A comparison of methionine, histidine and cysteine in copper(I)-binding peptides reveals differences relevant to copper uptake by organisms in diverse environments
Jeffrey T.
Rubino
a,
Michael P.
Chenkin
a,
Matthew
Keller
b,
Pamela
Riggs-Gelasco
b and
Katherine J.
Franz
*a
aDepartment of Chemistry, Duke University, P.O. Box 90346, Durham, NC 27708-0346, USA. E-mail: katherine.franz@duke.edu; Fax: +1 919-660-1605; Tel: +1 919-660-1541
bDepartment of Chemistry and Biochemistry, College of Charleston, 66 George St., Charleston, SC 29424, USA. E-mail: gelascop@cofc.edu; Fax: +1 843-953-1404; Tel: +1 843-953-7182
First published on 4th November 2010
Abstract
The N-terminal, extracellular regions of eukaryotic high affinity copper transport (Ctr) proteins vary in composition of the Cu(I) binding amino acids: methionine, histidine, and cysteine. To examine why certain amino acids are exploited over others in Ctrs from different organisms, the relative Cu(I) binding affinity and the dependence of binding on pH were examined for 3 peptides of the sequence MG2XG2MK, where X is either Met, His, or Cys. Cu(I) affinity was examined using an ascorbic acid oxidation assay, an electrospray ionization mass spectrometry technique, and spectrophotometric titration with a competitive Cu(I) chelator. The relative affinities of the peptides with Cu(I) reveal a trend whereby Cys > His > Met at pH 7.4 and Cys > Met > His at pH 4.5. Ligand geometry and metric parameters were determined with X-ray absorption spectroscopy. Susceptibility of the peptides to oxidation by hydrogen peroxide and copper-catalyzed oxidative conditions was evaluated by mass spectrometry. These results support hypotheses as to why certain Cu(I) binding amino acids are preferred over others in proteins expressed at different pH and exposed to oxidative environments. The results also have implications for interpreting site-directed mutagenesis studies aimed at identifying copper binding amino acids in copper trafficking proteins.
Introduction
Copper trafficking proteins include integral membrane proteins that transport copper across cell and organelle membranes, copper chaperones responsible for shuttling the cofactor to respective cuproenzymes, and resistance proteins utilized to prevent accumulation of toxic levels of copper.1–4 In contrast to the enzymes and proteins that utilize copper as a cofactor and therefore bind copper in coordination sites that prevent loss of the metal during redox cycling, copper trafficking proteins must provide labile sites that enable metal transfer. To accomplish metal selectivity and promote lability, these sites are often specific for copper in its reduced, +1 oxidation state and bind with either low affinity or with low coordination number so as to facilitate ligand exchange reactions for metal transfer. Methionine, an amino acid that is rarely found in the active sites of cuproenzymes, is emerging as a critical residue involved in mechanisms of copper transport.5 When methionine is found at a cuproprotein active site, it is typically a weakly coordinated axial ligand that plays a supporting role in addition to histidine and cysteine side chains, as in electron-transfer blue copper proteins, for example. The membrane-bound copper transport proteins of the Ctr1 family, however, contain methionine-rich sites, or “Mets motifs” that can accommodate Cu(I) binding in flexible, thioether-only binding sites of the form MX1–2MX1–2M (where X is a non-coordinating amino acid).6Ctr proteins are ubiquitously expressed in eukaryotes and have been identified in yeast, insects, mice, humans, and various plants.7–12 Ctr proteins do not utilize ATP as an energy source to transport copper across membranes.13 Instead, the transporter takes advantage of the favorable concentration gradient as intracellular copper is effectively sequestered. Ctr proteins are symmetrical homotrimers that form a pore at the interface of 3 α helix-containing subunits.14–16 The N-terminal, extracellular region of the yeast protein yCtr1 contains 30 methionines arranged in 8 Mets motifs.7 Interestingly, other organisms also utilize histidine or cysteine in concert with methionine in the N-terminal extracellular regions of Ctr proteins, presumably to recruit Cu(I) to the surface of the cell for transport down the concentration gradient.17,18
Each of these amino acids, methionine, histidine, and cysteine, exhibits different Cu(I) binding characteristics that can vary based on environmental conditions like pH and the presence of oxygen. These different characteristics appear to have been utilized during evolution to fine-tune the binding affinity of copper trafficking proteins under specific conditions, allowing them to function optimally.
Yeast acidify their surrounds to pH 4–5.19 As cysteine and histidine have ionizable sides chains with pKa values of 8 and 6, respectively, their ability to bind copper should diminish as the pH decreases. Cysteine is also susceptible to oxidation, forming disulfide bonds with neighboring cysteine residues. Mets motifs would therefore appear to be optimal for yeast because they exhibit an affinity for Cu(I) that is independent of pH, and they are less susceptible to oxidation.
Some green algae express cysteine in addition to methionine on the N-terminus of Ctr proteins. The best studied of these are the green alga Chlamydomonas versions of the protein containing six Cys-Met motifs, the most common being CX2MX2MX2CX5/6C.12 These algae often grow in hypoxic, nutrient-poor environments,20 where the low oxygen content results in the precipitation of Cu(I) as insoluble sulfide salts.21 To compensate for the low environmental levels of copper, Chlamydomonas Ctr evolved to bind the metal with an affinity 10–20 times greater than that of the yeast version of the protein.7,22,23 Methionine motifs, exhibiting micromolar KD values,6 alone would not be able to accommodate this high affinity of copper transport, requiring utilization of another copper binding amino acid. Oxidation of cysteine is less problematic considering the hypoxic environment.
Humans and other mammals express histidine and methionine on the cell surface portion of their Ctr proteins. In separate work, we have examined the copper-binding properties of model peptides containing histidine and methionine residues located on the N-terminus of human hCtr1 and characterized a Cu(II) binding site composed of an H2N-XXH amino terminal copper/nickel (ATCUN) motif as well as a high affinity Cu(I) site in a mixed coordination environment of histidine and methionine.24 These histidine and methionine motifs are found in the oxidative extracellular environment at physiological pH 7.4. Cysteine would not be suited for these conditions as it could easily be oxidized. At this pH, Cu(II) is not soluble in its aquated form, and is usually found stabilized in extracellular copper binding proteins such as ceruloplasmin and serum albumin. Histidine is likely required by hCtr1 as methionine motifs alone are presumably not strong enough to recruit copper from these carrier proteins. The methionine motifs likely play a more important role in copper acquisition in the small intestine, where the pH ranges from acidic to neutral pH (5–7.3).25 At acidic pH dietary copper is soluble and histidine is likely not the best Cu(I) ligand due to the protonatable side chain.
In previous work, we described two interesting characteristics of methionine motifs: they exhibit pH-independent Cu(I) binding with low-to-moderate affinity in the micromolar range, and they appear to be unaffected by metal catalyzed oxidation (MCO) reactions.6 In the current study, we examined a series of model peptides at different pH and various oxidative conditions in order to better understand the individual contributions to binding affinity and susceptibility to oxidation of methionine, histidine, and cysteine in a copper-binding motif (Table 1). These results further characterize the unique properties of methionine-only Cu(I) coordination, and reveal why nature evolved the use of specific copper binding amino acids for different environmental conditions.
| Peptide | Sequence |
|---|---|
| MG2MG2M | Ac-M-G-G-M-G-G-M-K-NH2 |
| MG2HG2M | Ac-M-G-G-H-G-G-M-K-NH2 |
| MG2CG2M | Ac-M-G-G-C-G-G-M-K-NH2 |
Material and methods
Peptide synthesis and purification
Peptides were synthesized using a Protein Technologies PS3 automated peptide synthesizer in 0.1 mmol scale on PAL-PEG-PS resin (Applied Biosystems). Standard Fmoc (9-fluorenylmethoxy-carbonyl)-protected amino acids (Chem-Impex and Novabiochem) were coupled in 20 min cycles with HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate) (Novabiochem) and N-methylmorpholine (NMM) (Acros) in N,N′-dimethylformamide (DMF) (Caledon). Fmoc protecting groups were removed by using 20% piperidine (Aldrich) in DMF. The N-termini of peptides were acetylated by using acetic anhydride (Aldrich) and NMM. Cleavage from resin and the removal of side chain protecting groups was accomplished by treating resin with a 10-mL mixture of 95% trifluoroacetic acid (TFA) (Sigma-Aldrich), 2.5% 1,2-ethandithiol (EDT) (Fluka), and 2.5% triisopropylsilane (TIS) (Aldrich) under nitrogen while shaking for 4 h. During the final hour of incubation additional EDT (100 μL) and 65 μL of bromotrimethylsilane (TMSBr) (Acros) were added to prevent methionine oxidation. Peptide was precipitated from solution by evaporating off TFA with a nitrogen stream, followed by three washes with diethyl ether (Caledon). Purification was accomplished by semi-preparative reversed-phase HPLC on a YMC C18 column with a linear 40 min gradient from 7 to 70% acetonitrile in water with 0.1% TFA. Purity was validated to be greater than 95% by analytical HPLC. Mass of each peptide was determined by ESI-MS. MG2MG2M calcd for C31H56N10O9S3: 808.3. Found: (M + H+) 809.6. MG2HG2M calcd for C32H54N12O9S2: 814.3. Found: (M + H+) 815.4. MG2CG2M calcd for C29H52N10O9S3: 780.3. Found: (M + H+) 781.3.Preparation of stock materials
Peptide stock solutions were prepared by adding 5–15 mg of lyophilized peptide to a pre-tared microcentrifuge tube containing 250–1000 μL of nanopure water. Concentration was verified by a UV-visible spectroscopic assay utilizing extinction coefficients generated by data from quantitative amino acid analysis (Dana Farber Cancer Institute, Boston, MA and AAA Laboratory, Mercer Island, WA). Absorption spectra were recorded in a 1-cm, 1-mL Starna masked quartz cuvette (Atascadero, CA) on a Varian (Palo Alto, CA) Cary 50 UV-vis spectrophotometer. Peptides in lyophilized and solution form were stored at −20 °C. Ascorbic acid (H2Asc) solution was prepared fresh daily from L-(+)ascorbic acid (Acros 99%) in nanopure water. Stock solutions of CuSO4·5H2O (Sigma) and [Cu(CH3CN)4]PF6 (Aldrich) were prepared in nanopure water, and in degassed acetonitirile, respectively. The concentration of [Cu(CH3CN)4]PF6 was confirmed by using bathocuproine disulfonic acid (BCS) (MP Biomedicals) (Cu(BCS)23−ε483 = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 M−1 cm−1).26 All experiments using [Cu(CH3CN)4]PF6 were conducted using a concentrated stock solution of [Cu(CH3CN)4]PF6 in acetonitrile to minimize the percentage of acetonitrile in the final solutions to 0.25–2%.
300 M−1 cm−1).26 All experiments using [Cu(CH3CN)4]PF6 were conducted using a concentrated stock solution of [Cu(CH3CN)4]PF6 in acetonitrile to minimize the percentage of acetonitrile in the final solutions to 0.25–2%.
Ascorbic acid oxidation assay
Experiments were conducted in a multi-well plate using a Perkin Elmer 1420 Multilabel Counter Victor3 V Model with attached automated injector system containing two pumps. Detailed experimental conditions were identical to those described previously.6 Briefly, the oxidation of 400 μM ascorbic acid at pH 4.5 by 5 to 16 μM CuSO4 in the presence of oxygen was monitored spectrophotometrically and the observed rate constant was correlated to the concentration of total copper ([Cu]T) to obtain a standard curve of kobsvs. [Cu]T. The assay was then conducted with 400 μM ascorbic acid and 8 μM CuSO4 in the presence of increasing concentrations (10–100 μM) of peptide in order to generate plots of kobsvs. [P]T. By comparison to the standard curve, these data provide values for the amount of catalytically available copper [Cu]F at each peptide concentration.Under these conditions, a 1:1 peptide copper binding stoichiometry is assumed as the concentration of peptide is considerably greater than the concentration of CuSO4, and similar conditions examined on ESI-MS only reveal masses associated with 1:1 complexes. The equilibrium expression in eqn (1) can be expressed as a dissociation constant, KD in eqn (2), where the peptide–Cu complex [PCu] and the concentration of free, or unbound peptide, [P]F, are defined by the mass balance equations in eqn (3) and (4). The kobsvs. [P]T plot is then fit to the expression in eqn (5) to yield the effective KD:
| P + Cu+ ⇄ PCu | (1) |
| KD = [P]F[Cu]F/[PCu] | (2) |
| [PCu] = [Cu]T − [Cu]F | (3) |
| [P]F = [P]T − [PCu] | (4) |
 | (5) |
Electrospray ionization mass spectrometry
Electrospray ionization mass spectrometry (ESI-MS) experiments were performed on an electrospray quadrupole ion trap mass spectrometer (1100 Series LC/MSD Trap, Agilent, Palo Alto, CA) as described previously.6 Tandem mass spectrometry (MS/MS) experiments were performed by isolating the ions at 4 m/z width and using 1.2 activation amplitude.For effective KD determination, a standard curve was generated by recording the intensity of the peak associated with free peptide at concentrations ranging from 2–10 μM in 5 mM H2Asc for studies at pH 4.5, and in 5 mM H2Asc in 10 mM ammonium acetate (NH4OAc) (Mallinckrodt) buffer for studies at pH 7.4. Dilute NH4OH (Fisher) was used to adjust H2Asc to 4.5, and the H2Asc/NH4OAc buffer pH to 7.4. The intensity of the free peptide peak was then recorded with increasing concentrations of CuSO4 ranging from 5–50 μM in 5 μM increments, while the peptide concentration was kept constant at 10 μM in 5 mM H2Asc or 5 mM H2Asc in 10 mM NH4OAc buffer. The effective KD was calculated according to eqn (2). The concentration of free peptide [P]F was determined by comparison to the standard curve. The concentration of the copper-peptide complex, [PCu], and free Cu, [Cu]F, were determined from rearranged mass balance eqn (4) and (3), respectively. For each peptide a [P]Fvs. [Cu]T graph was generated and fit to the quadratic equation (similar to eqn (5)) to yield the effective KD.
Spectrophotometric titrations of Cu(BCA)2
Concentrated solutions of peptides were titrated into solutions containing 15–25 μM [Cu(CH3CN)4]PF6 and 45–75 μM 2,2′-biquinoline-4,4′-dicarboxylic acid (BCA) (Fluka) in 10 mM MOPS (3-[N-morpholine]propane-sulfonic acid, Sigma >99.5%, titration) at pH 7.4 or 10 mM H2Asc at pH 4.5. BCA was prepared in Chelex-treated nanopure water, degassed with argon and made slightly basic with 1 M NaOH to promote dissolution. Stock MOPS and H2Asc buffers were prepared in Chelex-treated nanopure water, degassed with argon and brought to pH 7.4 and pH 4.5 using 1 M NaOH.Effective KD values were determined by monitoring the decrease in the characteristic Cu(BCA)2 complex absorbance at 562 nm (ε = 7900 M−1).27 The system of interest is shown in eqn (6), which shows the exchange reaction between BCA and P for Cu(I) binding. The overall equilibrium constant, or Kex, for the competition is shown in eqn (7), and is equal to the ratio of KPCu,the peptide–copper association constant, and β2, where β2 is the association constant for Cu(BCA)2 defined in eqn (8). Kex is determined experimentally by measuring [Cu(BCA)2] spectrophotometrically and determining the values of the remaining variables from the mass balance eqn (9)–(11). Once Kex is obtained, KPCu can be calculated using one of the reported β2 values for Cu(BCA)2.27,28 The values reported here utilized a β2 value 4.6 × 1014 M−2 determined by Rosenzweig,28 as the resulting KD values seem more biologically relevant. The constant reported by Wedd27 yields KD values that suggest a tighter binding affinity by 2–3 orders of magnitude.
 | (6) |
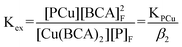 | (7) |
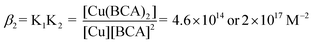 | (8) |
| [PCu] = [Cu]T − [Cu(BCA)2] | (9) |
| [P]F = [P]T − [PCu] | (10) |
| [BCA]F = [BCA]T − 2[Cu(BCA)2] | (11) |
Metal catalyzed oxidation reactions
MCO reactions were prepared with 100 μM peptide, (MG2MG2M, MG2HG2M, or MG2CG2M) with or without 100 μM CuSO4, 1 mM H2O2, and 5 mM H2Asc in 10 mM NH4OAc buffer, pH 7.4 and incubated at room temperature for 24 h before direct injection onto the ESI-MS. ESI-MS was used to identify oxidation products, and MS/MS was used to determine the sites of amino acid oxidation.X-Ray absorption spectroscopy
Solution samples of MG2HG2M and MG2CG2M were generated by mixing stock solutions of purified peptide, H2Asc, and CuSO4. Glycerol was added to minimize ice crystals from forming during rapid freezing in liquid nitrogen. Final concentrations were 4 mM peptide, 2 mM CuSO4, 5 mM H2Asc, and 30% glycerol for MG2MCG2M. For MG2HG2M, final concentrations were 1.2 mM peptide, 0.9 mM CuSO4, 5 mM H2Asc, and 30% glycerol. The final pH was 3.5 for the XAS samples. The preparation and results of fitting analysis for the MG2MG2M peptide were reported previously6 and spectra are shown here for comparison purposes. X-ray absorption spectra were measured at the Stanford Synchrotron Radiation Laboratory at beamline 7-3 using a 30 element solid state Ge detector (Canberra) to monitor the copper Kα fluorescence. The storage ring was operating in top-off mode with beam current maintained at 300 mA. The beamline optics include a mirror for harmonic rejection. Samples were maintained at 10 K in a continuous flow liquid helium Oxford Instruments cryostat. The spectrum of copper foil was measured simultaneously to serve as an internal energy calibration.Data were analyzed using EXAFSPAK. Single and multiple scattering models were generated using an interface to FEFF.29 A scale factor of 1 and a ΔE0 value of −17 eV were used based on calibration to model compounds and on optimization. Other fitting and data collection details were recently reported.6
Results
Model peptides listed in Table 1 were derived from the MG2MG2M scaffold peptide analyzed in previous work.6 The central methionine residue was replaced with the other copper binding amino acids, histidine and cysteine (MG2HG2M and MG2CG2M, respectively), in order to elucidate the effect each of the individual amino acids has on the Cu(I) binding affinity of a copper binding motif. Methionine seemed an appropriate scaffold to study the histidine and cysteine contributions as methionine presumably exhibits the weakest affinity for Cu(I). These affinities were examined at 2 biologically relevant pHs, 4.5, the pH to which yeast acidify their media, and 7.4, human physiological pH. A variety of techniques was utilized to determine effective KD values for these peptides, however not all techniques were used for each peptide due to the inherit limitations of each assay. These techniques included an ascorbic acid oxidation assay, which has been used to determine the values for Cu(I) binding peptides exhibiting KDs weaker than 1 × 10−6 M at pH 4.5.6,30 Accurate KD values cannot be determined for peptides with tighter affinities as the equilibrium on the P + Cu+ ⇄ PCu system lies too far to the right, which was the scenario for MG2CG2M. Peptides that bind Cu(II) cannot be studied with this assay due to the potential to form ternary complexes of peptide–Cu(II)–HAsc− that themselves are catalytically active, which negates the underlying assumption that [Cu]F is the catalytic species and can be used to calculate the effective KD of the peptides for Cu(I) (as described thoroughly in previous work6). This scenario was the case for MG2HG2M. A method utilizing ESI-MS, which can determine KD values in the same range as the ascorbic acid oxidation assay but can be conducted at variable pH, was also used.6,30 Again, accurate KD values cannot be determined for peptides with KD values lower than ∼1 × 10−6 M, as the equilibrium on the P + Cu+ ⇄ PCu system lies too far to the right, which was the scenario for MG2CG2M. Spectrophotometric titrations using the Cu(I) specific chelator BCA have been used in the literature to determine the binding affinity of a variety of Cu(I) binding peptides and proteins with KD values ranging 1 × 10−9–1 × 10−12 M.24,28,31,32 This technique was suitable to study MG2CG2M at both pH values due to its high affinity for Cu(I). MG2MG2M at both pHs and MG2HG2M at pH 4.5 could not effectively compete with BCA for Cu(I).Determination of peptide–Cu(I) KD values
Results of the determination of effective Cu(I) KD values for the model peptides, and the techniques used, are summarized in Table 2. To determine the effective binding affinity of MG2MG2M for Cu(I), the rate of copper-catalyzed ascorbate oxidation was determined as a function of peptide concentration, and the observed rate constant (kobs) was compared to a standard curve of kobsvs. [Cu]T. The concentration of the catalytically available copper in solution is directly proportional to kobs, which allows for the quantitation of free copper in solutions where peptide is present and a portion of the total copper is chelated and unavailable for catalysis. The plot of kobsvs. peptide concentration for MG2MG2M is shown in Fig. 1. Data were fit to eqn (5) to determine an effective KD for Cu(I) at 4.4(2) × 10−6 M at pH 4.5. | ||
| Fig. 1 k obs versus the concentration of total peptide added (P = MG2MG2M) for the copper catalyzed oxidation of ascorbic acid (8 μM CuSO4, 400 μM H2Asc, pH 4.5). The line is the fit of the data to eqn (5) with R = 0.99. | ||
:1 peptide–Cu(I) complexes with MG2MG2M, MG2HG2M, and MG2CG2M at pH 4.5 and 7.4 determined by ascorbic acid assay (AAA), mass spec assay (ESI), and competition assay (BCA)
| Peptide | K AAAD pH 4.5 (M)a | K ESID pH 4.5 (M)b | K BCAD pH 4.5 (M)c | K ESID pH 7.4 (M)d | K BCAD pH 7.4 (M)e |
|---|---|---|---|---|---|
a In 400 μM H2Asc.
b In 5 mM H2Asc.
c In 10 mM H2Asc.
d In 5 mM H2Asc, 10 mM NH4OAc.
e In 10 mM MOPS.*Could not be determined, binds Cu(II),  Could not be determined, exhibits too high a Cu(I) affinity, ◆ Could not be determined, exhibits too low a Cu(I) affinity. Values in parentheses represent the upper and lower limit detected using the indicated technique and conditions. Could not be determined, exhibits too high a Cu(I) affinity, ◆ Could not be determined, exhibits too low a Cu(I) affinity. Values in parentheses represent the upper and lower limit detected using the indicated technique and conditions. |
|||||
| MG2MG2M | 4.4(2) × 10−6 | 4(1) × 10−6 | ◆ | 4.8(7) × 10−6 | ◆ |
| MG2HG2M | * | 1.3(1) × 10−5 | ◆ | (> 1 × 10−6) | (<1 × 10−9) |
| MG2CG2M |

|

|
3.5(2) × 10−9 |

|
6(1) × 10−11 |
ESI-MS was used to determine KD values for MG2HG2M and MG2MG2M at pH 4.5, and MG2MG2M at pH 7.4. For these experiments, standard curves were generated for each peptide in order to correlate the intensity of the apo-peptide peak to concentration at each condition of interest. There is a direct correlation between the concentration of peptide in solution and the intensity of the peak associated with the mass of the peptide plus one proton (P + H+). If the concentration of peptide is kept constant and the concentration of copper increases, the intensity of the apo-peptide decreases and a peak associated with the mass of a peptide copper adduct emerges. It can therefore be assumed that the loss of the apo-peptide signal is the result of the formation of the peptide–copper complex. The concentration of the peptide–copper complex cannot be determined directly due to differences in ionization efficiencies of adducts and charge states. Once apo-peptide intensities are converted into concentration, the quadratic equation is fit to the data of [P]Fvs. [Cu]T plots to determine KD.
MG2MG2M exhibited KD values of 4(1) × 10−6 M and 4.8(7) × 10−6 M at pH 4.5 and 7.4, respectively. MG2HG2M exhibited a KD of 1.3(1) × 10−5 M at pH 4.5. At pH 7.4 MG2HG2M appears to exhibit a KD value weaker than 1 × 10−6 M, which as described earlier, is the lower limit of detectable binding affinity utilizing this method.
BCA is a Cu(I)-specific chelator that exhibits a strong absorbance at 358 nm (ε = 42900 M−1) and 562 nm (ε = 7900 M−1) upon formation of the [Cu(BCA)2]3− complex.27,33 Concentrated solutions of peptide were titrated into solutions of known Cu(BCA)2 concentration in an attempt to remove copper from the complex. Peptides that compete with BCA for Cu(I) cause a decrease in the absorbance of the characteristic peaks associated with the Cu(BCA)2 complex. The loss was quantitated using the known extinction coefficients and used to determine an effective KD for MG2CG2M at pH 7.4 and pH 4.5. Experimental results of the titration of up to 20 equivalents of MG2CG2M into 25 μM Cu(BCA)2 in 10 mM MOPS buffer at pH 7.4 are shown in Fig. 2. The insert shows that as the concentration of peptide increases, there is a decrease in absorbance of the peak at 562 nm until it reaches zero, where presumably all copper in solution is bound by the peptide. An effective KD value was determined at each point, the average being 6(1) × 10−11 M.
![Titration of up to 20 equivalents of MG2CG2M into Cu(BCA)2 at pH 7.4 (25 μM [Cu(CH3CN)4]PF6, 75 μM BCA, in 10 mM MOPS). Insert shows the decrease in the absorbance of the characteristic Cu(BCA)2 complex peak at 562 nm.](/image/article/2011/MT/c0mt00044b/c0mt00044b-f2.gif) | ||
| Fig. 2 Titration of up to 20 equivalents of MG2CG2M into Cu(BCA)2 at pH 7.4 (25 μM [Cu(CH3CN)4]PF6, 75 μM BCA, in 10 mM MOPS). Insert shows the decrease in the absorbance of the characteristic Cu(BCA)2 complex peak at 562 nm. | ||
The KD value for MG2CG2M at pH 4.5 was determined in the same fashion. Here, up to 100 equivalents of peptide were titrated into 15 μM Cu(BCA)2 in 10 mM H2Asc buffer at pH 4.5. The absorbance of the 562 nm peak was not reduced to zero suggesting that copper was not completely removed from the Cu(BCA)2 complex, even at the highest concentration of peptide tested (Fig. 3). MG2CG2M does appear to compete with Cu(BCA)2 for copper, as there is a continual decrease in the absorbance at 562 nm as more peptide is added to the system. The effective KD value was estimated to be 3.5(2) × 10−9 M.
![The absorbance of the characteristic Cu(BCA)2 peak at 562 nm of solutions containing 15 μM [Cu(CH3CN)4]PF6 and 45 μM BCA in 10 mM H2Asc buffer pH 4.5 upon addition of up to 100 equivalents of MG2CG2M.](/image/article/2011/MT/c0mt00044b/c0mt00044b-f3.gif) | ||
| Fig. 3 The absorbance of the characteristic Cu(BCA)2 peak at 562 nm of solutions containing 15 μM [Cu(CH3CN)4]PF6 and 45 μM BCA in 10 mM H2Asc buffer pH 4.5 upon addition of up to 100 equivalents of MG2CG2M. | ||
This experiment was also attempted for MG2HG2M at pH 7.4, but an effective KD could not be determined from the data. Here, up to 800 equivalents of peptide were titrated into 15 μM Cu(BCA)2 in 10 mM MOPS buffer at pH 7.4. Even in the presence of such an excessive amount of peptide the absorbance of 562 was barely reduced (Fig. 4). The decrease in the absorbance plateaus after the addition of 300 equivalents of peptide, suggesting an equilibrium has been reached and that MG2HG2M cannot effectively compete with BCA for copper. Because the binding affinity for Cu(I) of MG2HG2M at pH 7.4 appears to be tighter than what can be detected by the ESI-MS method but weaker than what can be detected by titration into Cu(BCA)2, the KD value must lie somewhere in the range of 1 × 10−6–1 × 10−9 M.
![The absorbance of the characteristic Cu(BCA)2 peak at 562 nm of solutions containing 15 μM [Cu(CH3CN)4]PF6 and 45 μM BCA in 10 mM MOPS buffer pH 7.4, upon titration with up to 800 equivalents of MG2HG2M.](/image/article/2011/MT/c0mt00044b/c0mt00044b-f4.gif) | ||
| Fig. 4 The absorbance of the characteristic Cu(BCA)2 peak at 562 nm of solutions containing 15 μM [Cu(CH3CN)4]PF6 and 45 μM BCA in 10 mM MOPS buffer pH 7.4, upon titration with up to 800 equivalents of MG2HG2M. | ||
One possibility for the behavior observed in Fig. 4 is the formation of a ternary complex of peptide, BCA, and Cu+ ion that also absorbs at 562 nm. Three different mass spectrometry techniques were employed in an attempt to find a mass associated with such a complex: ESI-MS, matrix assisted laser desorption ionization mass spectrometry (MALDI-MS), and direct analysis in real time mass spectrometry (DART-MS); however, no such peak was found. In a UV-vis experiment, [Cu(CH3CN)4]PF6 was incubated with excess peptide, and then titrated with less than 1 equivalent of BCA in an attempt to promote ternary complex formation. Comparison of spectra did not reveal any new features that could be attributed to the formation of a ternary complex. While no evidence for a ternary complex has been obtained, its existence cannot be ruled out.
Metal catalyzed oxidation reactions
Metal catalyzed oxidation reactions are often used to identify copper and iron binding sites, utilizing conditions similar to those in the ascorbic acid oxidation assay.34–40 Upon oxidation of Cu(I) to Cu(II) or Fe(II) to Fe(III), reactive oxygen species are generated at the sites of the metal in close proximity to metal binding ligands.41 Hence, the metal binding ligands are the residues most likely to succumb to oxidative modification. Interestingly, the Mets peptides in previous studies6 did not experience oxidation under these conditions even after prolonged exposure to ascorbate and copper sulfate. To understand this phenomenon, and to examine the susceptibility of methionine, histidine, and cysteine in copper binding sites to oxidation, model peptides were treated with H2O2 and a variety conditions often used in metal catalyzed oxidation reactions (Table 3).Metal catalyzed oxidation is usually specific, involving only those amino acids that interact with copper. As glycine has a very low propensity for oxidation, only oxidation products of methionine, histidine, and cysteine were expected. The oxidized histidine product observed was 2-imidazolone, unless specifically noted. The oxidized methionine product observed was methionine sulfoxide. Additions of 2 oxygen atoms to histidine or methionine to give 5-hydroxy-imidazolone or methionine sulfone were rare. Oxidation of cysteine resulted in the formation of a disulfide bond and a dimer of 2 peptide molecules with a total of 4 methionine residues.
ESI-MS was used to identify the masses of oxidation products, and MS/MS was used to determine the specifc amino acids oxidized. The MS/MS process results in the collisional decomposition of the peptide, breaking into well defined ions. The most common collisional products are the b and y ions that form upon cleavage of the peptide bond.42,43 These b and y ions are assigned numbers based on which peptide bond is broken, and the number of amino acids the ion contains. Another phenomenon observed in the ESI-MS is the collisional decomposition of methionine sulfoxide.44 This oxidative product of methionine is easily identified in ESI-MS spectra by the loss of 64 mass units due to liberation of methanesulfenic acid (CH3SOH) upon formation of vinyl glycine. In the following spectra the asterisk (*) denotes peaks associated with this loss of 64 mass units.
All three peptides were treated with condition A (H2O2) and incubated at room temperature for 24 h to determine which amino acid is more prone to oxidation by hydrogen peroxide. All three peptides were oxidized. ESI-MS analysis of treatment of MG2MG2M with condition A confirmed the generation of 2 main oxidation products, Pox and P2ox, along with a minor P3ox product containing 1, 2 or 3 O atoms, respectively (not shown). ESI spectra of treatment of MG2CG2M with condition A is shown in Fig. 5. Condition A induced the oxidative formation of a disulfide bond between 2 peptide molecules, with peaks corresponding to the mass-to-charge (m/z) value of the dimer (P2), with no detectable signal for the monomer. Oxidation products of the dimer include addition of 1, 2, and 3 O atoms. The m/z values and their assignments are given in the figure caption. Based on these results, methionine and cysteine residues are easily oxidized by H2O2, with cysteine being more susceptible to oxidation than methionine. The MG2CG2M peptide exists only as a dimer after treatment, suggesting complete cysteine oxidation, while some methionine residues remain unoxidized after treatment of both MG2CG2M and MG2MG2M.
 | ||
| Fig. 5 Mass spectrum of oxidation products of MG2CG2M treated with condition A (100 μM MG2CG2M, 1mM H2O2 in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h). Peaks associated with a disulfide bonded dimer are observed at m/z = 1559.5 (P2 + H+, z = 1) and 781.3 (P2 + 2H+, z = 2). Oxidation products of the dimer are also observed, 1O at 1575.5 (P2 ox + H+, z = 1) and 788.4 (P2 ox + 2H+, z = 2), 2O at 1591.5 (P2 2ox + H+, z = 1) and 796.4 (P2 ox + 2H+, z = 2), and 3O at 1607.5 (P2 3ox + H+, z = 1) and 804.4 (P2 3ox + 2H+, z = 2). | ||
Treatment of MG2HG2M with condition A (H2O2) generated intense Pox and P2ox products that correspond to the addition of 1 and 2 O atoms to the peptide, respectively. Fig. 6a shows the standard MS, with the m/z values and their assignments noted in the figure caption. MS/MS analysis was required to determine which specific amino acids were oxidized. The MS/MS of Pox is shown in Fig. 6b, prominent peaks include an oxidized full-length peptide with loss of CH3SOH (*Pox) along with unoxidized fragment ions b7, b6, b5, and b4 (see labeling diagram in Fig. 6), and oxidized *b7 and *b6 ions with loss of CH3SOH. The MS/MS of P2ox is shown in (c), prominent peaks include *P2ox and 2*P2ox species, which correspond to doubly oxidized peptides with the loss of 1 or 2 equivalents of CH3SOH. An oxidized *b6 ion is also observed. The observation that the only oxidized species detected are those that lose CH3SOH in the MS/MS identifies methionine as the site of oxidation and demonstrates that histidine is not prone to oxidation by H2O2.
 | ||
| Fig. 6 ESI-MS/MS spectra of oxidation products of MG2HG2M treated with condition A (100 μM MG2HG2M, 1mM H2O2 in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h). (a) standard MS, prominent peaks include peptide plus 1 proton at m/z = 815.4 (P + H+, z = 1), peptide plus 2 protons at 408.2 (P + 2H+, z = 2), and oxidation products including addition of 1O at 831.3 (Pox + H+, z = 1) and 416.2 (Pox + 2H+, z = 2), and 2O at 847.4 (P2ox + H+, z = 1) and 424.2 (P2ox + 2H+, z = 2). (b) MS/MS of Pox + H+, prominent peaks include Pox + H+ minus CH3SOH (*Pox + H+) at 767.4, b7, b6, b5, and b4 ions at 686.3, 539.1, 464.2, and 425.2, respectively, and oxidized b7 and b6 minus CH3SOH (*b7ox and *b6 ox, respectively) at 622.3 and 491.3. (c) MS/MS of P2ox+H+, prominent peaks include *P2ox + H+ at 783.4, a peak associated with the loss of 2 equivalents of CH3SOH (2*P2ox + H+) at 719.3, and *b6 ox ion at 491.3. ‘*’ indicates the loss of CH3SOH, −64 m/z. | ||
All three peptides were treated with condition B (H2Asc, CuSO4) as well as conditions C (H2Asc) and D (CuSO4) to serve as controls. Upon examination of these reactions after 24 h incubation at room temperature, only MG2HG2M treated with condition B was oxidized. No peaks associated with the masses of oxidation products for MG2MG2M or MG2CG2M were observed in the ESI-MS. Fig. 7 shows the ESI-MS spectra of the MG2HG2M peptide solution (a), including MS/MS of the different oxidation products (b–e) in an attempt to determine which amino acids were being oxidized, and a diagram of the y and b ions and their respective masses. The standard MS (a) includes peaks associated with masses for the unoxidized native peptide (P + H+) as well as oxidation products Pox, P2ox, and P3ox that correspond to the addition of 1, 2, or 3 O atoms, respectively. The observed m/z values and their assignments are given in the figure caption. The MS/MS of the unoxidized peptide gives b7, y7, b6, b5, and b4 fragment ions with m/z values matching those predicted in the diagram in Fig. 7. The pattern of oxidized ions observed in the MS/MS of Pox, P2ox, and P3ox (Fig. 7c–e) suggests a mixture of MCO products wherein either methionine or histidine has been oxidized. For example, b6 ox is the most intense peak observed in (c), but the absence of a corresponding *b6 ox that would correspond to the loss of CH3SOH suggests that b6 ox corresponds to a peptide fragment containing 2-imidazolone as opposed to methionine sulfoxide. In the MS/MS spectrum in (d), the presence of a *P2ox peak confirms methionine oxidation has occured, but the lack of a 2*P2ox suggests histidine, not methionine, is the second site of oxidation. Despite the low ion count detected in the ESI-MS, the presence of the 2*P3ox in MS/MS spectrum (e) indicates that both methionine residues are oxidized in addition to histidine. Taken together, these tandem mass spectral data show that oxidation of both methionine and histidine occurs in the MG2HG2M peptide exposed to a combination of copper and ascorbic acid.
 | ||
| Fig. 7 ESI-MS/MS spectra of oxidation products of MG2HG2M treated with condition B (100 μM MG2HG2M, 100 μM CuSO4, 5 mM H2Asc in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h). (a) Standard MS, prominent peaks include peptide plus 1 proton (P + H+) at m/z = 815.4, peptide plus 2 protons at 408.2 (P + 2H+, z = 2), and oxidation products including addition of 1O at 831.3 (Pox + H+, z = 1) and 416.2 (Pox + 2H+, z = 2), 2O at 847.4 (P2ox + H+, z = 1), and 3O at 863.3 (P3ox + H+, z = 1). (b) MS/MS of P+H+, peaks include P + H+ minus H2O (°P + H+) at 797.4, and b7, y7, b6, b5, and b4 ions with m/z matching those predicted in the diagram above. (c) MS/MS of Pox + H+, prominent peaks include °Pox + H+ at 813.3, *Pox + H+ at 767.4, b7 ox, y7 ox, and b6 ox, at 686.2, 658.3, and 555.2, respectively, *b7 ox at 622.2, and b6. (d) MS/MS of P2ox + H+, prominent peaks include °P2ox + H+ at 829.3, *Pox + H+ at 783.4, b7 2ox, y7 2ox, *b7 ox, b6 2ox, and *b6 2ox ions, at 702.2, 674.3, 638.2, 571.2, and 507.2, respectively. (e) MS/MS of P3ox + H+, prominent peaks include °P3ox + H+ at 845.3, *P3ox + H+ at 799.4, 2*P3ox+H+ at 735.3, and *b73ox, and b62ox ions at 654.2, and 571.2, respectively. ‘°’ indicates a loss of water, −18 m/z, ‘*’ indicates the loss of CH3SOH, −64 m/z. | ||
To further probe the susceptibility of methionine, histidine, and cysteine to oxidation in MCO reactions, iterations of conditions A (H2O2) were employed, including addition of H2Asc (condition E) and addition of CuSO4 (condition F). Addition of H2Asc resulted in less oxidation for all peptides. Results of the treatment of MG2MG2M with condition E are shown in Fig. 8a, shown in comparison to treatment with condition A (Fig. 8b). In comparison to spectrum (b), spectrum (a) has significantly fewer oxidation products; spectrum (b) has intense Pox and P2ox peaks, while spectrum (a) has only a weak Pox. These results suggest that ascorbic acid alone acts as an antioxidant, scavenging H2O2 before it can react on peptide methionine residues. This is in contrast to condition B (H2Asc, CuSO4) where in concert with CuSO4, ascorbic acid acts as a pro-oxidant.
 | ||
| Fig. 8 ESI-MS comparison of oxidation products of MG2MG2M treated with conditions E and A. (a) 100 μM MG2MG2M, 1mM H2O2, 5 mM H2Asc in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h, only one oxidation product is observed, Pox + H+ at 825.3 m/z. (b) 100 μM MG2MG2M, 1mM H2O2, in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h, multiple oxidation products are observed, Pox + H+ at 825.3 m/z and P2ox + H+ at 841.3 m/z. | ||
Addition of CuSO4 to H2O2 (condition F) resulted in the complete oxidation of copper binding amino acids in all peptides. MS/MS indicated the presence of 2 methionine sulfoxides and the 5-hydroxy-2-imidazolone (not shown). Results of the treatment of MG2HG2M with condition F (H2O2, CuSO4) are shown in Fig. 9a, in comparison to treatment with condition A (H2O2, Fig. 9b). The addition of CuSO4 significantly increased the number oxidation products, with intense peaks corresponding to the addition of 2–4 O atoms. The absence of unoxidized or even singly oxidized peptide suggests complete oxidation of the peptide, indicating CuSO4 acts as a pro-oxidant in concert with H2O2.
 | ||
| Fig. 9 ESI-MS comparison of oxidation products of MG2HG2M treated with conditions F and A. (a) 100 μM MG2HG2M, 100 μM CuSO4, 1mM H2O2 in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h, multiple oxidation products are observed: addition of 2O, P2ox + H+ at 847.4 and P2ox + 2H+ at 424.2, addition of 3O, P3ox + H+ 863.3, and addition of 4O, P4ox + H+ at 879.3. (b) 100 μM MG2HG2M, 1mM H2O2 in 10 mM NH4OAc buffer at pH 7.4 incubated for 24 h, only two oxidation products are observed, addition of 1O, Pox + H+ at 831.5 and Pox + 2H+ at 416.2, and addition of 2O, P2ox + H+ at 847.4 and Pox + 2H+ at 424.2. | ||
X-Ray absorption spectroscopy
The X-ray absorption near edge spectra for the three peptides are shown in Fig. 10. In order to compare the MG2CG2M and MG2HG2M samples directly to those of MG2MG2M reported previously,6 the samples were prepared with substoichiometric copper and excess ascorbic acid to ensure formation of Cu(I). The final pH of the solutions was 3.5. The presence of the prominent 1s-to-4p transition at 8984 eV in the edge of both the MG2MG2M and the MG2CG2M peptides indicates a Cu(I) oxidation state. The intensity of this feature is consistent with three coordinate ligation for these two peptides.45 The edge scan of MG2HG2M, however, is more complicated to interpret. While individual scans prior to averaging of the MG2CG2M peptide were identical (data not shown), the first scan of MG2HG2M was substantially different from the subsequent scans. There is an apparent photoreduction or other alteration of the sample after the exposure of the first scan (typically 45 min) for the MG2HG2M peptide–copper complex. In addition, the feature at 8984 becomes more intense after the first scan and a unique feature at 8997 eV decays after beam exposure. This feature can be seen in other published spectra of Met/His peptides and proteins.46–48 | ||
| Fig. 10 XANES spectra of MG2HG2M, first scan of 4 (blue); MG2HG2M, last scan of 4 (red); MG2CG2M (black); and MG2MG2M (green).6 Samples were prepared with 5 mM H2Asc to ensure Cu(I) formation. Final pH was 3.5 | ||
The EXAFS data and best fits to MG2HG2M and MG2CG2M are shown in Fig. 11. Table 4 summarizes the fitting trends. The MG2CG2M peptide can be modeled exclusively with a shell of sulfurs at 2.27 Å (Fit A, Table 4). This distance is shorter than the 2.31 Å distance that refined for the MG2MG2M peptide complex.6 When the coordination number is allowed to refine, it minimizes at a value of 3.1. The addition of a low-Z ligand to the first shell results in a lower fit quality and a fairly long distance for a three coordinate Cu(I) species. A carbon shell can be added at 3.3 Å without affecting the first shell analysis, but it does not offer a substantive improvement in fit quality.
 | ||
| Fig. 11 Unfiltered, unsmoothed EXAFS data and corresponding Fourier transforms for MG2CG2M (black line, top panel) and MG2HG2M (black line, bottom panel). Best one-shell fit for MG2CG2M (fit A from Table 4) and best three-shell fit for MG2HG2M (fit F from Table 4) are shown in red. | ||
| # | Model | Cu–S | Cu–N | Cu–C | F′ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R | CN | σ2 | R | CN | σ2 | R | CN | σ2 | |||
| Fits were to unfiltered, unsmoothed data from 1–13.1 Å−1. A scale factor of 1 and a ΔE0 value of -17 eV were used as fixed values for all shells. R (Å) and σ2 (Å2) were variables in all fits. Fits where coordination number (CN) was also freely varied are indicated with an asterisk (*). σ2 is scaled by 103 in the table. | |||||||||||
|
|||||||||||
| MG2CG2M | |||||||||||
| A | S | 2.27 | 3.1* | 6.1 | 0.125 | ||||||
| B | S, N | 2.27 | 2 | 3.8 | 2.06 | 1 | 4.6 | 0.199 | |||
|
|||||||||||
| MG2HG2M (all scans) | |||||||||||
| C | S | 2.28 | 3.1* | 8.8 | 0.541 | ||||||
| D | S, N | 2.29 | 2 | 5.4 | 1.99 | 1 | 6.0 | 0.451 | |||
| E | S, N | 2.29 | 2.6* | 6.9 | 1.96 | 1 | 6.6 | 0.430 | |||
| F | S, N, C | 2.29 | 2.5* | 6.8 | 1.97 | 1 | 6.5 | 3.35 | 2 | 6.6 | 0.392 |
| G | S, Nimid | 2.31 | 2 | 4.4 | 2.00 | 1.0 | 4.4 | 0.592 | |||
|
|||||||||||
| MG2HG2M (last three scans) | |||||||||||
| H | S, N | 2.29 | 2.5* | 6.7 | 1.97 | 1 | 7.2 | 0.520 | |||
|
|||||||||||
| MG2HG2M (first scan only) | |||||||||||
| I | S, N | 2.29 | 2.8* | 7.7 | 1.95 | 1 | 5.0 | 0.860 | |||
In contrast, the MG2HG2M EXAFS requires a low-Z ligand at 2.0 Å in addition to sulfur at 2.29 Å to model the data. With these parameters, the total coordination number refines to 3.6 (Fit E, Table 4). If the first scan of four is fit by itself, the optimal coordination number is slightly higher, at 3.8. If the last three scans with identical edges are averaged, the optimal coordination number is 3.5. This reduction in coordination number with beam exposure correlates with the increase in the 1s-to-4p feature. The Fourier transform of the MG2HG2M sample shows increased intensity from 2.5–3.5 Å (R + Δ), consistent with the presence of the rigid histidine ring. These outer shells from carbon backscattering can be fit using multiple scattering (Fit G, Table 4), however the carbon contributions from the methionines contribute to these features as well. The best single scattering fit with three shells (Fit F) is shown in Fig. 11. The refined Cu–S and Cu–N distances of 2.29 Å and 1.97 Å respectively are typical for Met rich proteins with mixed Met/His ligation.46–48
Discussion
Relative affinity and pH dependence of Cu(I) binding amino acids
In order to examine the individual contribution of methionine, histidine, and cysteine on the Cu(I) binding affinity in a binding site at acidic and physiological pH, the effective KD values for the 3 different model peptides were determined at pH 4.5 and pH 7.4 (Table 2). As expected, the KD values for peptides containing copper binding amino acids with protonatable side chains decreased with a decrease in the pH. From pH 7.4 to pH 4.5, the KD of MG2CG2M dropped 2 orders of magnitude, 6(1) × 10−11 to 3.5(2) × 10−9 M, and the KD of MG2HG2M dropped 1 or 4 orders of magnitude, from a value within the range between 1 × 10−6–1 × 10−9 M to 1.3(1) × 10−5 M. The peptide containing methionines as the only copper binding amino acid was unaffected by the decrease in pH, exhibiting a KD between 4–5 × 10−6 M at all examined conditions. This result was also expected as methionine is the only copper binding amino acid lacking a protonatable side chain. At pH 4.5 methionine therefore binds Cu(I) with greater affinity than histidine. The side chain of histidine has a pKa of 6.02. At pH 4.5, roughly 97% of the imidazole should be protonated, compared to 4% at pH 7.4. Thus, considerably weaker binding would be expected for the histidine-containing peptide at pH 4.5 compared to pH 7.4. Despite being weaker than methionine at this pH, the contribution of the histidine to the binding affinity is not negligible. A peptide containing the same Mets motif as MG2HG2M, yet lacking the central copper binding amino acid, (MTGNleKGMS) was determined to have an even weaker affinity (KD = 5(2) × 10−5 M) under the same conditions, utilizing the same method.30The XANES spectra of the MG2HG2M complex also indicate that histidine contributes along with methionine to Cu(I) binding, even at pH 3.5, since both S and N coordination are required to best fit the data. The instability of the sample in the X-ray beam is likely a consequence of both the diminished binding affinity of this peptide for copper at this pH, as well as this peptide's ability to bind both Cu(I) and Cu(II). The initial scan appears to have contributions from a species that is either more oxidized or of a higher coordination number or both. As X-ray exposure continues, it appears that the fourth ligand starts to dissociate. If there is a Cu(II) species present initially, it must be a minor component, given that there is no evidence of a 1s-to-3d pre-edge transition. Also, the refined distances do not change substantially as the sample photoreduces completely to Cu(I), indicating that the peptide–Cu(I) species predominates. It will be interesting in future work to recollect these spectra at pH 7.4 where the affinity of this particular peptide for Cu(I) is greater.
The decrease in the affinity of MG2CG2M for Cu(I) from pH 7.4 to pH 4.5 by two orders of magnitude is expected. The pKa of free cysteine is 8; therefore 75% of the side chain is protonated at pH 7.4 and 99.9% at pH 4.5. These percentages may be an over estimation as the pKa values for cysteine in copper binding motifs are often lower, around 6 (4% and 99% protonated at pH 7.4 and 4.5, respectively) some as even low as 3.5 (0.01% and 10% protonated at pH 7.4 and 4.5, respectively).49,50 The estimated KD value seems reasonable in light of biological data regarding a binding motif similar to our MG2CG2M model peptide. Ctr proteins from the green alga Chlamydomonas contain a cysteine and methionine rich copper binding CX2MX2MX2CX5/6C motif.12 Previous studies have indicated that Chlamydomonas grown under hypoxic conditions are able to reduce the concentration of copper in the media down to as low as subnanomolar levels (0.2–1.6 nM).12,22,51
These results can be used to justify a qualitative description of the trend of Cu(I) binding affinity for the individual copper binding amino acids that comprise binding sites observed in copper trafficking proteins at pH 7.4 and pH 4.5. Cysteine appears to bind Cu(I) more effectively than either histidine or methionine at both pHs, histidine binds Cu(I) more effectively than methionine at pH 7.4, while methionine binds Cu(I) more effectively than histidine at pH 4.5. The Cu–S distances refined in the EXAFS fitting are also consistent with this trend; the MG2CG2M peptide complex has the shortest Cu–S distance. The following trend therefore emerges: Cys > His > Met at pH 7.4, whereas Cys > Met > His at pH 4.5.
These results have interesting applications to biochemical techniques commonly used to identify potential copper binding amino acids in proteins. Site directed mutagenesis is often used to identify amino acids in the binding sites of copper trafficking proteins.17,52–54 These studies are regularly accompanied by the determination of the levels of either copper uptake or efflux, in order to confirm the suggested functionality of the amino acid. Typically, potential copper binding amino acids are replaced with non-binding analogues, for example serine for cysteine, alanine for histidine, and isoleucine for methionine are some of the most common mutations. Utilizing any one of these mutations in a copper trafficking protein would be expected to inhibit copper uptake or efflux. To confirm whether or not certain copper binding amino acids in a protein are actually involved in copper trafficking, they are often first replaced with a non-binding analogue to inhibit activity, and then replaced with an alternate copper binding amino acid to restore function. Considering the pH dependent relative binding affinities determined in this study, Cys > His > Met at pH 7.4 and Cys > Met > His at pH 4.5, such mutations may not always restore activity, and that phenotype may be misinterpreted as a false positive.
Copper trafficking pathways are highly evolved networks where binding affinities have been fine tuned to promote transfer of the metal to a recipient protein. These binding affinities are the result of the inclusion of specific copper binding amino acids organized to promote specific types of geometries. While a negative result is expected when mutating a copper binding amino acid for a non-binding analogue, it is not safe to assume that mutating the same amino acid to an alternative, non-native copper-binding amino acid will necessarily restore function. As the current study suggests, a Cys-to-His or Met mutation in a copper-binding motif will likely decrease its binding affinity. The same can be said for a His-to-Met mutation around physiological pH, and for a Met-to-His mutation at an acidic pH. These types of mutations could cause false positives, whereby the mutated protein of interest now exhibits a much lower Cu(I) binding affinity, and can no longer effectively accept Cu(I) from a donor protein. A Met or His-to-Cys, a Met-to-His at physiological pH, or a His-to-Met at acidic pH will likely cause an increase in the Cu(I) binding affinity, which could alter the protein's ability to promote copper transfer to a recipient protein.
Susceptibility of Cu(I) binding amino acids to oxidation by metal catalyzed oxidation reactions
A summary of the results of treatment of the three peptides with conditions commonly used in metal catalyzed oxidation reactions is shown in Table 3. Incubation of peptide with just ascorbic acid (condition C) or CuSO4 (condition D) in buffer did not induce oxidation. However, a combination of the two did (condition B), but only in the case of the histidine containing peptide, MG2HG2M. Oxidation via condition B appears to be dependent on the ability of the peptide to bind both Cu(I) and Cu(II). Histidine with the imidazole side chain is a borderline base, and much more likely to accommodate binding of the borderline acid Cu2+ than either the soft methionine thioether or cysteine thiolate. Thus, MG2HG2M is the only peptide examined that has the ability to bind both Cu(I) and Cu(II). This ability is critical considering that ROS are being generated by Fenton-Haber Weiss chemistry at the site of the copper atom (eqn (12)–(16)). Under these conditions, oxidation of Cu+ to Cu2+ can accompany the generation of O2˙−, H2O2, and ˙OH. Binding sites that accommodate CuI/II redox cycling create a cage for the generated ROS, keeping the ROS in close proximity to the copper binding amino acids, resulting in their oxidation.41 Oxidation is not observed for Cu(I) specific binding sites, as upon conversion of Cu+ to Cu2+ the peptide is released and diffuses away from copper and the generated ROS.| Cu(II)P + HAsc− → Cu(I)P + HAsc˙− | (12) |
| Cu(I)P + O2 → Cu(II)P + O2˙− | (13) |
| Cu(I)P + 2H+ + O2˙− → Cu(II)P + H2O2 | (14) |
| HAsc− + 2H+ + O2˙− → Asc + H2O2 | (15) |
| Cu(I)P + H2O2 → Cu(II)P + ˙OH + −OH | (16) |
Treatment with H2O2 (condition A) caused the oxidation of methionine and cysteine residues, but not histidine. This suggests that the H2O2 generated in eqn (14) is not the ROS responsible for the direct oxidation of histidine in condition B, leaving the possibility of a mechanism involving O2˙− or ˙OH. When comparing the number and intensity of oxidation products observed after treatment of MG2HG2M with condition B (H2Asc + CuSO4) vs. condition F (H2O2 + CuSO4) (Fig. 7a and 9a), the combination of H2O2 and CuSO4 generates a greater amount of oxidation products than the combination of H2Asc and CuSO4. This result suggests that the ˙OH generated in eqn (16) is the ROS responsible for direct oxidation of histidine. Fewer oxidation products are observed with the combination of H2Asc and CuSO4, as these conditions must first generate reactants (eqn (12) and (13)), prior to formation H2O2 (eqn (14) and (15)). While H2O2 is catalytically generated under these conditions, the presence of the high concentration of H2Asc may be scavenging H2O2, as was observed when comparing the amount of oxidation products of H2O2 (condition A) to the combination of H2Asc and H2O2 (condition E). These examples show the dual antioxidant/pro-oxidant nature of ascorbic acid.
The lack of peptide oxidation of MG2MG2M and MG2CG2M after treatment with condition B (H2Asc, CuSO4) suggests that Cu(I) specific binding sites may not be identifiable by MCO. While the literature is full of examples of MCO reactions identifying copper binding amino acids in peptides and proteins, none are Cu(I) specific.34–40 There are examples in the literature of copper binding sites that resist oxidation by MCO reactions, such as the ATCUN binding motif.55–57 These Cu(II) binding sites are believed to bind the metal in a coordination environment that prevents redox cycling by stabilizing Cu(II), thus preventing the generation of ROS. The lack of oxidation observed for MG2MG2M and MG2CG2M, on the other hand, is more likely the result of the peptides preventing redox cycling by stabilizing Cu(I), or by diffusing away from any ˙OH generated by Cu+/Cu2+ redox activity.
Implications for cellular copper uptake by different organisms
Combined, the results obtained from this study of model peptides elucidate an evolutionary preference for methionine, histidine, or cysteine utilization in the N-terminal regions of Ctr1 proteins of different organisms that have adapted to acquire copper in drastically different environments. While yeast import copper from acidic yet oxic environments, some green algae acquire this nutrient from acidic, copper-depleted and hypoxic environments, whereas mammalian Ctr1 proteins operate in both acidic and neutral oxic conditions. To accomplish the same function of copper acquisition under these different environmental conditions, yeast, green algae, and human Ctr1 proteins incorporate methionine, methionine/cysteine, and methionine/histidine motifs, respectively.The methionine motifs expressed by yeast on the cell surface portion of the protein allow for pH-independent accumulation of copper at the site of the Ctr1 pore in an oxidative environment. Histidine would not be suitable as the ability to bind copper is compromised at this pH as a result of the protonateable sidechain. Cysteine would also not be suitable under these conditions due to its susceptibility to oxidation and propensity to form disulfide bonds, also compromising the ability to bind copper.
The methionine/cysteine motifs expressed by green algae allow for high affinity copper uptake in environments where copper is scarce. Copper concentrations in these environments are so low that methionine motifs alone presumably do not have a high enough affinity for effective Cu(I) accumulation at the Ctr1 pore. Cysteine, which binds Cu(I) with a considerably greater affinity, is required for transport function under these conditions. Oxidation of cysteine to form disulfide bonds is not of concern as these algae grow in hypoxic environments, and histidine would be less suitable under such conditions since the low pH compromises its copper binding ability.
The methionine/histidine motifs expressed by humans and other mammals have been proposed to facilitate transfer of copper from extracellular proteins like cerulopasmin and serum albumin to the Ctr1 protein in the oxidative extracellular environment.24 Histidine is optimal for this function, as the methionine motifs do not exhibit a strong enough affinity to remove copper from these proteins. Methionine, on the other hand, may play a more critical role in copper uptake under acidic conditions, as would be found in the small intestine, for example.
Conclusions
This study characterized two of the unique properties of Mets motifs in greater detail, pH dependence of Cu(I) binding, and resistance to metal catalyzed oxidation, as first described in previous work.6 Determination of Cu(I) KD values for model peptides revealed the relative Cu(I) binding affinities for methionine, histidine, and cysteine at acidic and physiological pH (Cys > Met > His, pH 4.5 and Cys > His > Met, pH 7.4). These relative affinities are important to consider when using site directed mutagenesis to identify copper binding amino acids in copper trafficking proteins, as mutations may drastically affect a protein's Cu(I) binding affinity and compromise the trafficking network.Treatment of model peptides with H2O2 demonstrated that histidine is resistant to oxidation by this ROS. While methionine and cysteine are both susceptible to oxidation by H2O2, cysteine appears to be more so. Treatment of model peptides with MCO reactions revealed that histidine-containing peptides are more susceptible to oxidation, presumably via a caging effect as they bind both Cu(I) and Cu(II), whereas peptides comprised of methionine and cysteine that specifically bind Cu(I) are not. This observation suggests that copper-binding amino acids in Cu(I) specific proteins may not be identifiable using this method.
Understanding the differences among this series of seemingly simple 8 amino acid peptides helps reveal how biological systems adapt to acquire an essential nutrient like copper. The pH dependence on Cu(I) binding and the susceptibility to oxidation of peptides containing clustered Met/His/Cys motifs highlights how Nature uses the chemical differences among imidazole, thiolate, and thioether functional groups to optimize copper acquisition under very different environmental conditions.
Abbreviations
| BCA | bicinchoninic acid |
| Ctr | Copper transporter |
| MCO | Metal catalyzed oxidation |
| H2Asc | Ascorbic acid |
| ESI-MS | Electrospray ionization mass spectrometry |
| MS/MS | Tandem mass spectrometry |
| MOPS | 3-[N-morpholine]propane-sulfonic acid |
| XAS | X-ray absorption spectroscopy |
| EXAFS | Extended X-ray absorption fine structure |
| XANES | X-ray absorption near edge structure |
Acknowledgements
We thank the National Science Foundation (Grant CAREER 0449699 to KJF) and the National Center for Research Resources at the National Institutes of Health (P20 RR-016461 to the State of South Carolina) for funding these studies. Both KJF and PRG also thank the Camille and Henry Dreyfus Foundation. The Sloan Foundation also supports KJF. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.References
- C. Rensing and G. Grass, FEMS Microbiol. Rev., 2003, 27, 197–213 CrossRef CAS.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS.
- L. Banci, I. Bertini, K. S. McGreevy and A. Rosato, Nat. Prod. Rep., 2010, 27, 695–710 RSC.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS.
- A. V. Davis and T. V. O'Halloran, Nat. Chem. Biol., 2008, 4, 148–151 CrossRef CAS.
- J. T. Rubino, P. Riggs-Gelasco and K. J. Franz, J. Biol. Inorg. Chem., 2010, 15, 1033–1049 CrossRef CAS.
- A. Dancis, D. S. Yuan, D. Haile, C. Askwith, D. Eide, C. Moehle, J. Kaplan and R. D. Klausner, Cell, 1994, 76, 393–402 CrossRef CAS.
- H. Zhou and D. J. Thiele, J. Biol. Chem., 2001, 276, 20529–20535 CrossRef CAS.
- H. Zhou, K. M. Cadigan and D. J. Thiele, J. Biol. Chem., 2004, 279, 2332–2332 CAS.
- K. Kampfenkel, S. Kushnir, E. Babiychuk, D. Inze and M. Vanmontagu, J. Biol. Chem., 1995, 270, 28479–28486 CrossRef CAS.
- B. Zhou and J. Gitschier, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7481–7486 CrossRef CAS.
- M. D. Page, J. Kropat, P. P. Hamel and S. S. Merchant, Plant Cell, 2009, 21, 928–943 CrossRef CAS.
- J. Lee, M. M. O. Peña, Y. Nose and D. J. Thiele, J. Biol. Chem., 2002, 277, 4380–4387 CrossRef CAS.
- A. E. M. Klomp, J. A. Juijn, L. T. M. Van der Gun, I. E. T. Van den Berg, R. Berger and L. W. J. Klomp, Biochem. J., 2003, 370, 881–889 CrossRef CAS.
- S. G. Aller and V. M. Unger, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3627–3632 CrossRef CAS.
- C. J. De Feo, S. G. Aller, G. S. Siluvai, N. J. Blackburn and V. M. Unger, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4237–4242 CrossRef CAS.
- S. Puig, J. Lee, M. Lau and D. J. Thiele, J. Biol. Chem., 2002, 277, 26021–26030 CrossRef CAS.
- S. Puig and D. J. Thiele, Curr. Opin. Chem. Biol., 2002, 6, 171–180 CrossRef CAS.
- M. E. Van der Rest, A. H. Kamminga, A. Nakano, Y. Anraku, B. Poolman and W. N. Konings, Microbiol. Rev., 1995, 59, 304–322 CAS.
- E. H. Harris, in The Chlamydomonas Sourcebook, Academic Press, San Diego, 2 edn, 2009, vol. 1, pp. 1–24 Search PubMed.
- R. Osterber, Nature, 1974, 249, 382–383 CrossRef.
- K. L. Hill, R. Hassett, D. Kosman and S. Merchant, Plant Physiol., 1996, 112, 697–704 CrossRef CAS.
- C. M. Lin and D. J. Kosman, J. Biol. Chem., 1990, 265, 9194–9200 CAS.
- K. L. Haas, A. B. Putterman, D. R. White, D. J. Thiele and K. J. Franz, 2010, submitted.
- V. A. Gray and J. B. Dressman, Pharmacop. Forum, 1996, 22, 1943–1945 Search PubMed.
- Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, J. Am. Chem. Soc., 2004, 126, 3081–3090 CrossRef CAS.
- Z. G. Xiao, P. S. Donnelly, M. Zimmermann and A. G. Wedd, Inorg. Chem., 2008, 47, 4338–4347 CrossRef CAS.
- L. A. Yatsunyk and A. C. Rosenzweig, J. Biol. Chem., 2007, 282, 8622–8631 CrossRef CAS.
- A. L. Ankudinov and J. J. Rehr, Phys. Rev. B: Condens. Matter, 1997, 56, R1712–R1715 CrossRef CAS.
- J. Jiang, I. A. Nadas, M. A. Kim and K. J. Franz, Inorg. Chem., 2005, 44, 9787–9794 CrossRef CAS.
- Y. Xue, A. V. Davis, G. Balakrishnan, J. P. Stasser, B. M. Staehlin, P. Focia, T. G. Spiro, J. E. Penner-Hahn and T. V. O'Halloran, Nat. Chem. Biol., 2008, 4, 107–109 CrossRef CAS.
- I. R. Loftin, N. J. Blackburn and M. M. McEvoy, J. Biol. Inorg. Chem., 2009, 14, 905–912 CrossRef CAS.
- L. X. Chong, M. R. Ash, M. J. Maher, M. G. Hinds, Z. G. Xiao and A. G. Wedd, J. Am. Chem. Soc., 2009, 131, 3549–3564 CrossRef CAS.
- K. Uchida and S. Kawakishi, Arch. Biochem. Biophys., 1990, 283, 20–26 CrossRef CAS.
- J. D. Bridgewater, J. Lim and R. W. Vachet, Anal. Chem., 2006, 78, 2432–2438 CrossRef CAS.
- C. Schoneich, in Redox-Active Metals in Neurological Disorders, New York Acad Sciences, New York, 2004, vol. 1012, pp. 164–170 Search PubMed.
- J. Lim and R. W. Vachet, Anal. Chem., 2003, 75, 1164–1172 CrossRef CAS.
- R. C. Nadal, S. R. Abdelraheim, M. W. Brazier, S. E. J. Rigby, D. R. Brown and J. H. Viles, Free Radical Biol. Med., 2007, 42, 79–89 CrossRef CAS.
- R. C. Nadal, S. E. J. Rigby and J. H. Viles, Biochemistry, 2008, 47, 11653–11664 CrossRef CAS.
- E. A. Houghton and K. M. Nicholas, J. Biol. Inorg. Chem., 2009, 14, 243–251 CrossRef CAS.
- E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef CAS.
- K. L. Schey and E. L. Finley, Acc. Chem. Res., 2000, 33, 299–306 CrossRef CAS.
- K. Biemann and H. A. Scoble, Science, 1987, 237, 992–998 CrossRef CAS.
- X. Y. Jiang, J. B. Smith and E. C. Abraham, J. Mass Spectrom., 1996, 31, 1309–1310 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- K. Peariso, D. L. Huffman, J. E. Penner-Hahn and T. V. O'Halloran, J. Am. Chem. Soc., 2003, 125, 342–343 CrossRef CAS.
- P. D'Angelo, F. Pacello, G. Mancini, O. Proux, J. L. Hazemann, A. Desideri and A. Battistoni, Biochemistry, 2005, 44, 13144–13150 CrossRef CAS.
- F. Arnesano, L. Banci, I. Bertini, S. Mangani and A. R. Thompsett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3814–3819 CrossRef CAS.
- L. Zhou, C. Singleton and N. E. Le Brun, Biochem. J., 2008, 413, 459–465 CrossRef CAS.
- J. W. Nelson and T. E. Creighton, Biochemistry, 1994, 33, 5974–5983 CrossRef.
- S. Merchant, K. Hill and G. Howe, EMBO J., 1991, 10, 1383–1389 CAS.
- I. Bagai, W. Liu, C. Rensing, N. J. Blackburn and M. M. McEvoy, J. Biol. Chem., 2007, 282, 35695–35702 CrossRef CAS.
- S. M. Lee, G. Grass, C. Rensing, S. R. Barrett, C. J. D. Yates, J. V. Stoyanov and N. L. Brown, Biochem. Biophys. Res. Commun., 2002, 295, 616–620 CrossRef CAS.
- S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2003, 185, 3804–3812 CrossRef.
- D. Bar-Or, L. T. Rael, E. P. Lau, N. K. R. Rao, G. W. Thomas, J. V. Winkler, R. L. Yukl, R. G. Kingston and C. G. Curtis, Biochem. Biophys. Res. Commun., 2001, 284, 856–862 CrossRef CAS.
- Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 127, 8408–8415 CrossRef CAS.
- L. Perrone, E. Mothes, M. Vignes, A. Mockel, C. Figueroa, M. C. Miquel, M. L. Maddelein and P. Faller, ChemBioChem, 2009, 11, 110–118 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |