensor self-organized on silica nanoparticles: receptors and fluorophores are individually grafted to the surface.45 | ||
Following a similar strategy, a three-component self-assembled nanosensor was subsequently developed by Larpent and co-workers.46 The authors used polystyrene nanoparticles (with a diameter of 15–20 nm, prepared by polymerization in microemulsion) as a template, cyclam as a metal ion ligand and a BODIPY derivative as a fluorescent reporter. The hydrophobic dye was entrapped within the polymeric matrix by impregnation, while the ligand was covalently attached to the polymer backbone. Binding of the Cu(II) ions to the cyclam ligand results in a strong quenching of BODIPY fluorescence probably via an energy transfer process (FRET) from the dye to the metal ion complex. Although cyclam binds Zn(II), Ni(II) and other transition metal ions with high affinity, the sensor responds selectively to Cu(II). This peculiar kind of selectivity is due to the overlap of the absorption spectrum of the Cu complex with the emission spectrum of the dye, a necessary condition for FRET which is not observed in the other complexes. In this case, collective quenching was much more efficient than in the previous examples, with one single metal ion able to quench the emission of up to 44 dye molecules. This cooperative behaviour resulted in a very high sensitivity, the detection limit being in the nanomolar range.
An important evolution of the self-organization approach has been recently described by Martínez-Máñez, Sancenón and co-workers (Fig. 14).47 The authors reported a nanoparticle-based colorimetric sensor for organic carboxylates which relies on the polarity-controlled merocyanine-spiropyran equilibrium: the red cyanine form is the prevailing species in polar environments, while the pale yellow spiropyran form prevails in non-polar media. A thiourea derivative and a spirobenzopyran dye bearing trialkoxylane moieties were anchored to the surface of commercial silica nanoparticles with a diameter of 20 nm. Addition of organic carboxylates to a suspension of nanoparticles in water at pH 7 leads to a visible color change from red to pale yellow. The interaction of carboxylates with the thiourea groups induces a polarity change of the environment surrounding the dye units, causing the cyclization of the dye to the spiropyran form. No effects are observed upon addition of other inorganic anions. It must be underlined that this is one of the very few examples of self-organized sensors where a detection mechanism is at play that is different from emission quenching by the substrate.
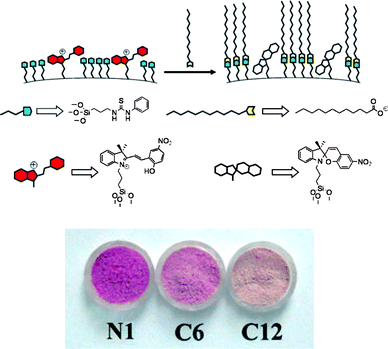 | ||
| Fig. 14 Working scheme of silica nanoparticle-based sensor for carboxylates. The nanoparticles are functionalized with thiourea (blue) and merocyanine (red). Addition of lipophilic carboxylates (yellow) results in the cyclization to the spirocyclic form and a color modulation from pink-red to pale yellow (lower, N1: functionalized nanoparticles; C6, hexanoate added, C12; dodecanoate added; adapted from ref. 47). | ||
As mentioned in the case of sensing quantum dots, nanoparticles may also act as a template for the formation of analyte binding sites with high affinity from crowded monodentate donors. One example is the ability of dye doped silica nanoparticle to act as Cu(II) sensors.48 In this case, silica itself provides the receptor units of the sensor: the network of acidic silanol groups on the surface forms the metal binding sites. Once bound to the silica network, Cu(II) ions are close enough to the fluorescent units entrapped in the particle to quench their emission, thus producing a measurable signal. The formation of nanoparticles by co-polimerization of tetraethoxysilane and a triethoxysilyl-dansyl derivative leads therefore, at the same time, to the formation of metal ion binding sites as well as to the covalent grafting of a fluorescent reporter in their proximity, thus giving rise to a simple conversion of fluorescent dyes into sensors.
This approach can be extended to the detection of other transition metals if the nature of the functional groups present on the particle surface is changed. 50 nm dansylamide doped silica nanoparticles coated with a thin layer of thiols can signal the presence of Pb(II) ions in millimolar concentration in water, through a quenching mechanism.49 This kind of on-off sensor could even be converted to a more useful ratiometric probe (Fig. 15). As discussed in the PEBBLE paragraph, this should be easily achieved in nanoparticle-based chemosensors by the addition of a second, substrate insensitive, dye that provides the reference signal. However, in the case of quenching substrates, such a strategy can be ineffective since the quencher does not distinguish between the two dyes and ultimately quenches both. Differentiation between the responses of two embedded dyes was obtained by confining the different components within spatially separated domains. We prepared 50 nm multilayer silica nanoparticles made of a 40 nm core doped with a methoxynaphthalene dye, a 7 nm thick insulating silica shell, a second 3 nm thick shell doped with a dansylamide derivative and a thiol-functionalized surface layer (Fig. 15). Pb(II) ions, bound to the binding sites spontaneously formed in the thiol layer, quench the emission of neighbouring dansylamide fluorophores in the outer shell, while the methoxynaphthalene reference dye, buried in the core and further protected by the insulating shell, remains unaffected (Fig. 15). This produces a ratiometric response to the presence of Pb(II) ions.
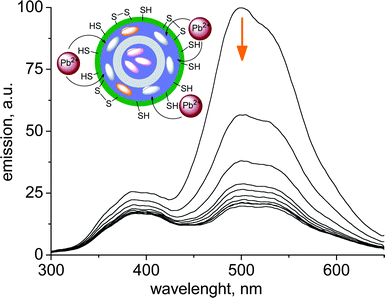 | ||
| Fig. 15 Ratiometric behavior of the fluorescence emission at different Pb(II) concentrations with multishell sensing silica nanoparticles. Inset: schematic working scheme of the sensor (adapted from ref. 49). | ||
A much more complex sensing scheme has been recently proposed by Marcos, Martinez-Manez and co-workers (Fig. 16) for the realization of fluorescent nanoparticles for heparin sensing.50 The surface of commercially available 20 nm silica nanoparticles was functionalized with thiol and diethylenetriamine moieties. When a squaraine dye is added to a solution of nanoparticles, the nucleophilic attack of thiols on the electron deficient four-member ring of the squaraine bleaches both the absorption and the emission of the dye. Heparin, a polyanionic polysaccharide with anticoagulant properties, binds to the polycationic surface of the nanoparticle, thus inhibiting the bleaching of squaraine. Heparine determination using squaraine emission has a detection limit of 2 micromolar and the performance of the sensing system is not affected by the presence of other anions and saccharides with the exception of polyanionic chondroitin-4-sulfate.
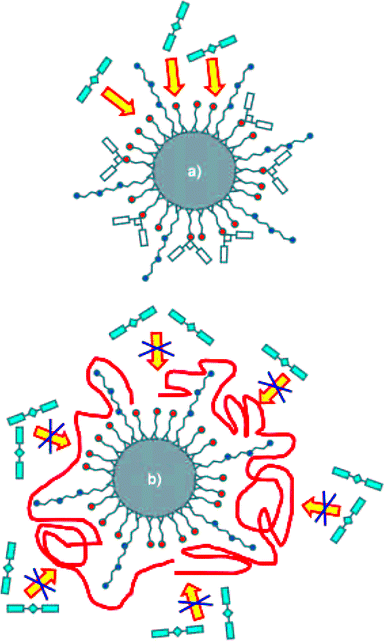 | ||
| Fig. 16 Sensing scheme for heparin signalling nanoparticles. (a) nanoparticles functionalized with thiols (orange) and dietilenetriamine (blue) react with squaraine (cyan) resulting in a non-emissive conjugate; (b) added heparin coordinates (red) the polyamines, thus inhibiting the thiol-squaraine reaction (reproduced form ref. 50). | ||
A different approach for the realization of silica nanoparticles-based chemosensors was explored by Lin and co-workers in 2004 (Fig. 17).51 Here the nanoparticles were designed to act as a nano-chromatographic system with fluorescence detection. Mesoporous silica nanospheres with an average pore diameter of 2.5 nm were prepared following a procedure developed by the authors, which involves the polymerization of 3-mercaptopropyl-trimethoxysilane in the presence of the template surfactant CTABr. The surface of the nanoparticles was functionalized with 5,6-epoxyhexyltriethoxysilane (EHTES) before removing the surfactant, which acts as a protecting group for the thiols in the mesopores. Treatment with boiling HCl/MeOH leads to the conversion of the epoxyhexyl groups to a 5,6-dihydroxyhexyl groups and to surfactant removal as well. The surface of the nanosphere is then coated with a 11 nm thick poly-(lactic acid) (PLA) shell and the thiol groups located within the pores are reacted with o-phthalaldehyde to obtain an o-phthalic hemithioacetal (OPTA) group. The addition of amines to a suspension of nanospheres originates a fluorescence emission as a consequence of their reaction with the o-phthalic hemithioacetal located in the pores to form fluorescent isoindole derivatives. Moreover, different substrates penetrate trough the PLA shell at different rates depending on their polarity. As a consequence, the addition of the amine-containing neurotransmitters dopamine and glutamic acid results in different rates of fluorescence increase allowing the selective detection of dopamine.
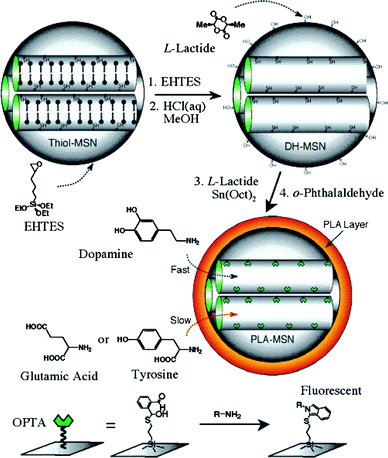 | ||
| Fig. 17 Schematic representation of the synthesis and working scheme of Lin et al.'s fluorescence sensor system for the detection of amine-containing neurotransmitters. EHTES: 5,6-epoxyhexyltriethoxysilane), —●: CTAB surfactant (reproduced from ref. 51). | ||
Conclusions
Many years after its conception, fluorescence sensing is still attracting great interest as a mean to the development of optical sensors and intracellular probes. Fluorescent chemosensors are fascinating tools for biological studies in vitro and even in vivo, needing only simple instrumentation (in some cases as simple as the naked eye) and granting high sensitivity and spatial resolution.Fluorescent nanoparticles, in particular, offer new opportunities and perspectives to this field. The superior emission properties of quantum dots and dye-doped polymeric particles warrant a substantial improvement of the performances of systems designed according to the “classical” strategies used for molecular chemosensors. Most interestingly, new sensing schemes can be invented by taking advantage of the ability of nanoparticles to organize molecular species in complex, even ordered, arrangements. Nanoparticles doped with multiple fluorophores can be used to achieve dual band emission, ratiometric sensing and signal amplification due to collective emission quenching or, in rare cases, to antenna effects. On the other hand, the organization of binding units on the particles surface leads to the spontaneous organization of high-affinity binding sites for the substrate. Finally, the spatial confinement of active units that can be achieved in particles with topologically defined structures enables the realization of sophisticated systems such as those described by Martinez-Manez et al.47,50 and Lin et al.51
The diverse examples reported in this review demonstrate the huge potential benefits disclosed by the use of fluorescent nanoparticles. However, a lot of work remains to be done. In particular, applications of these nanosensors to the real time monitoring of chemical species in live cells or organisms, which is the elective application of fluorescent probes, are rare and limited to PEBBLE systems. Some problems still need to be addressed. The delivery of nanoparticles into live cells is probably one of the most relevant issues: nanoparticles are much larger than molecular chemosensors and are not taken up by cells by passive mechanisms. Moreover, surface properties prone to trigger active uptake mechanisms, such as endocytosis, may not be compatible with the planned sensing scheme. As a consequence, the use of auxiliary delivery systems, such as gene guns or transfection agents may be needed. A second issue is the stability of nanosystems in biological media. Protein adhesion and coating removal may easily occur in such chemically diverse and crowded environments.
Another important point concerns sensing mechanisms. The most desirable behavior for a fluorescent probe is the ratiometric response, which is quite easily achieved in nanoparticles-based systems, using either dual fluorophore systems or the modulation of FRET processes. However, particularly in the case of self-organized agents, the detection of analytes often relies on their quenching ability. Many biologically relevant target are not quenchers, hence the effective use of such systems will require the design of new transduction mechanisms.
Of course, the strategies reported here do not exhaust the possibilities of fluorescent nanoparticles. On the contrary, the new perspectives disclosed by such systems will stimulate the fantasy and the synthetic ability of chemists and lead to the realization of ever more sophisticated and better performing systems.
References
- Selected reviews: (a) T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS; (b) L. Prodi, New J. Chem., 2005, 29, 20–31 RSC; (c) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS; (d) R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419–4476 CrossRef CAS; (e) K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116–127 RSC; (f) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS; (g) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef.
- A. W. Czarnik, Fluorescent Chemosensors for Ion and Molecule Recognition, American Chemical Society, Washington, DC, 1993 Search PubMed.
- A. P. de Silva, T. P. Vance, M. E. S. West and G. D. Wright, Org. Biomol. Chem., 2008, 6, 2468–2480 RSC.
- R. Y. Tsien, Biochemistry, 1980, 19, 2396–2404 CrossRef CAS.
- (a) M. Arduini, E. Rampazzo, F. Mancin, P. Tecilla and U. Tonellato, Inorg. Chim. Acta, 2007, 360, 721–727 CrossRef CAS; (b) F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Chem.–Eur. J., 2006, 12, 1844–1854 CrossRef CAS; (c) A. T. Wright and E. V. Anslyn, Chem. Soc. Rev., 2006, 35, 14–28 RSC; (d) S. L. Wiskur, H. Aït-Haddou, J. J. Lavigne and E. V. Anslyn, Acc. Chem. Res., 2001, 34, 963–972 CrossRef CAS; (e) L. Fabbrizzi, M. Lichelli and A. Taglietti, Dalton Trans., 2003, 3471–3479 RSC.
- (a) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS; (b) M. Arduini, F. Felluga, F. Mancin, P. Rossi, P. Tecilla, U. Tonellato and N. Valentinuzzi, Chem. Commun., 2003, 1606–1607 RSC; (c) R. Pohl, D. Aldakov, P. Kubat, K. Jursikova, M. Marquez and P. Anzenbacher, Chem. Commun., 2004, 1282–1283 RSC.
- R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579–591 RSC.
- S. M. Buck, H. Xu, M. Brasuel, M. A. Philbert and R. Kopelman, Talanta, 2004, 63, 41–59 CrossRef CAS.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362–1395 CAS.
- J. H. Li and J. Z. Zhang, Coord. Chem. Rev., 2009, 253, 3015–3041 CrossRef CAS.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- (a) F. Mancin, P. Tecilla and U. Tonellato, Optical Signalling with Silica Nanoparticles, in The Supramolecular Chemistry of Organic-Inorganic Hybrid Materials, R. Martinez-Manez and K. Rurack ed., Wiley, Hoboken, New Jersey, USA, 2010, pp. 351–376 Search PubMed; (b) S. Santra, J. S. Xu, K. M. Wang and W. H. Tan, J. Nanosci. Nanotechnol., 2004, 4, 590–599 CrossRef CAS.
- Y. F. Chen and Z. Rosenzweig, Anal. Chem., 2002, 74, 5132 CrossRef CAS.
- (a) C. Han and H. Li, Small, 2008, 4, 1344–1350 CrossRef CAS; (b) K. Konishi and T. Hiratani, Angew. Chem., Int. Ed., 2006, 45, 5191–5194 CrossRef CAS; (c) T. Jin, F. Fujii, H. Sakata, M. Tamura and M. Kinjo, Chem. Commun., 2005, 4300–4302 RSC; (d) W. J. Jin, M. T. Fernandez-Arguelles, J. M. Costa-Fernandez, R. Pereiro and A. Sanz-Medel, Chem. Commun., 2005, 883–885 RSC; (e) B. Tang, J. Niu, C. Yu, L. Zhuo and J. Ge, Chem. Commun., 2005, 4184–4186 RSC; (f) B. Chen, Y. Yu, Z. Zhou and P. Zhong, Chem. Lett., 2004, 33, 1608–1609 CrossRef CAS.
- D. B. Cordes, S. Gamsey and B. Singaram, Angew. Chem., Int. Ed., 2006, 45, 3829–3832 CrossRef CAS.
- A. Touceda-Varela, E. I. Stevenson, J. A. Galve-Gasion, D. T. F. Dryden and J. C. Mareque-Rivas, Chem. Commun., 2008, 1998–2000 RSC.
- S. Banerjee, S. Kara and S. Santra, Chem. Commun., 2008, 3037–3039 RSC.
- R. C. Mulrooney, N. Singh, N. Kaur and J. F. Callan, Chem. Commun., 2009, 686–688 RSC.
- M. G. Sandros, D. Gao and D. E. Benson, J. Am. Chem. Soc., 2005, 127, 12198–12199 CrossRef CAS.
- C. Y. Chen, C. T. Cheng, C. W. Lai, P. W. Wu, K. C. Wu, P. T. Chou, Y. H. Chou and H. T. Chiu, Chem. Commun., 2006, 263–265 RSC.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS.
- E. R. Goldman, I. L. Medintz, J. L. Whitley, A. Hayhurst, A. R. Clapp, H. T. Uyeda, J. R. Deschamps, M. E. Lassman and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 6744–6751 CrossRef CAS.
- R. Freeman, L. Bahshi, T. Finder, R. Gill and I. Willner, Chem. Commun., 2009, 764–766 RSC.
- I. L. Medintz, A. R. Clapp, J. S. Melinger, J. R. Deschamps and H. Mattoussi, Adv. Mater., 2005, 17, 2450–2455 CrossRef CAS.
- P. T. Snee, R. C. Somers, G. Nair, J. P. Zimmer, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320–13321 CrossRef CAS.
- (a) Y. E. K. Lee, R. Smith and R. Kopelman, Annu. Rev. Anal. Chem., 2009, 2, 57–76 Search PubMed; (b) S. M. Buck, Y. E. L. Koo, E. Park, H. Xu, M. A. Philbert, M. A. Brasuel and R. Kopelman, Curr. Opin. Chem. Biol., 2004, 8, 540–546 CrossRef CAS , and references therein.
- H. Xu, J. W. Aylott, R. Kopelman, T. J. Miller and M. A. Philbert, Anal. Chem., 2001, 73, 4124–4133 CrossRef CAS.
- Y. E. L. Koo, Y. F. Cao, R. Kopelman, S. M. Koo, M. Brasuel and M. A. Philbert, Anal. Chem., 2004, 76, 2498–2505 CrossRef CAS.
- (a) E. Schmalzlin, B. Walz, I. Klimant, B. Schewe and H. G. Lohmannsroben, Sens. Actuators, B, 2006, 119, 251–254 CrossRef; (b) E. Schmalzlin, J. T. van Dongen, I. Klimant, B. Marmodee, M. Steup, J. Fisahn, P. Geigenberger and H. G. Lohmannsroben, Biophys. J., 2005, 89, 1339–1345 CrossRef CAS; (c) Y. F. Cao, Y. E. L. Koo and R. Kopelman, Analyst, 2004, 129, 745–750 RSC.
- A. Burns, P. Sengupta, T. Zedayko, B. Baird and U. Wiesner, Small, 2006, 2, 723–726 CrossRef CAS.
- E. Pringsheim, D. Zimin and O. S. Wolfbeis, Adv. Mater., 2001, 13, 819–822 CrossRef CAS.
- M. G. Brasuel, T. J. Miller, R. Kopelman and M. A. Philbert, Analyst, 2003, 128, 1262–1267 RSC.
- O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657–2669 RSC.
- A. Graefe, S. E. Stanca, S. Nietzsche, L. Kubicova, R. Beckert, C. Biskup and G. J. Mohr, Anal. Chem., 2008, 80, 6526–6531 CrossRef CAS.
- Q. Chang, L. Zhu, G. Jiang and H. Tang, Anal. Bioanal. Chem., 2009, 395, 2377–2385 CrossRef CAS.
- J. N. Anker and R. Kopelman, Appl. Phys. Lett., 2003, 82, 1102–1104 CrossRef CAS.
- R. M. Jones, L. D. Lu, R. Helgeson, T. S. Bergstedt, D. W. McBranch and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14769–14772 CrossRef CAS.
- M. Montalti, L. Prodi and N. Zaccheroni, J. Mater. Chem., 2005, 15, 2810–2814 RSC.
- S. Bonacchi, E. Rampazzo, M. Montalti, L. Prodi, N. Zaccheroni, F. Mancin and P. Teolato, Langmuir, 2008, 24, 8387–8392 CrossRef CAS.
- P. Teolato, E. Rampazzo, M. Arduini, F. Mancin, P. Tecilla and U. Tonellato, Chem.–Eur. J., 2007, 13, 2238–2245 CrossRef CAS.
- S. Kim, H. E. Pudavar and P. N. Prasad, Chem. Commun., 2006, 2071–2073 RSC.
- C. F. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741–2745 CrossRef CAS.
- (a) M. A. Cejas and F. M. Raymo, Langmuir, 2005, 21, 5795–5802 CrossRef CAS; (b) F. M. Raymo and M. A. Cejas, Org. Lett., 2002, 4, 3183–3185 CrossRef CAS.
- (a) E. Brasola, F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Chem. Commun., 2003, 3026–3027 RSC; (b) E. Rampazzo, E. Brasola, S. Marcuz, F. Mancin, P. Tecilla and U. Tonellato, J. Mater. Chem., 2005, 15, 2687–2696 RSC.
- (a) R. Méallet-Renault, R. Pansu, S. Amigoni-Gerbier and C. Lampent, Chem. Commun., 2004, 2344–2345 RSC; see also: (b) M. Frigoli, K. Ouadahi and C. Larpent, Chem.–Eur. J., 2009, 15, 8319–8330 CrossRef CAS; (c) M. Frigoli, K. Ouadahi and C. Larpent, Chem.–Eur. J., 2009, 15, 8319–8330 CrossRef CAS.
- P. Calero, E. Aznar, J. M. Lloris, M. D. Marcos, R. Martinez-Manez, J. V. Ros-Lis, J. Soto and F. Sancenon, Chem. Commun., 2008, 1668–1670 RSC.
- (a) M. Arduini, S. Marcuz, M. Montalti, E. Rampazzo, F. Mancin, S. Gross, L. Armelao, P. Tecilla and U. Tonellato, Langmuir, 2005, 21, 9314–9321 CrossRef CAS; (b) M. Montalti, L. Prodi, N. Zaccheroni, G. Battistini, S. Marcuz, F. Mancin, E. Rampazzo and U. Tonellato, Langmuir, 2006, 22, 5877–5881 CrossRef CAS.
- M. Arduini, F. Mancin, P. Tecilla and U. Tonellato, Langmuir, 2007, 23, 8632–8636 CrossRef CAS.
- E. Climent, P. Calero, M. D. Marcos, R. Martinez-Manez, F. Sancenon and J. Soto, Chem.–Eur. J., 2009, 15, 1816–1820 CrossRef CAS.
- D. R. Radu, C. Y. Lai, J. W. Wiench, M. Pruski and V. S. Y. Lin, J. Am. Chem. Soc., 2004, 126, 1640–1641 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |