Electrochemistry in nanoscopic volumes
Tao
Li
ab and
Wenping
Hu
*a
aBeijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: huwp@iccas.ac.cn
bGraduate University of Chinese Academy of Sciences, Beijing, 100039, China
First published on 21st October 2010
Abstract
Exploration of electrochemical properties in ultrasmall volumes is still an emerging area. It is not only of great importance for the fundamental research, but also endowed with practical significance in the area of bioanalysis and medicine. Microelectrodes with superior electrochemical characteristics and versatile configurations are suitable tools for the investigation in confined geometries, and remarkable progress involving both preparation methods and theoretical interpretation has been made during the last few decades. Despite this success, electrochemical studies in nanoscopic volumes are still highly challenging due to the less predictable situations in very limited spatial and temporal domains, as well as difficulty in micromanipulation at the nanoscale. In this mini-review, we will summarize the main strategies for this topic, briefly look through the recent advances, and specifically introduce the design and application of a new kind of on-chip ultrasmall electrochemical cells based on micro- and nanogap electrodes, which are prepared by photolithographic method with volume ranging from femtolitre to attolitre. Finally, the limits of current systems and the future perspectives of this field are discussed.
 Tao Li | Tao Li grew up in Shandong province, China. He received his B.S. degree (2005) in the School of Materials and Engineering, BeiHang University. Then he joined the Institute of Chemistry, Chinese Academy of Sciences as a Ph.D. candidate. He focuses on nanogap electrodes in electrochemistry and molecular electronics. |
 Wenping Hu | Wenping Hu is a Professor of the Institute of Chemistry, Chinese Academy of Sciences. He received his Ph.D. from the Institute in 1999. Then he joined Osaka University and Stuttgart University as a research fellow of Japan Society for the Promotion of Sciences and Alexander von Humboldt, respectively. In 2003 he worked at Nippon Telephone and Telegraph, and then returned to the Institute. He focuses on molecular electronic materials and devices. |
Introduction
Miniaturization of existing techniques for higher efficiency devices has always been the developing trend of science and technology. In the area of electrochemistry, to be more specific, the study of electrochemical responses in nanoscopic volumes (in this article defined as being in or below the nanolitre range) has been a very intriguing topic for both electrochemists and biologists in recent years.1,2 On one hand, from the standpoint of theoretical research, microelectrodes have pushed the boundary of electrochemistry into the nanoscale region since its initial introduction in the early 1980s.3,4 It has been experimentally recorded that measurement in tiny volumes may bring about significant difference of responses from that conducted in bulk environments.5,6 Several diffusion-based explanations have been made to clarify the corresponding observations. However, no unanimous conclusions have been drawn so far, attributed to the complicated factors involved in the redox processes within constricted geometries. It is necessary to understand more about this scientific issue, to serve as a beneficial supplement for electrochemical theory and accordingly broaden its scope. On the other hand and more importantly, from the point of view of practical applications, many analytes released from single biological cells are electrochemically active and can be detected at moderate potentials.7,8 Therefore understanding the complex physical processes of living organisms, such as the neurotransmission process, calls for effective testing protocols at trace level. Furthermore, the advancement of nanoscience and nanotechnology has enabled the preparation of various types of microelectrodes suited for the analysis in confined spaces.9,10 To address these issues, more and more efforts have been devoted to this topic during the last few decades, involving preparation methods of microsensors,11–13 modeling and studying of volume-limited natures,14,15in vivo detection,16,17 cellular function simulations,18etc.According to the aspects mentioned above, we organize this mini-review in three main sections. In the first section we start with a brief introduction of the superior electrochemical properties of microelectrodes and, based upon this, summarize the main strategies for investigating electrochemical responses in nanoscopic volumes. Then in the second section we give an overview of the progress that has been made in this area both with artificial model systems and living cells, including the research of constraining effect on the diffusion profile to the electrode, electrochemical determination of exocytosis of neurotransmitters and other biochemically relevant molecules, detection of individual molecules with scanning electrochemical microscopy (SECM), and so on. In the last section we introduce a new protocol of electrochemical chip for applications. These ultrasmall electrochemical cells, ranging from femtolitre to attolitre, are devised based on micro- and nanogap electrodes and can be conveniently manufactured in a mass-producible way by photolithographic methods. Electrochemical characterization revealed the spatial constrains to microelectrode voltammetry and a positive feedback effect was observed attributing to the generator/collector-like interactions between the close-by working and reference/counter microelectrodes. This novel kind of device is found to be a very suitable tool for theoretical study and electroanalysis applications. It also offers a more approachable insight for single molecule electrochemistry in the near future.
Microelectrodes and the manipulation in nanoscopic domains
Microelectrodes are a kind of microscopic sensors with micrometre or even nanometre dimensions (typically smaller than 25 μm) that are employed in electrochemical applications. Compared with their macroscopic counterparts, microelectrodes have been proved to possess many superior advantages such as enhanced mass transport, short timescales, improved faradic-to-capacitive signal and decreased deleterious effects of solution resistance, etc.19–22 On this basis, microelectrodes have opened new possibilities for electrochemical study and measurements in previously inaccessible spatial and temporal domains.23,24 Various types of microelectrodes, from single nanoprobes to well-defined arrays,25–30 have been specifically prepared, characterized, and applied to electroanalysis at trace level,31 measurement in highly resistive media,32 research on fast electrode kinetics,33 and of course, electrochemical study in nanoscopic volumes.Microelectrodes with the above-mentioned advantages, especially the great spatial and temporal resolution, are suitable for exploring the nature of the nanoscopic world which remains unknown to a great extent, particularly in biological systems. Despite of the favorable properties, it is still a very challenging work for the manipulation of microelectrodes in confined geometries, asking for delicate designs and advanced technologies for manufacturing more sophisticated structures. Currently, the strategies of working in nanoscopic volumes mainly fall into two categories: (i) positioning the top ends of microelectrodes into target domains (Fig. 1a and 1b); (ii) preparing volume-limited structures with integrated functional electrodes (Fig. 1c and 1d).
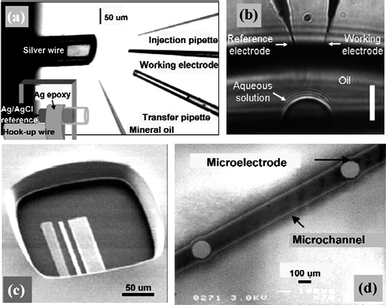 | ||
| Fig. 1 Representative ways of manipulating microelectrodes in nanoscopic domains. (a) Manipulation of pipette and microelectrode into the open end of picolitre-volume vials. A mineral oil cap was placed on the vial to prevent solution evaporation. Image reprinted with permission from ref. 35. (b) Insertion of needle-type microelectrodes into microdroplets with volumes down to a few picolitres. The scale bar is 10 μm. Image reprinted with permission from ref. 15. (c) Integration of microelectrodes into microchambers fabricated by photolithographic method. Image reprinted with permission from ref. 60. (d) Microchannels with integrated micro-working, counter and reference electrodes prepared by plasma etching. Image reprinted with permission from ref. 54. | ||
The former represents a straightforward way for which needle-type microelectrodes with fine tips are the best candidates and the “target domains” could be model systems such as microfabricated vials34,35 and nanodroplets,6,15 or living biological systems (mostly single cells).16,36,37Carbon fiber sealed in a glass capillary38 or carbon fiber nanoelectrodes39 are commonly used because of their admirable electrochemical properties and ease of processing, like sharpening and etching. By using multibarrel glass capillaries, carbon fiber microelectrode arrays40 can also be fabricated which are applicable for spatially sensing chemical changes in tight spaces, such as studying exocytosis from different regions of the single-cell surfaces. In addition to carbon fibers, laser-pulled Pt-disk nanoelectrodes,13 cathodic electrophoretic paint-coated tungsten nanowires,12nanotube-based Au nanoprobes15etc. are also reported. For the above-mentioned “model systems”, what is equally important is the microfabrication and filling of solution containers or formation of sample droplets with nano- and picolitre volumes. Wightman et al.35 described the preparation of picolitre vials from fused-silica capillaries that could provide a transparent container suitable for the observation and manipulation of samples and electrodes. An electrochemically chloridized silver wire was inserted into the capillary to serve as the reference electrode, leaving a void space whose volume was determined by the insertion depth of the silver wire (Fig. 1a).
A more controllable way for defining container volumes was reported by Ewing et al.34 who utilized lithographically fabricated templates to imprint microvials in polystyrene. These transparent microvials ranging from 300 to 0.4 pL can be easily viewed with transmission optical microscopy, which is required for handling extremely small volumes. On appropriate substrates and under specific protections, microdroplets can also be stably formed without a container (Fig. 1b).6,15 As ultrasmall droplets have very large surface-to-volume ratios, necessary measurements should be taken to prevent quick evaporation. A water-saturated heptane6 or mineral-oil layer,15,35,37 or addition of humectants such as glycerol34 are often utilized as protective methods, which are also applicable to the filled microvials and so forth. The experiments are generally performed on the stage of a microscope equipped with micromanipulators for positioning the electrodes and microinjectors. It is noteworthy that not only the working electrodes, but also miniaturized reference electrodes should be manipulated into the confined spaces for measurements. Carefully-treated Ag/AgCl wires6,35 usually play this role. More innovatively, several groups41–43 specially designed probe tips with integrated working/reference electrodes as positional microcells for nanoscopic analysis. For example, Gao et al.43 reported the electrochemical single-cell analysis with a positionable dual microelectrode. In this way it was found that the 20 or 100 nL of detection volume was suitable for peroxidase determination in single cells.
Scanning electrochemical microscopy (SECM)44 is one of the scanning probe microscopies first introduced by Bard and his coworkers in 1980s. It can obtain three-dimensional images of sample surfaces by scanning microelectrode tips and simultaneously detecting local electrochemical activities. Compared with the micromanipulators described above, SECM could offer much higher precision for positioning probes and enable electrochemical measurements in distinct areas of a sample at very close distances (several nanometres). Furthermore, with well-characterized nanotips that are much smaller than a cell, electrochemical probes can even penetrate the membrane without apparent damage to do intracellular measurements. Therefore SECM is a highly desirable technique for microelectrode manipulation in ultrasmall volumes, such as detection inside of a cell and in intact tissue.45–50 As an example, Sun et al.51 have conducted spatially resolved electrochemical measurements in cultured human breast cells by SECM methods and demonstrated the possibility of studying redox properties at the subcellular level (Fig. 2).
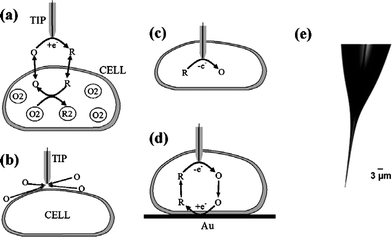 | ||
| Fig. 2 Schematic diagrams of the SECM experiments with single cells (a–d) and an optical micrograph of a typical nanotip used in such experiments (e). (a) The tip is positioned in the solution close to the cell surface. Positive feedback is due to bimolecular electron transfer between hydrophobic redox mediator (O/R) and cell-bound redox moieties (O2/R2). (b) The lipid cell membrane is impermeable for a hydrophilic redox mediator. Negative feedback is due to the hindered diffusion of redox species to the tip electrode. (c) Nanoelectrode voltammetry inside the cell. (d) Positive feedback is produced by mediator regeneration by way of electron transfer at the underlying Au surface. Images reprinted with permission from ref. 51. | ||
The limitation of probe-manipulating scheme is that it involves too much manual attendance, very probably resulting in a variation of experimental conditions (e.g., position of microelectrodes) or even personal errors. Accordingly another electrochemical measurement strategy was developed concerning with volume-limited structures with integrated functional electrodes. They are generally formed on chips (e.g., silicon or glass substrates) using thin- or thick-film processes by microfabrication method, such as photolithography,11,52,53plasma etching,54 screen printing and laser ablation techniques,55,56 which make batch fabrication possible, leading to a cost reduction. Patterning of these structures can be diverse, among which microchambers (Fig. 1c) and microflow channels (Fig. 1d) with embedded microelectrodes are two main types. Murray et al.57 first reported the fabrication of three-electrode electrochemical cells on Si/SiO2 wafers utilizing three coplanar gold-film electrodes as working, counter and pseudoreference electrodes and demonstrated that such microcells could be considered as disposable electroanalytical devices. Cooper and coworkers11,58–61 developed this concept and fabricated a series of microchambers with subnanolitre volume as miniaturized electrochemical sensors for research on cell biology.
Microfluidic devices represent a category of miniaturized total analysis system (μTAS),62,63 and have attracted extensive attention due to their ability of integrating sample preparation, injection, separation, derivatization, and detection into a single miniaturized device. Electrochemical detection method with advantages such as sensitivity, selectivity and ease of miniaturization, is ideally suited for microfluidic format. On this basis, microchip capillary electrophoresis/electro-chemistry hybrid systems have been well developed and very small amounts (10−21 to 10−15 mol) of analytes associated with pL to nL volumes and nM to μM concentrations can be reliably analyzed.64–67 Significant progress of electrochemical techniques for lab-on-a-chip applications, especially in the area of bioanalysis, has been achieved and thoroughly reviewed.68–75 Limited by the capacity of this review, we will not discuss this topic further.
Volume-limited nature of electrochemical response and single molecule electrochemistry in nanoscopic domains
Microelectrode voltammetry in bulk solutions has been well studied based on radial (three-dimensional) diffusion regime which results in sigmoid steady-state response, rather than peak-shaped behavior recorded with macroelectrodes at slow scan rates.21 This notable feature enables time-independent currents to be easily monitored and therefore facilitates the measurements under conditions where variations in potential are disadvantageous.3 In constrained spaces, however, it is natural to image that diffusion pathways to the surface of working electrode would be inevitably bounded and disrupted. Therefore it is in doubt whether the condition of radial diffusion regime can be fulfilled as that in bulk solutions. In allusion to this scientific issue, several groups5,6,14,15 studied the volume-limited nature of electrochemical responses in nanoscopic domains. Sample-volume and scan-rate dependent features were observed and discussed.Ewing et al.5 performed electrochemical experiments with 5-μm-diameter carbon fiber microelectrode in volumes as small as 1 pL defined by lithographically fabricated microvials.34 It was found that the experimental parameters such as scan rate, vial size and sample concentration would all make influence on the voltammetric behaviors. Peak-shaped voltammograms deviating from bulk microelectrode behavior were recorded at slow scan rates in smaller microvials (16 pL or less) (Fig. 3), attributed to the depletion of redox species and restricted diffusion pattern in confined spaces. Similar work was done by Kashyap et al.6 carried out in picolitre droplets formed on the bottom of a polystyrene dish under water-saturated heptane. A carbon fiber microdisk working electrode and a reference electrode with a miniature junction were both inserted into the studied droplet, functioning as an electrochemical microcell. Major differences in voltammetric and amperometric microelectrode behavior in picolitre domains were recorded compared to the behavior in nanolitre volumes and bulk solution. The author interpreted this phenomenon as disruption of some diffusion pathways to the working electrode surface by sample boundaries or even the shank and tip of the microscopic reference. Rossier et al.14 studied the electrochemical measurement in polymer microchannels with integrated microelectrodes. They also found that the walls of the micro-channels prevented the lateral expansion of the diffusion field and a linear diffusion field can develop within the channel, resulting in partial or even complete depletion of the reactants. This quasi-thin-layer cell behavior resembled that described by Ewing and Kashyap. It was also reported in other studies15 that in ultrasmall volumes redox-active molecules can be regenerated and available to both working and reference microelectrodes, which subsequently dominated the electrochemical reaction at the reference electrode and established a stable and self-adjusting reference potential.
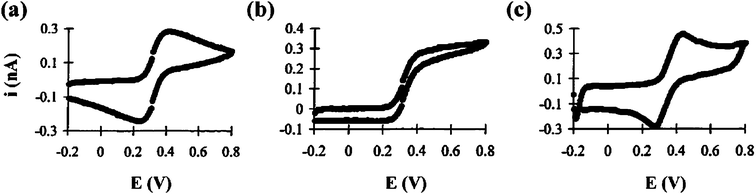 | ||
| Fig. 3 Cyclic voltammograms of 1.0 mM ferrocenecarboxylic acid in a 16 pL vial. The scan rates are (a) 0.1, (b) 1.0, and (c) 10 V s−1. Carbon fiber working electrodes are 5 μm in diameter. Images reprinted with permission from ref. 5. | ||
Due to the complex conditions that may affect the electrochemical properties in highly confined volumes, it is not easy to draw definite conclusions, such as in what dimensions a deviation from steady state can be observed. However, based on the experimental observations, it can be summarized that radial diffusion mode is the prerequisite for steady state, which is easy to fulfil with microelectrodes in bulk conditions. However, when the sample volume is decreased to a certain extent that, either (sometimes both) the boundaries of the sample prevent a full development of three-dimensional diffusion pattern around the working electrode (e.g., microchannels reported by Rossier et al.), or the confined amount of redox species is inadequate to maintain a fast transport rate to the working electrode (e.g., depletion in microvials reported by Ewing et al.), steady state would not be completely accomplished. Moreover, “microscopic” is also a relative concept. The dimensions of microelectrodes with respect to the sample volume as well as their relative positions to sample boundaries are also important factors. Furthermore, in very small sample volumes, the distance between working and reference microelectrodes is very likely limited. In this case, the redox species can be easily regenerated (positive feedback) and thereafter affect the diffusion mode to the working electrodes, resulting in novel electrochemical responses.
Another measurement assay for studying volume-limited electrochemical properties is the fabrication of microdisk electrodes slightly recessed within their insulation sheath, termed nanopore electrodes.10,26 By etching the surface of a nanoelectrode, a nanometre sized cavity can be formed, whose volume can be ranging from several hundred attolitres76 to as small as <1 zeptolitre.77 In such a small volume space, in principle a certain number of molecules, even only one, can be trapped and analyzed. Electroanalytical chemistry at single molecule level represents the ultimate sensitivity in trace chemical analysis, and the effects that are averaged out in the environment of a large number of molecules can be discovered in this way. Bard and coworkers78,79 first published their research on electrochemical detection of single molecules carried out in an SECM. The single-molecule trapping was achieved by making the tip of a small Pt disk electrode (∼10–20 nm) encased and slightly recessed in a wax sheath contact with a conductive substrate in a solution containing an electroactive species and excess inert supporting electrolyte. The trapped molecule shuttled back and forth between the tip and substrate, and repeatedly underwent electron-transfer reactions. This process contributed to a positive feedback effect that can provide a ∼10-million-fold amplification of current signal. The estimated single molecule current was ∼1.6 pA which was readily measurable and not far from actual observations. Recently the procedure for single molecule experiments was further optimized in which the recessed electrodes as SECM tips were immersed in a mercury pool77 instead of contacting with solid substrates (e.g., indium tin oxide (ITO)) to form a thin layer cell (Fig. 4). In this way the deleterious effects on the protruding shroud geometries can be excluded, which is appealing for mathematical modeling and treatment. This kind of ultrasmall cell can contain from one to a few thousand redox molecules and high quality steady-state voltammograms of ≥1 molecules were obtained for a variety of neutral and charged redox species and different concentrations of supporting electrolyte. The analysis of such voltammograms yields information about mass transfer, adsorption, electron transfer kinetics, and double-layer effects on the nanoscale.
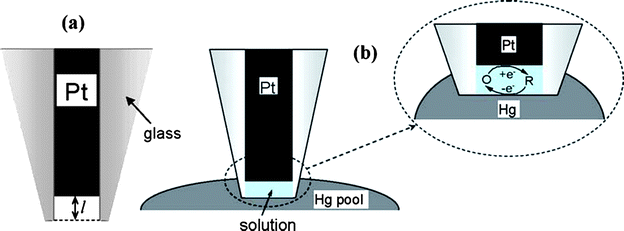 | ||
| Fig. 4 Schematic representation of a recessed nanoelectrode (a) and thin layer cell (TLC) prepared by immersing an etched Pt nanoelectrode into the pool of Hg (b). The inset shows a thin water layer trapped inside the glass sheath. In the nanometre-sized electrochemical TLC, the total number of redox molecules can be varied between one and a few hundred. The oxidized form of the mediator is reduced at Pt UME and reoxidized at the Hg surface. Images reprinted with permission from ref. 77. | ||
Electrochemical bioanalysis in nanoscopic domains
In nature, many chemical messengers involved in biological processes are electrochemically detectable. For the specific research on living organisms in microscopic dimensions, such as a single cell whose activity can be easily masked by a large number of cells, spatial and temporal resolution, as well as sensitivity of the electrodes should all be optimized. In this sense, microelectrodes with cellular or subcellular spatial resolution and the ability of conducting submicrosecond measurements, are particularly useful tools for the investigation of biological systems, and great progress has been achieved in this area over the past few decades.80 The following are some of the representative results.Exocytosis is a process during which molecules stored within secretory vesicles inside a cell are released to the extracellular space by fusion of the vesicle membrane with the cell's plasma membrane. Many of these chemical messengers, for example neurotransmitters released from neurons, are electrochemically active. Therefore by positioning a microsensor in close proximity to or directly in contact with the target cell, secretory events at the cell surface can be conveniently monitored by electrochemical methods.
Exocytosis of neurotransmitters, such as catecholamines (dopamine, epinephrine, and norepinephrine), serotonin, and histamine, has been extensively studied since the pioneering work by Adams81 in 1970s. In these experiments carbon fiber microelectrodes with good biocompatibility are often used as microsensors. Constant-potential amperometry7,82–85 and fast-scan cyclic voltammetry86–89 are the two most widely used techniques, satisfying quantitative measurement and real-time monitoring of secreted molecules respectively. Thanks to the great efforts that have been made, detection of neurotransmitters released from a single vesicle90,91 in the attomole92 to zeptomole level,93 as well as identification of different neurotransmitters co-released from a certain type of cells,85,94 have been achieved. In some cases, modification measures should be taken to improve the sensitivity and selectivity of carbon fiber electrodes, which is not only helpful for the detection of neurotransmitters,84,95 but also necessary for the determination of other chemical messengers, such as insulin8,37,96,97 and ascorbic acid.16 Alternatively, with regard to the high adsorption nature and brittleness of carbon fiber electrodes, recently a new type of boron-doped diamond microelectrode98 was developed and has proved to be advantageous to perform in vivo selective detection of dopamine in mouse brain. In addition to obtain quantitative information of chemical analytes released viaexocytosis, study on spatial heterogeneity of exocytotic events,99 which could provide valuable information for understanding the mechanism of neural secretion, is equally important and more challenging to manipulate. Hafez et al.100 electrochemically imaged the opening of individual exocytotic fusion pores in chromaffin cells with high time resolution using electrochemical detector arrays that consisted of four platinum microelectrodes (Fig. 5). By positioning a single cell on top of the array electrodes, exocytosis of single vesicles containing catecholamines was detected as time-resolved oxidation current. The temporal and spatial variation of current signals recorded by individual addressable microelectrodes determined the position of individual release events. Foot signals that indicated fusion pore formation and expansion were also observed. Similarly, Zhang et al.40 reported the preparation of individually addressable carbon microelectrode arrays and their application to spatially and temporally resolve neurotransmitter release from single pheochromocytoma (PC 12) cells, the results of which proved the feasibility of this method for detecting the subcellular heterogeneity in single-cell exocytosis.
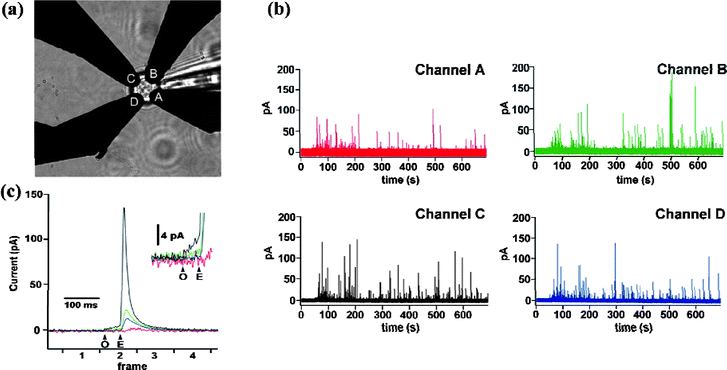 | ||
| Fig. 5 Electrochemical detector (ECD) array recording from a chromaffin cell. (a) Light microscope image of a chromaffin cell placed on top of an ECD array, with four electrodes labeled A through D. (b) Amperometric recordings from the four electrodes (channel A, red trace; channel B, green trace; channel C, black trace; channel D, blue trace) recorded from a single bovine chromaffin cell mechanically stimulated by a patch pipette in calcium-containing external solution. (c) One exocytotic event from the experiment of (b) shown on an expanded time scale. Arrows mark start and end of the foot signal (black trace) indicating fusion pore opening (O) and expansion (E). (Inset) Foot signal on expanded current scale. Images reprinted with permission from ref. 100. | ||
As mentioned above, exocytosis is a very complex process that involves formation of a small fusion pore at the vesicle-plasma membrane interface, followed by expansion of the pore for release of neurotransmitter. The process of exocytosis has been extensively studied.101–104 However, there are too many interfering factors when living cells are used, which make it difficult for further understanding exocytosis. Accordingly, Ewing's group18 described the use of liposome-lipid nanotube networks to create an artificial cell model to mimic the later stages of exocytosis. The experimental results were very similar to what was observed during exocytosis in living cells, which proved that this type of artificial cell model can be used to examine fundamental aspects of exocytosis with many experimental parameters well under control. They also used this liposome-based system to characterize the amperometric response observed for the release of catechol during artificial exocytosis in the nanometre environment between the electrode and the membrane. Their results provided a framework to evaluate the quantitative ability of amperometric experiments to measure exocytosis from different sized vesicles and a model for quantification of the response observed for larger vesicles.105 Based on paired-band microelectrode assemblies, Amatore et al.106 mimicked five basic steps of neuronal transmission using artificial neurons and demonstrated that these simple systems can be adapted to perform as logical gates with AND or OR functionalities.
Compared with detections performed in extracellular space, analysis of the intracellular redox state is more challenging. Mosharov et al.107 introduced a technique termed intracellular patch electrochemistry (IPE) which combined electrochemical methods with electrophysiological methods for intracellular measurement (Fig. 6), by which they for the first time conducted direct measurements of cytosolic oxidizable molecules in single mammalian cells. SECM equipped with well characterized nanotips is another suitable way for spatially resolved quantitative experiments inside living cells.51 In this manner the mass/charge transfer rate across the cell membrane, evaluation of membrane potential and measurement of redox properties with a nanoscale resolution can all be realized.
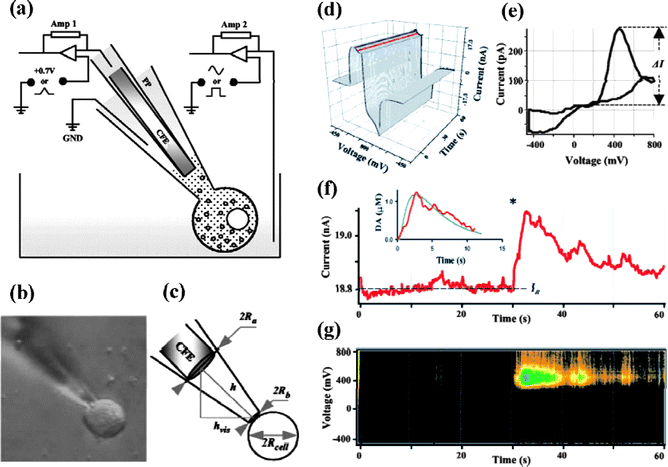 | ||
| Fig. 6 (a) Schematic diagram of the intracellular patch electrochemistry (IPE) setup. (b) An example of a photograph obtained before each recording and used to calculate the dilution of molecules inside the patch pipette (PP). (c) Estimation of the cytosolic volume, Vcell, and the volume inside the patch pipette, Vpipette, that contributed to the dilution of intracellular content. (d–g) Analysis of CV recordings from a single rat chromaffin cell. (d) A representative recording shown as a three-dimentional matrix of data. Red and blue lines represent samplings of the current at 400 and 0 mV, respectively. (e) Voltammogram of catecholamines released during the same recording. (f) Current sampled at 400 mV oxidation potential. The asterisk indicates a time when the patch was disrupted by suction and a whole-cell configuration was attained. The inset shows a random walk fit (green) of the sampled current trace (red) using 15 MΩ access resistance, 8 μm cell radius, 60 μm distance between the CFE and the pipette tip, and 6.7 μM cytosolic catecholamine concentration. (g) Pseudo-three-dimensional representation of approximate voltammograms of intracellular metabolites encountering the CFE during the recording, in which the intensity of color (“Planet Earth” color table) indicates the current at a given voltage and time. Images reprinted with permission from ref. 107. | ||
On-chip ultrasmall electrochemical cells based on micro- and nanogap electrodes
Nanogap electrodes (namely, a pair of electrodes with a nanometre gap) are fundamental building blocks for the preparation of nanodevices to examine material properties at nanometre or even molecular scale.108–110 Our group have worked on nanogap electrodes for several years and used them effectively to study optoelectronic properties of materials.111–113 It has been confirmed that nanogap electrodes can provide highly confined systems with ultrahigh sensitivity. If the great advantages of nanogap electrodes can be advanced into electrochemical microcells, together with the mass production ability of photo- or electron-beam lithography for desirable configured devices, it is highly possible to introduce (i) a new protocol of electrochemical chip for applications, (ii) a new tool for electroanalytical applications, and (iii) a more approachable insight for single molecule electrochemistry. Evoked by the above-mentioned inspiration and pioneering achievements described by Murray57 and Cooper11 (see previous sections), we developed a cost-effective, facile, and mass-producible way to prepare electrochemical microcells on chips based on micro- and nanogap electrodes with volumes ranging from femtolitre to attolitre.114Schematic view of the fabrication procedure is shown in Fig. 7a. Patterning of microelectrodes and formation of central cells are two key steps. Resist film is utilized as an insulating layer and the cell volumes are calculated according to the portion of resist that is controllably etched away. Two types of electrochemical microcells have been fabricated. One is prepared by photolithography with 140 femtolitre and embedded three-finger microelectrodes (i.e., a pair of Au microelectrodes and an Ag microelectrode), referred to as ‘Type I’. The other one is electron-beam lithographically fabricated with even smaller volume ranging from 200 to ∼14 attolitre and four-finger nanoelectrodes (i.e., a pair of Au nanoelectrodes and a pair of Pt nanoelectrodes), referred to as ‘Type II’.
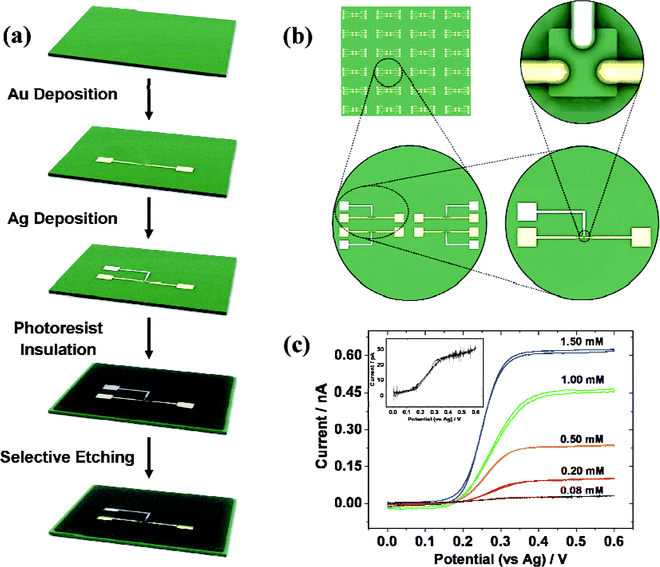 | ||
| Fig. 7 Schematic view of fabrication procedure (a) and (b) configuration of on-chip femtolitre electrochemical microcells. (c) Cyclic voltammograms recorded with femtolitre electrochemical cells at the scan rate of 50 mV s−1 in 0.01 M phosphate-buffered solution (PBS) containing 1.5 mM, 1.0 mM, 0.5 mM, 0.2 mM, and 0.08 mM FcCOOH, respectively; (inset) voltammogram recorded in 0.08 mM FcCOOH with magnified current axes. Images reprinted with permission from ref. 114. | ||
The lay out of ‘Type I’ microcells is shown in Fig. 7b. Four cells are designed as a group to save space, and arrays of these groups are patterned on the Si/SiO2 chip substrates. Ninety-six cells can be fabricated once a time on a 2 inch diamter wafer. The wafer would be cut into small chips before use and there is one chamber on each chip. The depth of the chamber is about 1.4 μm, then the volume can be calculated to be as small as 140 femtolitre (10 μm × 10 μm × 1.4 μm). It can be seen that the chips are well-defined and easy handling, with individually addressable Au and Ag microelectrodes whose dimension is generally below 4 μm, which guarantees radical (three-dimensional) diffusion regime under slow potential scan rates. The microcells are characterized by cyclic voltammetry by positioning a droplet of ∼1 μL redox solution (FcCOOH) onto the aperture of the cell using a micropipette to mimic bulk conditions. Two Au microelectrodes were used as working and counter electrodes respectively, and Ag microelectrode acted as the pseudoreference electrode. A series of steady-state voltammograms as a function of FcCOOH concentration at a scan rate of 50 mV s−1 were recorded with ‘Type I’ microcells (Fig. 7c). It is obvious that the voltammograms exhibited a uniform sigmoid shape with little hysteresis between the forward and reverse scans, which indicated the completeness of steady state attributing to the enhanced mass transport and a good insulating condition between the photoresist and electrode fraction. The steady-state limiting currents increased with FcCOOH concentration and showed a good linearity. The detection limit of FcCOOH can be calculated as 0.035 mM, based on a signal-to-noise ratio of 3. These results demonstrate the capability of reusable chip devices in developing analytical applications. It was also found that the limiting plateau current was apparently smaller when using a Ag/AgCl wire as reference/counter electrode, which can be interpreted as a positive feedback effect attributing to the generator/collector-like interactions115 between the close-by working and reference/counter microelectrodes.
The configuration of ‘Type II’ cells is shown in Fig. 8. Four kinds of cells with different sizes are fabricated, and the volumes of the chambers can be calculated as 200, 50, 32, and ∼14 attolitres, respectively (Fig. 8a-e). The depth of the cells and the thickness of the electrodes are characterized by atomic force microscopy (AFM) (Fig. 8f). It provides direct evidence that even with such small dimensions, the ‘Type II’ cells can be accurately fabricated. Compared with ‘Type I’ cells, with an Au nanoelectrode as the working electrode and a Pt nanoelectrode as the pseudo-reference/counter electrode, peak-shaped curves, rather than sigmoidal voltammograms, were obtained with ‘Type II’ cells (Fig. 8g). The experimental results resemble the volume-limited nature that is discussed in previous sections, although in our case CVs were not conducted in a completely sealed environment. In a control experiment, by substituting the Ag/AgCl wire described above for the reference/counter electrodes in the cell, with the Au nanoelectrode still acting as the working electrode, sigmoidal voltammograms were obtained, shown in the inset of Fig. 8g. The magnitude of limiting current (∼45 pA) roughly agreed with the value calculated for hemispherical electrodes in bulk solutions. Based on the experimental observations, we propose a model to clarify the difference in voltammetric responses. When an external counter/reference electrode is used, oxidation and reduction processes occur inside and outside the cell respectively. The concentration gradient facilitates the mass transfer between the working electrodes and the outside bulk solution, and disruption of radial diffusion pathways by the side walls dose not play a significant role. The radial diffusion mode becomes dominant and the condition resembles what is known about microelectrode voltammetry in bulk solution. In contrast, as the ‘Type II’ cells are at least 3 orders of magnitude smaller than ‘Type I’ cells, a more apparent effect of spatial constrains on voltammetric behavior appears for the former. On one hand, the nanoscale aperture of the cell would act as a resistant factor that makes it difficult for the redox species to diffuse into/out of the cell; on the other hand, when both the working and counter/reference electrodes are the nanoelectrodes integrated at the bottom of the vials, the reactant would be more inclined to undergo redox process back and forth in between the two electrodes. The result is that the reaction domain of interest is mostly confined inside the cell and the diffusion profile is bounded by the walls of the cell to a great extent, like a quasi-thin-layer cell. Larger contributions from planar diffusion result in peak-shaped voltammograms. There is one thing noteworthy that there is not much difference of experimental set-up between Type I and Type II cells, while the voltammetric responses are different. This means for this novel kind of devices, by down scaling the cell from micrometre to nanometre range, the space confinement effect would gradually dominate the diffusion behavior, from mostly spherical to planar diffusion.
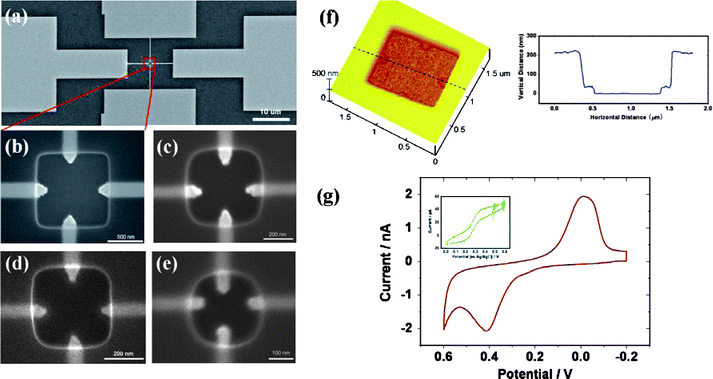 | ||
| Fig. 8 (a–e) SEM images of attolitre electrochemical microcells with designed parameters (cell volume and nanoelectrode diameter): (a) 200 attolitre, 200 nm; (b) magnified image of (a); (c) 50 attolitre, 100 nm; (d) 32 attolitre, 80 nm; (e) 18 attolitre, 60 nm. Electrodes on the horizontal and vertical direction are made of Au and Pt, respectively. (f) AFM image and vertical information of cross section of the 200 attolitre cell. (g) Cyclic voltammogram recorded in a drop of 1 mM FcCOOH (∼1 μL) with the 200 attolitre cell at 50 mV s−1; (inset) voltammogram recorded by substituting an Ag/AgCl wire for the Pt nanoelectrode in the 200 attolitre cell. Images reprinted with permission from ref. 114. | ||
In summary, a cost-effective, facile, and mass-producible method of lithographically manufactured electrochemical microcells based micro- and nanogap electrodes is presented. These simple-structured on-chip microsystems can be easily prepared with volumes as small as 140 femtolitre to 14 attolitre. These new types of ultrasmall cells have proved advantageous in analytical applications and novel properties such as positive feedback effect and volume-limited nature are useful experimental evidences for electrochemical study in nanoscopic domains.
Conclusions
We have briefly reviewed the advances of electrochemical measurements in nanoscopic volumes, in which both the fundamentals of electrochemistry in volume-limited domains and practical applications such as bioanalysis are introduced. Among these aspects, preparation of applicable microelectrodes and appropriate detection methods are two key points for electrochemical analysis in highly confined spaces. Needle-type microelectrodes with sharp tips are very suitable tools for working in nanoscopic domains, especially the bioanalysis of living cells. It would be more powerful and controllable if combined with SECM techniques. However, the precision manufacturing of these tiny probes is a very challenging work. The dimension of the tips would be hard to control in pulling, etching or other processes. The productivity is also relatively low. Correspondingly, the introduction of semiconductor technologies has enabled preparation of more sophisticated structures in a mass-producible way, of which microfabricated vials or channels with integrated microelectrodes can define exact sample volumes for electrochemical measurements without too much manual attendance, although not as flexible as single probes. Furthermore, highly integrated on-chip devices are promising to be the next generation of total analysis microsensors due to their miniaturized structures without compromise of sensitivity and selectivity. If a better combination with the great advantage of microelectrochemical technologies can be made, electrochemical measurements with ultrasmall sample volumes would very probably be raised to a new level.References
- R. M. Wightman, Science, 2006, 311, 1570–1574 CrossRef CAS.
- E. R. Travis and R. M. Wightman, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 77–103 CrossRef CAS.
- R. M. Wightman, Anal. Chem., 1981, 53, 1125A–1134A CrossRef CAS.
- S. Pons and M. Fleischmann, Anal. Chem., 1987, 59, 1391A–1399A.
- R. A. Clark and A. G. Ewing, Anal. Chem., 1998, 70, 1119–1125 CrossRef CAS.
- R. Kashyap and M. Gratzl, Anal. Chem., 1998, 70, 1468–1476 CrossRef CAS.
- P. S. Cahill, Q. D. Walker, J. M. Finnegan, G. E. Mickelson, E. R. Travis and R. M. Wightman, Anal. Chem., 1996, 68, 3180–3186 CrossRef.
- C. A. Aspinwall, J. R. T. Lakey and R. T. Kennedy, J. Biol. Chem., 1999, 274, 6360–6365 CrossRef CAS.
- C. G. Zoski, Electroanalysis, 2002, 14, 1041–1051 CrossRef CAS.
- R. W. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1997, 69, 253–258 CrossRef CAS.
- Y. Qiao, J. Chen, X. L. Guo, D. Cantrell, R. Ruoff and J. Troy, Nanotechnology, 2005, 16, 1598–1602 CrossRef CAS.
- B. B. Katemann and T. Schuhmann, Electroanalysis, 2002, 14, 22–28 CrossRef CAS.
- J. S. Rossier, M. A. Roberts, R. Ferrigno and H. H. Girault, Anal. Chem., 1999, 71, 4294–4299 CrossRef CAS.
- K. S. Yum, H. N. Cho, H. Hu and M. F. Yu, ACS Nano, 2007, 1, 440–448 CrossRef CAS.
- M. N. Zhang, K. Liu, L. Xiang, Y. Q. Lin, L. Su and L. Q. Mao, Anal. Chem., 2007, 79, 6559–6565 CrossRef CAS.
- B. J. Venton, H. Zhang, P. A. Garris, P. E. M. Phillips, D. Sulzer and R. M. Wightman, J. Neurochem., 2003, 87, 1284–1295 CrossRef CAS.
- A.-S. Cans, N. Wittenberg, R. Karlsson, L. Sombers, M. Karlsson, O. Orwar and A. Ewing, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 400–404 CrossRef CAS.
- R. M. Wightman, Science, 1988, 240, 415–420 CrossRef CAS.
- M. Montenegro, M. A. Queiros and J. L. Daschbach, Microelectrodes: Theory and Applications, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1991 Search PubMed.
- A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd ed; John Wiley & Sons, Inc.: New York, 2001 Search PubMed.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1993, 32, 1268–1288 CrossRef.
- S. L. Chen and A. Kucernak, Electrochem. Commun., 2002, 4, 80–85 CrossRef CAS.
- C. Amatore and E. Maisonhaute, Anal. Chem., 2005, 77, 303A–311A CAS.
- C. J. Slevin, N. J. Gray, J. V. Macpherson, M. A. Webb and P. R. Unwin, Electrochem. Commun., 1999, 1, 282–288 CrossRef CAS.
- B. Zhang, Y. H. Zhang and H. S. White, Anal. Chem., 2004, 76, 6229–6238 CrossRef CAS.
- J. Koehne, J. Li, A. M. Cassell, H. Chen, Q. Ye, H. T. Ng, J. Han and M. Meyyappan, J. Mater. Chem., 2004, 14, 676–684 RSC.
- I. Heller, J. Kong, H. A. Heering, K. A. Williams, S. G. Lemay and C. Dekker, Nano Lett., 2005, 5, 137–142 CrossRef CAS.
- D. Krapf, M. Y. Wu, R. M. M. Smeets, H. W. Zandbergen, C. Dekker and S. G. Lemay, Nano Lett., 2006, 6, 105–109 CrossRef CAS.
- Y. H. Lanyon, G. De Marzi, Y. E. Watson, A. J. Quinn, J. P. Gleeson, G. Redmond and D. W. M. Arrigan, Anal. Chem., 2007, 79, 3048–3055 CrossRef CAS.
- P. Bertoncello, J. P. Edgeworth, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2007, 129, 10982–10983 CrossRef CAS.
- M. Ciszkowska and Z. Stojek, J. Electroanal. Chem., 1999, 466, 129–143 CrossRef CAS.
- M. V. Mirkin, L. O. S. Bulhoes and A. J. Bard, J. Am. Chem. Soc., 1993, 115, 201–204 CrossRef CAS.
- R. A. Clark, P. B. Hietpas and A. G. Ewing, Anal. Chem., 1997, 69, 259–263 CrossRef CAS.
- P. Troyer and R. M. Wightman, Anal. Chem., 2002, 74, 5370–5375 CrossRef CAS.
- M. Heien, A. S. Khan, J. L. Ariansen, J. F. Cheer, P. E. M. Phillips, K. M. Wassum and R. M. Wightman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10023–10028 CrossRef CAS.
- R. T. Kennedy, L. Huang, M. A. Atkinson and P. Dush, Anal. Chem., 1993, 65, 1882–1887 CrossRef CAS.
- R. S. Kelly and R. M. Wightman, Anal. Chim. Acta, 1986, 187, 79–87 CrossRef CAS.
- W. Z. Wu, W. H. Huang, W. Wang, Z. L. Wang, J. K. Cheng, T. Xu, R. Y. Zhang, Y. Chen and J. Liu, J. Am. Chem. Soc., 2005, 127, 8914–8915 CrossRef.
- B. Zhang, K. L. Adams, S. J. Luber, D. J. Eves, M. L. Heien and A. G. Ewing, Anal. Chem., 2008, 80, 1394–1400 CrossRef CAS.
- T. W. Spaine and J. E. Baur, Anal. Chem., 2001, 73, 930–938 CrossRef CAS.
- F. Turcu, A. Schulte and W. Schuhmann, Anal. Bioanal. Chem., 2004, 380, 736–741 CrossRef CAS.
- H. Gao, M. H. Zhao, X. L. Zhang and W. R. Jin, Anal. Chem., 2006, 78, 231–238 CrossRef CAS.
- M. V. Mirkin and A. J. Bard, Scanning Electrochemical Microscopy, Dekker, New York, 2001 Search PubMed.
- N. Baltes, L. Thouin, C. Amatore and J. Heinze, Angew. Chem., Int. Ed., 2004, 43, 1431–1435 CrossRef CAS.
- A. Hengstenberg, A. Blochl, I. D. Dietzel and W. Schuhmann, Angew. Chem., Int. Ed., 2001, 40, 905–908 CrossRef CAS.
- R. T. Kurulugama, D. O. Wipf, S. A. Takacs, S. Pongmayteegul, P. A. Garris and J. E. Baur, Anal. Chem., 2005, 77, 1111–1117 CrossRef CAS.
- M. Liebetrau, H. M. Miller and J. E. Baur, Anal. Chem., 2003, 75, 563–571 CrossRef.
- M. Gonsalves, A. L. Barker, J. V. Macpherson, P. R. Unwin, D. O'Hare and C. P. Winlove, Biophys. J., 2000, 78, 1578–1588 CrossRef CAS.
- J. Mauzeroll, A. J. Bard, O. Owhadian and T. J. Monks, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17582–17587 CrossRef CAS.
- P. Sun, F. O. Laforge, T. P. Abeyweera, S. A. Rotenberg, J. Carpino and M. V. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 443–448 CrossRef CAS.
- P. Chen, B. Xu, N. Tokranova, X. J. Feng, J. Castracane and K. D. Gillis, Anal. Chem., 2003, 75, 518–524 CrossRef CAS.
- F. Dias, G. Dernick, V. Valero, M. G. Yong, C. D. James, H. G. Craighead and M. Lindau, Nanotechnology, 2002, 13, 285–289 CrossRef CAS.
- J. S. Rossier, C. Vollet, A. Carnal, G. Lagger, V. Gobry, H. H. Girault, P. Michel and F. Reymond, Lab Chip, 2002, 2, 145–150 RSC.
- J. C. Ball, D. L. Scott, J. K. Lumpp, S. Daunert, J. Wang and L. G. Bachas, Anal. Chem., 2000, 72, 497–501 CrossRef CAS.
- J. C. Ball, J. K. Lumpp, S. Daunert and L. G. Bachas, Electroanalysis, 2000, 12, 685–690 CrossRef CAS.
- L. Morita, M. L. and R. W. Murray, Anal. Chem., 1988, 60, 2770–2775 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1998, 70, 1164–1170 CrossRef CAS.
- T. Yasukawa, A. Glidle, J. M. Cooper and T. Matsue, Anal. Chem., 2002, 74, 5001–5008 CrossRef CAS.
- X. X. Cai, A. Glidle and J. M. Cooper, Electroanalysis, 2000, 12, 631–639 CrossRef CAS.
- X. X. Cai, N. Klauke, A. Glidle, P. Cobbold, G. L. Smith and J. M. Cooper, Anal. Chem., 2002, 74, 908–914 CrossRef CAS.
- S. C. Terry, J. H. Jerman and J. B. Angell, IEEE Trans. Electron. Devices, 1979, 1980.
- Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef.
- R. S. Martin, A. J. Gawron, S. M. Lunte and C. S. Henry, Anal. Chem., 2000, 72, 3196–3202 CrossRef CAS.
- R. P. Baldwin, T. J. Roussel, M. M. Crain, V. Bathlagunda, D. J. Jackson, J. Gullapalli, J. A. Conklin, R. Pai, J. F. Naber, K. M. Walsh and R. S. Keynton, Anal. Chem., 2002, 74, 3690–3697 CrossRef CAS.
- T. Woolley, K. Q. Lao, A. N. Glazer and R. A. Mathies, Anal. Chem., 1998, 70, 684–688 CrossRef CAS.
- L. A. Legendre, J. M. Bienvenue, M. G. Roper, J. P. Ferrance and J. P. Landers, Anal. Chem., 2006, 78, 1444–1451 CrossRef CAS.
- J. Khandurina and A. Guttman, J. Chromatogr., A, 2002, 943, 159–183 CrossRef CAS.
- J. Wang, Electroanalysis, 2005, 17, 1133–1140 CrossRef CAS.
- L. Nyholm, Analyst, 2005, 130, 599–605 RSC.
- F. Sassa, K. Morimoto, W. Satoh and H. Suzuki, Electrophoresis, 2008, 29, 1787–1800 CrossRef CAS.
- D. Erickson and D. Q. Li, Anal. Chim. Acta, 2004, 507, 11–26 CrossRef CAS.
- U. Bilitewski, M. Genrich, S. Kadow and G. Mersal, Anal. Bioanal. Chem., 2003, 377, 556–569 CrossRef CAS.
- N. A. Lacher, K. E. Garrison, R. S. Martin and S. M. Lunte, Electrophoresis, 2001, 22, 2526–2536 CrossRef CAS.
- W. R. Vandaveer, S. A. Pasas, R. S. Martin and S. M. Lunte, Electrophoresis, 2002, 23, 3667–3677 CrossRef CAS.
- P. Sun, Anal. Chem., 2010, 82, 276–281 CrossRef CAS.
- P. Sun and M. V. Mirkin, J. Am. Chem. Soc., 2008, 130, 8241–8250 CrossRef CAS.
- F.-R. F. Fan and A. J. Bard, Science, 1995, 267, 871–874 CAS.
- F.-R. F. Fan, J. Kwak and A. J. Bard, J. Am. Chem. Soc., 1996, 118, 9669–9675 CrossRef CAS.
- W. Schuhmann and A. Schulte, Angew. Chem., Int. Ed., 2007, 46, 8760–8777 CrossRef CAS.
- N. Adams, Anal. Chem., 1976, 48, 1126A–1138A CAS.
- C. Dugast, M. F. Suaud-Chagny and F. Gonon, Neuroscience, 1994, 62, 647–654 CrossRef CAS.
- H. Chow, J. Klingauf and E. Neher, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12765–12769 CrossRef.
- E. Hochstetler, M. Puopolo, S. Gustincich, E. Raviola and R. M. Wightman, Anal. Chem., 2000, 72, 489–496 CrossRef CAS.
- E. V. Mosharov and D. Sulzer, Nat. Methods, 2005, 2, 651–658 CrossRef CAS.
- K. Pihel, T. J. Schroeder and R. M. Wightman, Anal. Chem., 1994, 66, 4532–4537 CrossRef CAS.
- B. P. Jackson and R. M. Wightman, Brain Res., 1995, 674, 163–166 CrossRef CAS.
- A. Hermans, R. B. Keithley, J. M. Kita, L. A. Sombers and R. M. Wightman, Anal. Chem., 2008, 80, 4040–4048 CrossRef CAS.
- K. P. Troyer and R. M. Wightman, J. Biol. Chem., 2002, 277, 29101–29107 CrossRef CAS.
- D. J. Leszczyszyn, J. A. Jankowski, O. H. Viveros, E. J. Diliberto, J. A. Near and R. M. Wightman, J. Biol. Chem., 1990, 265, 14736–14737 CAS.
- R. M. Wightman, J. A. Jankowski, R. T. Kennedy, K. T. Kawagoe, T. J. Schroeder, D. J. Leszczyszyn, J. A. Near, E. J. Diliberto and O. H. Viveros, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10754–10758 CAS.
- D. Bruns and R. Jahn, Nature, 1995, 377, 62–65 CAS.
- K. Chen, G. O. Luo and A. G. Ewing, Anal. Chem., 1994, 66, 3031–3035 CrossRef.
- K. Pihel, S. Hsieh, J. W. Jorgenson and R. M. Wightman, Anal. Chem., 1995, 67, 4514–4521 CrossRef CAS.
- P. Hashemi, E. C. Dankoski, J. Petrovic, R. B. Keithley and R. M. Wightman, Anal. Chem., 2009, 81, 9462–9471 CrossRef CAS.
- L. Huang, H. Shen, M. A. Atkinson and R. T. Kennedy, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9608–9612 CAS.
- C. A. Aspinwall, L. Huang, J. R. T. Lakey and R. T. Kennedy, Anal. Chem., 1999, 71, 5551–5556 CrossRef CAS.
- A. Suzuki, T. A. Ivandini, K. Yoshimi, A. Fujishima, G. Oyama, T. Nakazato, N. Hattori, S. Kitazawa and Y. Einaga, Anal. Chem., 2007, 79, 8608–8615 CrossRef CAS.
- J. Schroeder, J. A. Jankowski, J. Senyshyn, R. W. Holz and R. M. Wightman, J. Biol. Chem., 1994, 269, 17215–17220.
- I. Hafez, K. Kisler, K. Berberian, G. Dernick, V. Valero, M. G. Yong, H. G. Craighead and M. Lindau, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13879–13884 CrossRef.
- R. H. Chow, L. Vonruden and E. Neher, Nature, 1992, 356, 60–63 CrossRef CAS.
- R. G. W. Staal, E. V. Mosharov and D. Sulzer, Nat. Neurosci., 2004, 7, 341–346 CrossRef CAS.
- G. A. Detoledo, R. Fernandezchacon and J. M. Fernandez, Nature, 1993, 363, 554–558 CrossRef.
- L. A. Sombers, H. J. Hanchar, T. L. Colliver, N. Wittenberg, A.-S. Cans, S. Arbault, C. Amatore and A. G. Ewing, J. Neurosci., 2004, 24, 303–309 CrossRef CAS.
- A.-S. Cans, N. Wittenberg, D. Eves, R. Karlsson, A. Karlsson, O. Orwar and A. Ewing, Anal. Chem., 2003, 75, 4168–4175 CrossRef CAS.
- C. Amatore, L. Thouin and J. S. Warkocz, Chem.–Eur. J., 1999, 5, 456–465 CrossRef CAS.
- E. V. Mosharov, L. W. Gong, B. Khanna, D. Sulzer and M. Lindau, J. Neurosci., 2003, 23, 5835–5845 CAS.
- M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252–254 CrossRef CAS.
- S. Kubatkin, A. Danilov, M. Hjort, J. Cornil, J. L. Bredas, N. Stuhr-Hansen, P. Hedegard and T. Bjornholm, Nature, 2003, 425, 698–701 CrossRef CAS.
- F. Chen, J. Hihath, Z. F. Huang, X. L. Li and N. J. Tao, Annu. Rev. Phys. Chem., 2007, 58, 535–564 CrossRef CAS.
- W. P. Hu, H. Nakashima, K. Furukawa, Y. Kashimura, K. Ajito, Y. Q. Liu, D. B. Zhu and K. Torimitsu, J. Am. Chem. Soc., 2005, 127, 2804–2805 CrossRef CAS.
- W. P. Hu, J. Jiang, H. Nakashima, Y. Luo, Y. Kashimura, K. Q. Chen, Z. Shuai, K. Furukawa, W. Lu, Y. Q. Liu, D. B. Zhu and K. Torimitsu, Phys. Rev. Lett., 2006, 96, 027801 CrossRef.
- T. Li, W. P. Hu and D. B. Zhu, Adv. Mater., 2010, 22, 286–300 CrossRef CAS.
- T. Li, L. Su, W. P. Hu, H. L. Dong, Y. F. Li and L. Q. Mao, Anal. Chem., 2010, 82, 1521–1526 CrossRef CAS.
- B. Fosset, C. A. Amatore, J. E. Bartelt, A. C. Michael and R. M. Wightman, Anal. Chem., 1991, 63, 306–314 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |