Re(CO)3(H2O)3+ binding to lysozyme: structure and reactivity
Sarah L.
Binkley
a,
Thomas C.
Leeper
*a,
Roger S.
Rowlett
b,
Richard S.
Herrick
c and
Christopher J.
Ziegler
*a
aDepartment of Chemistry, University of Akron, Akron OH 44325-3601, USA. E-mail: tleeper@uakron.edu; ziegler@uakron.edu
bDepartment of Chemistry, College of the Holy Cross, Worcester, MA 01610, USA. E-mail: rherrick@holycross.edu
cDepartment of Chemistry, Colgate University, 13 Oak Drive, Hamilton, NY 13346, USA. E-mail: rrowlett@colgate.edu
First published on 1st August 2011
Abstract
The reaction of Re(CO)3(H2O)3+ with hen egg white lysozyme in aqueous solution results in a single covalent adduct. Both NMR spectroscopy and single crystal X-ray diffraction show that the rhenium tricarbonyl cation binds to His15via replacement of one of the coordinated water molecules. The formation of this adduct does not greatly affect the structure of the protein.
Introduction
The biological chemistry of organometallic rhenium compounds has received increasing attention over the past two decades.1,2 Although rhenium is not a naturally occurring biological element, two research areas have sparked interest in the medicinal chemistry of this element, and specifically the Re(CO)3+ moiety. First, rhenium is a useful cold analogue of technetium.3–5 The isotope 99mTc is the most commonly used radionuclide in clinics, but technetium is a synthetic element with no stable isotopes. A current focus of researchers is creating new imaging agents from 99mTc(CO)3(H2O)+, because of the ability to convert 99mTcO4− to 99mTc(CO)3(H2O)+ (Fig. 1) using a procedure suitable for a clinical setting.3,6 The chemistry of the analogous Re(CO)3+ group closely resembles that of 99mTc(CO)3+, and thus it use provides a non-radioactive method for the design of new radiological imaging agent candidates. To this end, an extensive and still growing library of rhenium containing compounds has been established.7 Second, a nuclide of rhenium is becoming an excellent candidate for the creation of therapeutic radiopharmaceuticals. A 188W/188Re generator elutes carrier free 188ReO4− suitable for use in a hospital setting, with similar ease to the molybdenum column.8188Re is especially attractive as a therapeutic radiopharmaceutical nuclide as it has a relatively short half life (18 h), a therapeutic beta emission (β− of 2.1 MeV) and a γ emission of 155 keV that can be used to monitor the therapy with SPECT or PET. Critical to this effort, a new kit,9,10 similar to the kit for 99mTcO4−, is comercially available for the conversion of 188ReO4− to 188Re(CO)3(H2O)3+, (Fig. 1) opening up the possibility of creating 188Re(CO)3 derivatives in a clinical setting. The development of this kit is strong reinforcement for the ultimate goal of creating paired 99mTc/188Re compounds that target specific tumor receptors as theranostic radiopharmaceucticals. Such compounds should be able to simultaneously image tumors with high precision and sensitivity while providing locally concentrated radiation-based therapeutic options.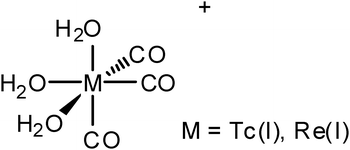 | ||
| Fig. 1 Structure of the M(CO)3(H2O)3+ cations. | ||
In addition to the development of diagnostic and therapeutic compounds, complexes containing the Re(CO)3+ moiety are currently being used to measure biological electron transfer. Specifically Re(CO)3(diimine)+ derivatives bound to a specific amino acid residue have been used as photoinduced electron transfer agents. Upon photoexcitation, the rhenium center undergoes a metal to ligand charge transfer (MLCT) transition, resulting in a transient Re(II) species.11,12 Most notably, Re(CO)3(diimine)+ fragments have been appended to the periphery of proteinsviahistidine groups to investigate the effects of protein structure on electron transfer kinetics.13
Accordingly, there has been increasing interest in the coordination chemistry of rhenium with biological molecules. In part, this chemistry has been driven as a result of the development of bifunctional chelating agent (BFCA)-based drug design strategies. Over the past few years, the binding of the Re(CO)3+ unit to nucleic acids and nucleotides14–18 as well as amino acids and polypeptides19–22 has been investigated. In our recent work, for example, we have observed that Re(CO)3(H2O)3+ forms discrete complexes with N-acetyl histidine and histidylhistidine, and the latter complex is stable in the presence of biological nucleophiles.23 However, there has been little work on rhenium–protein complexes as models for technetium-based imaging agents or rhenium-based therapeutic agents. Investigations of protein binding would provide useful information on possible biological processing of rhenium and technetium drugs, assist with the development of new protein-based drug candidates, and improve the synthesis of protein–rhenium diimine adducts for electron transfer studies.
Previously, we found that crystals of the protein hen egg white lysozyme (HEWL) grown from a solution containing Re(CO)3(H2O)3+ deliver a structure where the cation binds to a single amino acid side chain at the periphery of the proteinviaNε2 of the His15 imidazole ring.24 This imidazole replaces a water in the Re(CO)3(H2O)3+ cation, affording a monosubstituted complex. This selective binding can be readily observed by both IR spectroscopy and mass spectrometry.
In this report, we continue our structural study on the interaction between the Re(CO)3(H2O)3+ cation and HEWL. In addition to providing more detail from our previous structural characterization, we were able to generate the complexed protein by using the standard crystal soaking approach,25 and a solution study performed by NMR spectroscopy; in each case we observe structures identical to the structure previously reported, demonstrating that the mode of binding is independent of the mode of crystal growing and of whether the structure is packed in a crystal or dissolved in solution. In each case, the ligation of Re(CO)3(H2O)2+ to the periphery of lysozyme has minimal effect on the overall structure of the protein. The NMR experiments show that the timescale for rhenium binding to the periphery of lysozyme generally agrees with the known substitution kinetics of the Re(CO)3(H2O)3+ ion.26–29 Further we comment on the bonding of the Re(CO)3(H2O)2+ ion and note ways it could be enhanced.
Experimental
Materials and methods
Triple re-crystallized, lyophilized hen egg white lysozyme (HEWL) was obtained from MP Biomedicals. All reagents and solvents were purchased from Sigma, Aldrich, Acros Organics or Strem and used without further purification. Solutions of [Re(CO)3(H2O)3]Br were prepared as previously described30 and evaporated to dryness to produce a solid compound previously identified as [Re(CO)3(H2O)3][Re2(CO)6(μ2-Br)3]·6H2O.31IR spectra were recorded on a Nicolet NEXUS 870 FT-IR Esp and Perkin Elmer Spectrum One FT-IR spectrometers. Mass spectrometric analyses were carried out on a Bruker Reflex III MALDI-TOF at the Mass Spectrometry and Proteomics Facility at the Ohio State University in Columbus OH. Rhenium elemental analysis was carried out viaICP-OES at the University of Illinois at Urbana-Champaign Microanalysis Laboratory.Mass spectrometry experiments
Lysozyme was dissolved in nanopure water and reacted with aqueous solutions of [Re(CO)3(H2O)3]Br in nanopure water at molar equivalents of 0, 0.1, 0.5, 1, 2, or 3 Re![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein at a final protein concentration of 1 mg/mL. The samples were incubated at 4–10 °C for 24 h and then analyzed by MALDI-TOF-MS.
:protein at a final protein concentration of 1 mg/mL. The samples were incubated at 4–10 °C for 24 h and then analyzed by MALDI-TOF-MS.
Equilibrium dialysis experiments
Lysozyme was dissolved in 0.05 M sodium phosphate buffer (pH = 5.8) to make solutions with protein concentrations of 1000, 300, 100, 30, 10 and 3 μM. 3 mL of each of the preceding solutions were injected into Slide-A-Lyzer® dialysis cassettes. The protein solutions were dialyzed for 72 h against 1 μM [Re(CO)3(H2O)3]Br in 500 mL of 0.05 M sodium phosphate buffer (pH = 5.8). The protein solutions and samples of the corresponding dialyte were sent for rhenium elemental analysis.Protein–metal complex crystallization
50 mg of lysozyme were dissolved in 750 μL of sterile, nanopure water. [Re(CO)3(H2O)3]Br was dissolved in water and added to the lysozyme solution to produce a final lysozyme:Re ratio of 1:5. Sterile, nanopure water was added to a final volume of 1 mL and the resulting solution was incubated at 4–10 °C overnight. Crystals were obtained via the hanging drop method using a reservoir solution of 0.05 M MES buffer (pH = 5.5) and 0.8 M NaCl. The drop was composed of 4 μL of the reservoir solution and 4 μl of the incubated protein–metal solution. Large, tetragonal crystals appeared in 72 h. Crystals were cryo-preserved by soaking for a few minutes in mother liquor plus 30% glycerol and flash cooling in liquid nitrogen.
Re(CO)3(H2O)3+ Soaking experiments
Lysozyme crystals were obtained via the hanging drop method, using a reservoir solution of 0.05 M MES buffer (pH = 5.5) and a drop composed of 4 μL lysozyme solution (50 mg ml−1) and 4 μL reservoir solution. Large, tetragonal crystals appeared in 24 to 48 h. The crystals were then transferred to a 6 μL drop containing 0.10 M MES, 1.6 M NaCl, and 5 molar equivalents of [Re(CO)3(H2O)3]Br. The drop was re-suspended over the original reservoir solution and incubated for 5 days at room temperature. Crystals were cryo-preserved by soaking for a few minutes in mother liquor plus 30% glycerol and flash cooling in liquid nitrogen.Glyoxime soaking experiments
Lysozyme (50 mg ml−1) and 3 molar equivalents of aqueous [Re(CO)3(OH2)3]Br were allowed to incubate at 4 °C for 12 h. The resulting solution was crystallized via the hanging drop method. The drop was composed of 6 μl of the protein solution and 6 μL of the reservoir solution. The reservoir solution was composed of 0.05 M MES buffer (pH = 5.0) with 0.8 M NaCl. Crystals appeared within 48–72 h. Large, single domain crystals were harvested with a loop and transferred to a 10 μL drop that contained 10 molar equivalents of glyoxime in 0.1 M MES buffer (pH = 5.0) with 1.6 M NaCl. The new drop was then sealed over the original well solution and allowed to react over a period of 4 days. At that point, the crystals had acquired a yellow to yellow-brown hue. They were harvested and cryoprotected in a solution of the soaking buffer to which 30% (w/v) glucose had been added.X-Ray diffraction
X-Ray diffraction data to 1.6 Å resolution were collected at 1.54 Å (Oxford Diffraction Gemini R) at 110 K using the 2 kW Enhance Ultra Cu source and was integrated and scaled using CrysalisPro.32 Each crystal structure was solved by molecular replacement (Phaser33) using PDB 6LYZ and subjected to several cycles of restrained refinement using Refmac534 and rebuilding in Coot.35 The refined molecular replacement solution contained clearly identifiable electron density for Re(CO)3(H2O)2+ near Nε2 of His15. A set of refinement restraints were manually created for the Re(CO)3 fragment in which the carbonyl ligands were disposed in a facial arrangement with orthogonal (90°) C–Re–C bond angles, and Re–C and C–O bond lengths of 1.8 Å and 1.2 Å, respectively. The protein and rhenium tricarbonyl fragment was further refined in Refmac5, then solvent molecules and a chloride ion were modeled to account for remaining electron density. Data collection and refinement statistics are summarized in Table 1.| a Parentheses indicate information for highest resolution shell. b Based on ideal values from Engh and Huber.50 c MolProbity analysis.51 | |
|---|---|
| Data collection statistics | |
| Wavelength | 1.54 Å |
| Space Group | P43212 |
| Cell parameters | a = b = 78.71 Å, c = 36.99 Å |
| Resolution | 11.99–1.60 Å (1.66–1.60 Å)a |
| Unique reflections | 15847 (1591) |
| Redundancy | 22.4 (16.9) |
| Completeness | 99.7 (100) |
| R sym | 0.091 (0.670) |
| I/σI | 32.6 (2.78) |
| Refinement statistics | |
| Reflections in test set | 794 |
| R work (%) | 18.1 (25.8) |
| R free (%) | 21.5 (31.9) |
| Rmsd from idealb | |
| Bond distance (Å) | 0.015 |
| Bond angle (°) | 1.55 |
| Ramachandran plot outliers (%)c | 0.00 |
Nuclear magnetic resonance experiments
A solution of 0.05 M sodium phosphate, pH 5.8, was made using D2O, evaporated to dryness and then re-hydrated with D2O to produce the buffer. This buffer was then used to make solutions of lysozyme and [Re(CO)3(H2O)3]Br. Aliquots of these two solutions were combined to make samples containing 5 mM lysozyme with 0, 0.4, 1, or 3 molar equivalents of [Re(CO)3(H2O)3]Br. Samples were equilibrated for several hours prior to measurement. Proton 1D spectra and 2D 1H/13C HSQC spectra36 were collected on each of these pre-equilibrated solutions. HSQC spectra were collected with 80 complex pairs, 128 scans, a 1.2 s recycle delay, a 43 ppm carbon carrier, and sweep widths of 12000 and 15000 Hz (1H and 13C, respectively) on a 750 MHz Varian Inova spectrometer equipped with a triple resonance cold-probe. Aromatic resonances were aliased in the carbon dimension to improve resolution. For the 24 h time-course experiment, a three molar equivalent ratio of Re(CO)3(H2O)3+ to HEWL was employed with a sample of both species freshly dissolved in D2O. Immediately after dissolution, a 1H 1D spectrum time-course was collected as a pseudo-2D dataset on a Varian Inova 400 MHz spectrometer with 128 scans, 8196 points, and 2 s recycle delays. Initial time points of 300, 1200, and 2100 s between experiments were used, followed by 18 spectra separated by 3600 s each. Note that for the 1D spectra significant numbers of non-exchanged indole and backbone amide protons remain in these samples even after one or two days in D2O. Proton 1D spectra were processed and overlain in iNMR (http://www.inmr.net) while 2D datasets and 24 h pseudo-2D dataset were processed with NMRPIPE37 and visualized with SPARKY (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). Chemical shift values from the 24 h time-course were fit to an exponential function and plotted using IgorPro (Wavemetric, Inc.)
Lysozyme-Re(CO)3+ IR spectroscopy
HEWL crystals were grown after exposure to [Re(CO)3(H2O)3]Br as described above via the hanging drop method. Crystals were then harvested, washed, dissolved in methanol, and the resultant solution evaporated to produce a thin film on the diamond anvil of the FT-IR.Results and discussion
Since the first report of its synthesis by Alberto in 1994,4 the Re(CO)3(H2O)3+ ion has been shown to form stable complexes with a variety of ligand types upon substitution of one or more of the bound water molecules. The water molecules on Re(CO)3(H2O)3+ are labile, and many examples of Re(CO3)L3 compounds have been isolated, including biologically relevant complexes where L includes amines, imidazoles, thiolates, thioethers, alcohols and carboxylates.2 More recently, we have shown that Re(CO)3(H2O)3+ will react with amides in peptides to form N-bound deprotonated chelates.23 Thus, from a coordination chemistry standpoint, the Re(CO)3(H2O)3+ ion can potentially react with many amino acid side chains, and could even react and bind to the peptide backbone in a protein.When considering the protein lysozyme, there are a variety of potential metal binding sites at the periphery of the protein, and the external lysines have been an oft-used target.38,39 However, recent efforts have focused on the single peripheral imidazole ring at His15.40 Fontecilla-Camps et al. observed the binding of a Mn(CO)3(H2O)2+ unit to the His15 site of lysozyme,41 forming a protein adduct where the metal adopts an octahedral geometry. The coordination sphere in this protein complex consists of three facial carbonyl units, two water molecules and the imidazole ring of the histidine. Sadler et al. have reported an organometallic Ru-lysozyme complex.42 More recently, Romao et al. presented a communication on the soaking of lysozyme crystals with fac-Ru(CO)3Cl(κ2-H2NCH2COO).43 The metal primarily formed an adduct at the His15 site analogous (80% occupancy) to that seen with the Mn(I) chemistry except with two ciscarbonyls instead of three facial carbonyls. Additionally, ruthenium binding is also observed at two carboxylate side chains from amino acids Asp18 and Asp52 with 50% and 40% occupancies, respectively.
We carried out an initial evaluation using MALDI-TOF mass spectrometry to determine whether the Re(CO)3(H2O)3+ ion binds to lysozyme. We exposed lysozyme solutions (1 mg mL−1protein) in pure water to several stoichiometries of the Re(CO)3(H2O)3+ ion, and the observed mass spectra are shown in Fig. 2. Unmodified lysozyme shows a peak at approximately 14300 m/z, in agreement with its experimentally determined molecular weight.44 As the ratio of Re(CO)3(H2O)3+ ion to protein increases, a peak that corresponds to an increase in mass of approximately 270 appears and increases in intensity. This mass change corresponds to the weight of the Re(CO)3+ unit, a clear indicator that the Re(CO)3(H2O)3+ ion is interacting with the protein, possibly via coordination of one or more amino acid side chains.
 | ||
| Fig. 2
MALDI mass spectra of (a) native lysozyme, (b) 1:0.1, (c) 1:0.5, (d) 1:1, (e) 1:2 and (f) 1:3 native lysozyme: Re(CO)3(H2O)3+ reaction solution. An asterisk marks where the mass of the rhenium adduct appears. | ||
We also carried out a preliminary investigation into the binding of rhenium to lysozyme by using equilibrium dialysis. Samples of varying HEWL concentration were dialyzed against a solution of the Re(CO)3(H2O)3+ ion in phosphate buffer at pH 5.8. Total rhenium concentrations inside and outside of the dialysis bag were determined by ICP-MS. These experiments with Re(CO)3(H2O)3+ revealed a coarsely determined Kd of about 50 μM, consistent with the NMR-observed fast exchange behavior reported below. There has been some kinetics studies on simple ligands binding to Re(CO)3(H2O)3+ and the system most similar to the histidine side chain is the pyrazine system studied by Alberto et al.27 However, the analogous Kd for pyrazine and Re(CO)3(H2O)3+ (based on the reported Ka) is in the range of 10−2–10−3. The discrepancy in Kd can be rationalized several ways. First, the pyrazineKa was determined in a higher concentration of electrolyte (I = 1 M) than was used in our protein experiments, where the concentration of phosphate was only 0.05 M. In addition, the Re(CO)3(H2O)3+ can readily interact with anionic residues or side chain heteroatoms on the surface of lysozyme prior to ligation, which could result in an increased affinity. Lastly, the pyrazine experiments were conducted at low pH, and we would expect the presence of deprotonated acid residues to increase the affinity of the Re(CO)3(H2O)3+ ion. In particular, one acidic residue, Asp87, is within 4 Å of the rhenium atom in this structure (vide infra).
Using NMR we have probed the nature of Re(CO)3(H2O)3+ interaction with HEWL. 2D 1H–13C HSCQ spectra were collected on 24 h equilibrated complexes with various ratios of freshly prepared Re(CO)3(H2O)3+ solution. For the most part, chemical shifts of the protein were unchanged from published values.45 Only residue His15 shows appreciable chemical shift changes (Fig. 3, Insets). Thus only one specific site in HEWL is metallated by rhenium under these conditions. Additionally, the gradual chemical shift changes observed as a function of Re(CO)3(H2O)3+ concentration suggest fast chemical exchange between free metal ion and protein bound metal ion with regard to the NMR chemical shift timescale.
 | ||
| Fig. 3
13C HSQC spectra collected on 5 mM HEWL titrated with Re(CO)3(H2O)3+ at 0:1 (black), 0.4:1 (green), 1:1 (blue), 3:1 (red) ratios. | ||
We also collected 1H 1D spectra on these complexes and the aromatic/exchangeable proton region is shown in Fig. 4. Slow and incomplete H2O to D2O exchange permitted observation of amide and tryptophan indole resonances that shift upon metalation. All of the peaks move with apparent fast exchange with regard to chemical shift, although the peaks shift around surprisingly slowly relative to the timing of rhenium addition. The two carbon-bound, and hence non-solvent exchangeable, proton resonances from H15 that shift and broaden upon Re(CO)3(H2O)3+ addition are also labeled. The remaining resonances shift and broaden due to the combined process of slow deuterium exchange and rhenium binding. Mapping these changes onto the crystal structure of unmodified HEWL (Fig. 5) shows that majority of the changes correlated to two places, the binding site and two tryptophan indoles nearby. These later changes are probably a result of slight changes in the H2O to D2O exchange properties of these residues. The sites confirmed by NMR to be high occupancy sodium and chloride, rather than low occupancy rhenium, binding sites are indicated by the small blue spheres.
 | ||
| Fig. 4 1D spectra collected at 400 Mhz on 4 mM lysozyme freshly dissolved in 100% D2O (red), and the same sample with 2 equivalents of Re(CO)3(H2O)3+ after 24 h (black). | ||
 | ||
| Fig. 5 Amide chemical shift changes mapped on to the HEWL surface structure. Tentatively assigned residues that experience amide or indole chemical shift changes upon metalation are rendered purple. His15 experiences both amide and 13C imadizole shift changes (the only residue with changes in the 13C HSQC) and is colored cyan. | ||
To elaborate upon the rate of metalation, we collected a 24 h time-course, monitoring the chemical shift changes in protein resonances upon immediate introduction of an excess of Re(CO)3(H2O)3+ to HEWL (Fig. 6). In spite of the apparently “fast” chemical shift changes observed above, changes in the specra of HEWL took at least an hour to finish. This was surprising due to the fast exchange behavior and, as will be discussed below, indicates that Re(CO)3(H2O)3+ metalation is not a simple on/off (i.e. bimolecular) mechanism. In addition, the water exchange rates as investigated by Merbach et al. are clearly relevant to these processes.26
 | ||
| Fig. 6 Time dependent chemical shift changes for H15 HE1 show unusually slow “fast-exchange” behavior, i.e. one peak shifting between starting and ending chemical shifts. (a) Interferogram of the time dependence of chemical shift changes upon metalation. (b) Non-linear fit to y = y0 + A*exp−Tau*x of the boxed region in panel A. y0 = 3556.8 ± 0.07 Hz, A = −6.15 ± 0.23, Tau−1 = 1.22 ± 0.13 Hours. | ||
We were able to grow crystals from HEWL solutions exposed to Re(CO)3(H2O)3+ at 4° overnight using the hanging drop method. The concentration of the HEWL was 50 mg mL−1protein in 0.05 M MES buffer, pH 5.5, and 0.8 M NaCl. Large tetragonal crystals suitable for X-ray diffraction structure elucidation formed after 48–72 h. It is important to note that adduct formation was carried out in solution prior to crystal growth; typically, metal ligation is achieved only by soaking crystals in reagent solutions. We also observed that rhenium adducts could be produced via soaking methods; the structural parameters for these crystals were identical to those grown from solution.
The IR spectra of these rhenium-modified HEWL crystals were investigated to determine if we could observe the C–O stretching frequencies of the facial Re-tricarbonyl unit. In the 1800–2200 cm−1 region, unmodified lysozyme is transparent, but Re(CO)3+ compounds exhibit bands corresponding the the a1 and e modes of pseudo-C3v symmetry. Rhenium modified lysozyme crystals show ν(CO) stretching bands that match these modes, shown in Fig. 7. These modes do not correspond to those of free Re(CO)3(H2O)3+ ion, which has a1 and e bands at 2036 and 1916 cm−1.4 These rhenium-modified lysozyme crystals exhibit a splitting of the a1 band, which we show (vide infra) may result from multiple rotamers of the Re(CO)3+ unit in the solid state.
 | ||
| Fig. 7 IR spectra of the carbonyl stretching region of unmodified lysozyme (dashed line) and Re(CO)3(H2O)3+ modified lysozyme (solid line). | ||
We collected X-ray data on a crystal grown from the conditions described above to 1.6 Å resolution. A ribbon diagram of the structure of the rhenium protein adduct is shown in Fig. 8 along with a superposition of both native (PDB code 6LYZ) and rhenium-modified HEWL. The rhenium binds to the His15 imidazole side chain, and as expected, the Re(CO)3(H2O)3+ ion has lost a water molecule, forming a imidazole bound Re(CO)3(H2O)2+ fragment. In agreement with the observed NMR data, coordination of the rhenium to the His15 site does not greatly modify the structure of the protein. The RMS deviation between the α-carbons of the two protein models is 0.27 Å. As can be seen by superposition (Fig. 8), the most obvious structural accommodations for the rhenium complex binding is a flipping of the imidazole of His15 and a slight deviation in backbone position of this residue.
 | ||
| Fig. 8 Top: Ribbon diagram of HEWL with Re(CO)3(H2O)2+ bound to its unique site at Nε2 of His15. Hydrogen bond between Asp87 and water W1 of Re(CO)3(H2O)2+ is depicted as a magenta dashed line. Bottom: A superposition of both native (PDB code 6LYZ) and rhenium-modified HEWL. | ||
Fig. 9 shows the Fo − Fc omit map of the lysozyme–Re(CO)3(H2O)2+ adduct, contoured at 2.5σ. The occupancy of the Re(CO)3(H2O)2+ fragment in the model is less than 1.0 and was modeled at 0.60, consistent with the MALDI-MS data that indicates partial derivatization of the protein. The refined Re–O bond distance for water W2 is somewhat long (2.6 Å compared to the expected 2.2 Å as observed for W1), suggesting that there are at least two significantly populated rotamers of the Re(CO)3(H2O)2+ adduct, related by a 90° rotation about the Re–N bond. In the final model, however, only the principal rotamer was modeled. Partial occupancy of a CO ligand at the W2 position results in the Re–O bond having an artificially long Re–O bond distance. The observation that the electron density of the Re(CO)3(H2O)2+ fragment is well-ordered, rather than averaged, because of free rotation about the Re–N axis, is possibly due to the stabilizing hydrogen bond between Oδ2 of Asp87, and a water ligand (W1 in Fig. 9) on the rhenium. The Asp87–Oδ2–water distance (2.5 Å) in the X-ray structure is consistent with this interpretation. In the principal rotamer, Asp87 interacts with W1; in the minor rotamer, Asp87 interacts with W2. The Re–N distance of 2.2 Å compares well to related small molecule structures. 46
 | ||
| Fig. 9 F o − Fc omit map of the lysozyme–Re(CO)3(H2O)2+ adduct, contoured at 2.5σ. Arg14 has weak electron density and has alternate conformations, and does not appear to interact with Re(CO)3(H2O)2+. | ||
In addition to collecting X-ray data on crystals grown from lysozyme-rhenium reaction solutions, we also investigated the soaking of lysozyme crystals with the Re(CO)3(H2O)3+ ion. This reaction is analogous to the soaking experiments carried out by Fontecilla-Camps and coworkers with Mn(CO)3(H2O)3+ ion.41 We were able to collect data that was identical to that of the solution-based reaction crystals, with rhenium modification at His15. In addition, we also investigated soaking lysozyme–rhenium crystals with a chelating diimine, glyoxime. Glyoxime and other dioximes have been shown to avidly bind to Re(CO)3+ centers, and are structurally analogous to the diimine systems investigated by Gray et al.13 However, exposure of lysozyme–rhenium crystals to glyoxime leached the rhenium metal center away from the protein, regenerating unmodified lysozyme.
Conclusions
Using both solution methods (NMR) and crystallography, we have characterized the Re(CO)3(H2O)3+ complex of hen egg white lysozyme and found that the surface histidine binding site is the preferred and exclusive site of ligation. The significance of the consistency of these two methods suggests that this complex is probably relevant to both in situ and in vivo preparation of rhenium–protein complexes. In particular, the use of histidine, a nearly ubiquitously surface exposed amino-acid, for metalation suggests that many targets beyond simply lysozyme are available to rhenium complexation. Thus the general nature of this ligation may be invaluable in implementing this species to be a generic contrast agent probe.The reversible bonding of Re(CO)3(H2O)2+ to His15, as indicated by the fact that soaking the metalated crystal with glyoxime completely removed the bound rhenium, suggests that vicinal surface-bound histidines might be necessary to create complexes that might withstand biological conditions. Alternatively, His-tags23,47or the use of recombinant DNA techniques could produce suitable binding environments. We are currently looking for suitable candidates.
The time constant (τ) for the initial exchange of Re(CO)3(H2O)3+ onto HEWL is 1.2 h, i.e. with a rate of ∼2 × 10−4s−1 (Fig. 4), whereas fast exchange behavior on the chemical shift timescale (Fig. 1 and 2) is usually observed for states with lifetimes of ∼100 μsec. The eight orders of magnitude difference can only be explained if binding of rhenium to the protein is not simply a two-state process, but is limited due to a long-lived initially unreactive species. We hypothesize that it is the dissociative interchange from Re(CO)3(H2O)3+ that is the rate limiting since previous work suggested that this process has a similar magnitude rate (7.6 × 10−4 M−1s−1) for the dimethyl sulfide monitored formation of Re(CO)3(H2O)2(DMS)+ from the triaqua compound.48 This is significant because it suggests that protein metalation is highly dependent upon the residence time of coordinated waters. Modulation of the residence time of a single water may be achievable by substitution of the other two waters with different ligands such as 2,2′-bipyridine or similar diimines.49 This should suppress dissociation of the remaining water and prolong its residence time. Modulation of the residence time of the remaining water, and hence speed of protein metalation, may be important for engineering rhenium to be a viable imaging probe or antineoplastic agent.
Acknowledgements
T.C.L. and C.J.Z. would like to acknowledge the University of Akron for financial support and C.J.Z. would like to acknowledge a grant from the National Institutes of Health (R15 GM 083322) for support of this work. R.S.H. thanks the Research Corporation (CC6663/6616) for research support. This work was also supported in part by a grant from the National Science Foundation to R.S.R. (CHE-0819686).Notes and references
- R. Alberto, R. Schibli, R. Waibel, U. Abram and A. P. Schubiger, Coord. Chem. Rev., 1999, 190–192, 901–919 CrossRef CAS.
- R. S. Herrick, in New Developments in Organometallic Research, Nova Science Publishers, Inc., New York, 2006, pp. 117–49 Search PubMed.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135–3136 CrossRef CAS.
- R. Alberto, A. Egli, U. Abram, K. Hegetschweiler, V. Gramlich and P. A. Schubiger, J. Chem. Soc., Dalton Trans., 1994, 2815–2820 RSC.
- S. Liu and D. S. Edwards, Chem. Rev., 1999, 99, 2235–2268 CrossRef CAS.
- R. Schibli and P. A. Schubiger, Eur. J. Nucl. Med. Mol. Imaging, 2002, 29, 1529–1542 CrossRef CAS.
- C. C. Romão and B. Royo, in Comprehensive Organometallic Chemistry III, ed. H. C. Robert and D. M. P. Mingos, Elsevier, Oxford, 2007, pp. 855–960 Search PubMed.
- B. M. Coursey, J. M. Calhoun, J. Cessna, D. D. Hoppes, F. J. Schima, M. P. Unterweger, D. B. Golas, A. P. Callahan, S. Mirzadeh and F. F. Knapp, Jr., Radioact. Radiochem., 1990, 1(38), 42–39 Search PubMed.
- P. Kozminski, E. Gniazdowska, L. Fuks and S. Kowalska, Appl. Radiat. Isot., 2011, 69, 436–442 CrossRef CAS.
- L. Fuks, E. Gniazdowska, P. Kozminski, M. Lyczko, J. Mieczkowski and J. Narbutt, Appl. Radiat. Isot., 2009, 68, 90–95.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- J. Hawecker, J. M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- A. M. Blanco-Rodriguez, M. Busby, C. Gradinaru, B. R. Crane, A. J. Di Bilio, P. Matousek, M. Towrie, B. S. Leigh, J. H. Richards, A. Vlcek, Jr. and H. B. Gray, J. Am. Chem. Soc., 2006, 128, 4365–4370 CrossRef CAS.
- K. M. Adams, P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2007, 46, 9172–9181 CrossRef CAS.
- F. Zobi, O. Blacque, R. K. O. Sigel and R. Alberto, Inorg. Chem., 2007, 46, 10458–10460 CrossRef CAS.
- F. Zobi, B. Spingler and R. Alberto, ChemBioChem, 2005, 6, 1397–1405 CrossRef CAS.
- F. Zobi, O. Blacque, H. W. Schmalle, B. Spingler and R. Alberto, Inorg. Chem., 2004, 43, 2087–2096 CAS.
- F. Zobi, B. Spingler, T. Fox and R. Alberto, Inorg. Chem., 2003, 42, 2818–2820 CrossRef CAS.
- K. A. Stephenson, S. R. Banerjee, O. O. Sogbein, M. K. Levadala, N. McFarlane, D. R. Boreham, K. P. Maresca, J. W. Babich, J. Zubieta and J. F. Valliant, Bioconjugate Chem., 2005, 16, 1189–1195 CrossRef CAS.
- K. A. Stephenson, J. Zubieta, S. R. Banerjee, M. K. Levadala, L. Taggart, L. Ryan, N. McFarlane, D. R. Boreham, K. P. Maresca, J. W. Babich and J. F. Valliant, Bioconjugate Chem., 2004, 15, 128–136 CrossRef CAS.
- H. He, M. Lipowska, X. Xu, A. T. Taylor and L. G. Marzilli, Inorg. Chem., 2007, 46, 3385–3394 CrossRef CAS.
- H. He, M. Lipowska, X. Xu, A. T. Taylor, M. Carlone and L. G. Marzilli, Inorg. Chem., 2005, 44, 5437–5446 CrossRef CAS.
- R. S. Herrick, C. J. Ziegler and A. Gambella, Eur. J. Inorg. Chem., 2010, 3905–3908 CrossRef CAS.
- S. L. Binkley, C. J. Ziegler, R. S. Herrick and R. S. Rowlett, Chem. Commun., 2010, 46, 1203–1205 RSC.
- A. M. Hassell, G. An, R. K. Bledsoe, J. M. Bynum, H. L. Carter, III, S. J. J. Deng, R. T. Gampe, T. E. Grisard, K. P. Madauss, R. T. Nolte, W. J. Rocque, L. Wang, K. L. Weaver, S. P. Williams, G. B. Wisely, R. Xu and L. M. Shewchuk, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, D63, 72–79 CAS.
- P. V. Grundler, L. Helm, R. Alberto and A. E. Merbach, Inorg. Chem., 2006, 45, 10378–10390 CrossRef CAS.
- P. V. Grundler, B. Salignac, S. Cayemittes, R. Alberto and A. E. Merbach, Inorg. Chem., 2004, 43, 865–873 CrossRef CAS.
- B. Salignac, P. V. Grundler, S. Cayemittes, U. Frey, R. Scopelliti, A. E. Merbach, R. Hedinger, K. Hegetschweiler, R. Alberto, U. Prinz, G. Raabe, U. Koelle and S. Hall, Inorg. Chem., 2003, 42, 3516–3526 CrossRef CAS.
- U. C. Meier, R. Scopelliti, E. Solari and A. E. Merbach, Inorg. Chem., 2000, 39, 3816–3822 CrossRef CAS.
- N. Lazarova, S. James, J. Babich and J. Zubieta, Inorg. Chem. Commun., 2004, 7, 1023–1026 CrossRef CAS.
- R. S. Herrick, C. J. Ziegler, A. Cetin and B. R. Franklin, Eur. J. Inorg. Chem., 2007, 1632–1634 CrossRef CAS.
- Agilent, Agilent Technologies Ltd., Yarnton, England, 2010.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS.
- G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185–189 CrossRef CAS.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS.
- M. Salmain, B. Caro, F. Le Guen-Robin, J. C. Blais and G. Jaouen, ChemBioChem, 2004, 5, 99–109 CrossRef CAS.
- M. Salmain, J. C. Blais, H. Tran-Huy, C. Compain and G. Jaouen, Eur. J. Biochem., 2001, 268, 5479–5487 CrossRef CAS.
- A. Casini, G. Mastrobuoni, C. Temperini, C. Gabbiani, S. Francese, G. Moneti, C. T. Supuran, A. Scozzafava and L. Messori, Chem. Commun., 2007, 156–158 RSC.
- M. Razavet, V. Artero, C. Cavazza, Y. Oudart, C. Lebrun, J. C. Fontecilla-Camps and M. Fontecave, Chem. Commun., 2007, 2805–2807 RSC.
- I. W. McNae, K. Fishburne, A. Habtemariam, T. M. Hunter, M. Melchart, F. Wang, M. D. Walkinshaw and P. J. Sadler, Chem. Commun., 2004, 1786–1787 RSC.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romao and M. J. Romao, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS.
- Lysozyme, ed. P. Jolles, Academic Press, New York, NY, 1960 Search PubMed.
- H. Schwalbe, S. B. Grimshaw, A. Spencer, M. Buck, J. Boyd, C. M. Dobson, C. Redfield and L. J. Smith, Protein Sci., 2001, 10, 677–688 CrossRef CAS.
- B. R. Franklin, R. S. Herrick, C. J. Ziegler, A. Cetin, N. Barone and L. R. Condon, Inorg. Chem., 2008, 47, 5902–5909 CrossRef CAS.
- R. Waibel, R. Alberto, J. Willuda, R. Finnern, R. Schibli, A. Stichelberger, A. Egli, U. Abram, J.-P. Mach, A. Pluckthun and P. A. Schubiger, Nat. Biotechnol., 1999, 17, 897–901 CrossRef CAS.
- L. Helm, Coord. Chem. Rev., 2008, 252, 2346–2361 CrossRef CAS.
- P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2966–2974 CrossRef CAS.
- R. A. Engh and R. Huber, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, A47, 392–400 CrossRef.
- V. B. Chen, W. B. Arendall, III, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, D66, 12–21 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |