Novel benzyl-substituted N-heterocyclic carbene–silver acetate complexes: synthesis, cytotoxicity and antibacterial studies†
Siddappa
Patil
,
Anthony
Deally
,
Brendan
Gleeson
,
Helge
Müller-Bunz
,
Francesca
Paradisi
and
Matthias
Tacke
*
Conway Institute of Biomolecular and Biomedical Research, Centre for Synthesis and Chemical Biology (CSCB), UCD School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: matthias.tacke@ucd.ie
First published on 6th December 2010
Abstract
From the reaction of 1-methylimidazole (1a), 4,5-dichloro-1H-imidazole (1bII) and 1-methylbenzimidazole (1c) with p-cyanobenzyl bromide (2a), non-symmetrically substituted N-heterocyclic carbene (NHC) [(3a–c)] precursors, 5,6-dimethyl-1H-benzimidazole (1d) and 4,5-diphenyl-1H-imidazole (1e) with p-cyanobenzyl bromide (2a) and benzyl bromide (2b), symmetrically substituted N-heterocyclic carbene (NHC) [(3d–f)] precursors were synthesised. These NHC-precursors were then reacted with silver(I) acetate to yield the NHC–silver complexes (1-methyl-3-(4-cyanobenzyl)imidazole-2-ylidene)silver(I)acetate (4a), (4,5-dichloro-1-(4-cyanobenzyl)-3-methyl)imidazole-2-ylidene)silver(I)acetate (4b), (1-methyl-3-(4-cyanobenzyl)benzimidazole-2-ylidene)silver(I)acetate (4c), (1,3-bis(4-cyanobenzyl)5,6-dimethylbenzimidazole-2-ylidene) silver(I) acetate (4d), (1,3-dibenzyl-5,6-dimethylbenzimidazole-2-ylidene) silver(I) acetate (4e) and (1,3-dibenzyl-4,5-diphenylimidazol-2-ylidene) silver(I) acetate (4f) respectively. Three NHC-precursors 3c–e and four NHC–silver complexes 4b and 4d–f were characterised by single crystal X-ray diffraction. Preliminary in vitro antibacterial activity of the NHC-precursors and NHC–silver complexes was investigated against Gram-positive bacteria Staphylococcus aureus, and Gram-negative bacteria Escherichia coli using the qualitative Kirby–Bauer disk-diffusion method. NHC–silver complexes have shown very high antibacterial activity compared to the NHC-precursors. All six NHC–silver complexes were tested for their cytotoxicity through MTT based in vitro tests on the human renal-cancer cell line Caki-1 in order to determine their IC50 values. NHC–silver complexes 4a–f were found to have IC50 values of 6.2 (±1.0), 7.7 (±1.6), 1.2 (±0.6), 10.8 (±1.9), 24.2 (±1.8) and 13.6 (±1.0) μM, respectively. These values represent improved cytotoxicity against Caki-1, most notably for 4c, which is a three times more cytotoxic than cisplatin (IC50 value = 3.3 μM) itself.
1. Introduction
N-Heterocyclic carbenes (NHCs) are stable singlet carbenes that can act as excellent two electron donor ligands towards almost any element in the periodic table. They coordinate strongly to late transition metals and heavy main group elements, but are also known to bind to early transition metals and the lanthanoids. Öfele1 and Wanzlick2 successfully synthesised the first N-heterocyclic carbene transition metal complexes of chromium and mercury in 1968. The isolation of the first free carbene by Arduengo in 1991 set the scene for an ever-growing interest and advancement in the field of N-heterocyclic carbene chemistry.3 Shortly thereafter, the use of these ligands in organometallic chemistry, particularly in catalysis dramatically increased.4,5 Very recently, NHCs found an application in NHC–silver complexes exhibiting antimicrobial activity, in particular for the possible treatment of cystic fibrosis (CF) and chronic lung infections6–8 and maybe in the treatment of cancer.9Silver salts have enjoyed a long history as antimicrobial agents and have proved to exhibit low toxicity for humans. Thus most of the biomedical studies on NHC–silver complexes have been conducted on their antimicrobial properties. After a thorough search of the literature, it was evident that only a handful of research groups have investigated the antimicrobial and antifungal properties of NHC–silver complexes. Young's research group have reported antimicrobial activity of NHC–silver complexes derived from 1H-imidazole, 4,5-dichloro-1H-imidazole and xanthines against a panel of highly resistant pathogens recovered from the respiratory tract of cystic fibrosis (CF) patients.6,8,10 Another important contribution by the Ghosh research group led to the synthesis and antimicrobial evaluation of NHC–silver complexes derived from 1-benzyl-3-tert-butylimidazole.11 More recently Young's and colleagues have also shown that in vitro and murine efficacy and toxicity studies of nebulized methylated caffeine–silver(I) complex (SCC1), for treatment of pulmonary infections.12
Nowadays, compounds of wide structural diversity are used as therapeutic agents for cancer treatment. The discovery of the antitumour properties of cisplatin by Rosenberg13 has proven that not only drugs composed of the basic elements of the periodic table like carbon, oxygen, hydrogen and nitrogen but also those containing heavy elements may be toxic for tumour cells. Cisplatin, along with its second generation analogues, represent the most widely used chemotherapeutic agents.14 Clinical trials are not only restricted to the platinum element but also extended to ruthenium, iron, and titanium compounds.15–18
Recently, silver complexes have been reported to have anticancer activity in vitro. Egan has reported that silver complexes of coumarin derivatives possess anticancer activity against certain types of cancer.19 Zhu has reported that silver carboxylate dimers possess anticancer activity against human carcinoma cells.20 McKeage has shown phosphine complexes of silver to be active anticancer agents, even against cisplatin resistant cell lines.21 Youngs and coworkers have reported anticancer activity of NHC–silver complexes derived from 4,5-dichloro-1H-imidazole against the human cancer cell lines OVCAR-3 (ovarian), MB157 (breast), and Hela (cervical).9 These silver complexes have been shown to be very stable and can be synthesized efficiently. We have recently reported the anticancer and antibacterial activity of symmetrically p-methoxybenzyl-substituted and benzyl-substituted N-heterocyclic carbene–silver complexes. All the reported NHC–silver complexes have shown medium to high anticancer and antibacterial activity.22
Within this paper we present a new series of non-symmetrically and symmetrically p-cyanobenzyl- or benzyl-substituted N-heterocyclic carbene–silver acetate complexes, their synthesis, cytotoxicity and antibacterial studies.
2. Experimental
2.1 General considerations
All the solvents used were of analytical grade and were used without further purification. 4,5-Dichloro-1H-imidazole, 1-methylimidazole, 1-methylbenzimidazole, 5,6-dimethyl-1H-benzimidazole, 4,5-diphenyl-1H-imidazole, p-cyanobenzyl bromide, benzyl bromide, silver acetate, methyl iodide and K2CO3 were purchased from Sigma-Aldrich Chemical Company and were used without further purification. NMR spectra were measured on a Varian 400 MHz spectrometer. Chemical shifts are reported in ppm and are referenced to TMS. IR spectra were recorded on a Perkin Elmer Paragon 1000 FT-IR Spectrometer employing a KBr disc. UV–vis spectra were recorded on a Unicam UV4 Spectrometer. Electron spray mass spectrometry (MS) was performed on a quadrupole tandem mass spectrometer (Quattro Micro, Micromass/Water's Corp., USA), using solutions made up in 50% dichloromethane and 50% methanol. MS spectra were obtained in the ES+ (electron spray positive ionisation) mode for compounds 1b, 3a–f and 4a–f. CHN analysis was done with an Exeter Analytical CE-440 Elemental Analyser. Ag was estimated by spectrophotometric method (Atomic absorption spectra 55B Varian), while Cl, Br and I were determined in mercurimetric titrations. Crystal data were collected using an Oxford Diffraction SuperNova diffractometer fitted with an Atlas detector. 3c was measured with Mo-Kα (0.71073 Å), all others with Cu-Kα (1.54184 Å). 3c and 3e were collected at room temperature, all others at 100 K. An at least twice redundant dataset was collected, assuming that the Friedel pairs are not equivalent. For 3c a pseudo-empirical absorption correction based on redundant reflections was performed by the program SADABS.23 For all others an analytical absorption correction based on the shape of the crystal was performed.24 The structures were solved by direct methods using SHELXS-9725 and refined by full matrix least-squares on F2 for all data using SHELXL-97.25 The hydrogen atoms of the water molecules in 3c and 3e were located in the difference fourier map. Their thermal displacement parameters were fixed to 1.5 times the equivalent one of the parent oxygen atom. O–H bond lengths were restrained to be 0.82 Å using DFIX. In 3d the hydrogen attached to C(9) was located in the difference fourier map and allowed to refine freely due to its involvement in hydrogen bonding. All other hydrogen atoms were added at calculated positions and refined using a riding model. Their isotropic temperature factors were fixed to 1.2 times (1.5 times for methyl groups) the equivalent isotropic displacement parameters of the carbon atom the H-atom is attached to. Anisotropic thermal displacement parameters were used for all non-hydrogen atoms. Suitable crystals of 3c–e were formed from the slow evaporation of a saturated methanol solution, while crystals of 4b and 4d–f were grown in a saturated dichloromethane solution with slow infusion of pentane. Further details about the data collection are listed in Table 1, as well as reliability factors.| Identification code | 3c | 3d | 3e | 4b | 4d | 4e | 4f |
|---|---|---|---|---|---|---|---|
| Empirical formula | C16 H16 N3 O Br | C25 H21 N4Br | C23 H25 N2 O Br | C14 H12 N3 O2 Cl2Ag | C27 H23 N4 O2Ag | C25 H25Ag N2 O2 | C62 H54 N4 O4Ag2 |
| Molecular formula | [C16 H14 N3]+ [Br]− × H2 O | [C25 H21 N4]+[Br]− | [C23 H25 N2]+[Br]− × H2 O | C14 H12 N3 O2 Cl2Ag | C27 H23 N4 O2Ag | C25 H25Ag N2 O2 | C62 H54 N4 O4Ag2 |
| Formula weight | 346.23 | 457.37 | 425.36 | 433.04 | 543.36 | 493.34 | 1134.83 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n (#14) | P21/n (#14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2) (#2) |
P21/c (#14) | P2/n (#13) | P 21/c (#14) | C2/c (#15) |
| Unit cell dimensions/Å (°) | a = 7.0820(2) | a = 7.26900(4) | a = 9.9811(3) | a = 4.40268(4) | a = 15.3823(1) | a = 11.7885(3) | a = 23.9368(7) |
| b = 15.9788(3) | b = 12.62118(6) | b = 10.0794(4) | b = 16.7370(2) | b = 7.83949(7) | b = 47.3901(8) | b = 9.5997(2) | |
| c = 13.7653(3) | c = 23.4502(1) | c = 11.7361(3) | c = 21.1545(2) | c = 19.8044(2) | c = 7.8129(2) | c = 25.2845(7) | |
| α = 90 | α = 90 | α = 67.423(3) | α = 90 | α = 90 | α = 90 | α = 90 | |
| β = 90.326(2) | β = 97.0346(5) | β = 82.718(3) | β = 91.612(1) | β = 96.7996(7) | β = 99.281(2) | β = 119.297(4) | |
| γ = 90 | γ = 90 | γ = 76.645(3) | γ = 90 | γ = 90 | γ = 90 | γ = 90 | |
| Volume/Å3 | 1557.68(6) | 2135.205(18) | 1059.78(6) | 1558.21(3) | 2371.40(4) | 4307.60(17) | 5066.9(2) |
| Z | 4 | 4 | 2 | 4 | 4 | 8 | 4 |
| Density/Mg m−3 (calculated) | 1.476 | 1.423 | 1.333 | 1.846 | 1.522 | 1.521 | 1.488 |
| Absorption coefficient/mm−1 | 2.641 | 2.764 | 2.743 | 13.624 | 7.075 | 7.692 | 6.625 |
| F(000) | 704 | 936 | 440 | 856 | 1104 | 2016 | 2320 |
| Crystal size/mm | 0.3 × 0.2 × 0.2 | 0.1924 × 0.1119 × 0.0597 | 0.2009 × 0.1821 × 0.1689 | 0.3880 × 0.0694 × 0.0443 | 0.1983 × 0.0912 × 0.0700 | 0.2133 × 0.1538 × 0.1219 | 0.1491 × 0.0309 × 0.0169 |
| Theta range for data collection | 3.47 to 24.73° | 3.80 to 76.88° | 4.08 to 76.49° | 3.37 to 76.32° | 3.45 to 70.99° | 3.73 to 76.95° | 4.01 to 76.89° |
| Index ranges | −8 ≤ h ≤ 8, | −9 ≤ h ≤ 9, | −12 ≤ h ≤ 12, | −5 ≤ h ≤ 5, | −18 ≤ h ≤ 18, | −14 ≤ h ≤ 14, | −27 ≤ h ≤ 30, |
| −18 ≤ k ≤ 18, | −15 ≤ k ≤ 15, | −12 ≤ k ≤ 12, | −20 ≤ k ≤ 20, | −8 ≤ k ≤ 9, | −59 ≤ k ≤ 59, | −12 ≤ k ≤ 12, | |
| −16 ≤ l ≤ 16 | −29 ≤ l ≤ 29 | −14 ≤ l ≤ 14 | −25 ≤ l ≤ 26 | −24 ≤ l ≤ 22 | −9 ≤ l ≤ 9 | −31 ≤ l ≤ 31 | |
| Reflections collected | 22628 | 43690 | 23699 | 13661 | 31155 | 47192 | 24441 |
| Independent reflections | 2655 [R(int) = 0.0345] | 4494 [R(int) = 0.0220] | 4420 [R(int) = 0.0617] | 3244 [R(int) = 0.0346] | 4589 [R(int) = 0.0582] | 9032 [R(int) = 0.0288] | 5254 [R(int) = 0.0327] |
| Completeness to θmax | 99.60% | 99.80% | 99.00% | 99.00% | 100.00% | 99.40% | 98.20% |
| Max. and min. Transmission | 0.6202 and 0.4613 | 0.882 and 0.703 | 0.747 and 0.705 | 0.607 and 0.129 | 0.744 and 0.521 | 0.563 and 0.367 | 0.905 and 0.608 |
| Data/restraints/parameters | 2655/2/197 | 4494/0/277 | 4420/2/252 | 3244/0/201 | 4589/0/310 | 9032/0/547 | 5254/0/326 |
| Goodness-of-fit on F2 | 1.04 | 1.061 | 1.062 | 1.167 | 1.085 | 1.155 | 1.069 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0271, | R 1 = 0.0220, | R 1 = 0.0349, | R 1 = 0.0384, | R 1 = 0.0377, | R 1 = 0.0518, | R 1 = 0.0233, |
| wR2 = 0.0664 | wR2 = 0.0593 | wR2 = 0.1016 | wR2 = 0.0893 | wR2 = 0.1022 | wR2 = 0.1010 | wR2 = 0.0551 | |
| R indices (all data) | R 1 = 0.0317, | R 1 = 0.0235, | R 1 = 0.0366, | R 1 = 0.0393, | R 1 = 0.0406, | R 1 = 0.0533, | R 1 = 0.0273, |
| wR2 = 0.0688 | wR2 = 0.0601 | wR2 = 0.1032 | wR2 = 0.0897 | wR2 = 0.1048 | wR2 = 0.1015 | wR2 = 0.0564 | |
| Largest diff. peak and hole | 0.425 and −0.615 e.Å−3 | 0.311 and −0.331 e.Å−3 | 0.605 and −0.519 e.Å−3 | 2.310 and −0.888 e.Å−3 | 1.171 and −0.602 e.Å−3 | 3.974 and −2.735 e.Å−3 | 0.348 and −0.581 e.Å−3 |
CCDC 777915 (for 3c), 777917 (for 3d), 777916 (for 3e), 777918 (for 4b), 777919 (for 4d), 777921 (for 4e) and 777920 (for 4f) respectively, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
2.2 Synthesis
1H NMR (δ ppm DMSO-d6, 400 MHz): 9.27 (s, 1H, NCHN), 7.90 (d, J = 8.2 Hz, 2H, CHCyanobenzyl), 7.80 (s, 1H, CHImid), 7.74 (s, 1H, CHImid), 7.58 (d, J = 8.2 Hz, 2H, CHCyanobenzyl), 5.55 (s, 2H, CH2), 3.85 (s, 3H, N–CH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 140.6, 137.5, 133.3, 129.5, 124.6, 122.8, 118.8, 111.8 (NCN + CN + CImid + CCyanobenzyl), 51.6 (CH2), 36.4(CH3).
IR absorptions (KBr, cm−1): 3368 (s), 3135 (w), 3067 (w), 3018 (s), 2975 (w), 2857 (w), 2230 (s), 1610 (w), 1578 (s), 1535 (s), 1420 (m), 1164 (s), 1020 (w), 824 (w), 761 (m), 652 (w), 622 (m), 550 (s).
UV-Vis (CH3OH, nm): λ 225 (ε 10512), λ 268 (ε 3846), λ 366 (ε 1191).
MS (m/z, QMS-MS/MS): 198.32 [M+–Br].
Micro Analysis Calculated for C12H12N3Br (278.15): Calcd.: C, 51.87%; H, 4.34%; N, 15.10%; Br, 28.72%; Found: C, 51.76%; H, 4.11%; N, 15.12%; Br, 28.70%.
1H NMR (δ ppm CDCl3, 400 MHz): 7.68 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 7.46 (s, 1H, NCHN), 7.25 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 5.17 (s, 2H, CH2).
13C NMR (δ ppm CDCl3, 100 MHz, proton decoupled): 139.6, 134.5, 132.9, 127.6, 127.0, 118.0, 113.6, 112.7 (NCN + CN + CCl + CCyanobenzyl), 49.0 (CH2).
IR absorptions (KBr, cm−1): 3450 (w), 3114 (m), 2230 (s), 1522 (m), 1476 (s), 1388 (m), 1342 (w), 1251 (s), 1174 (w), 1111 (w), 1022 (w), 984 (m), 863 (w), 821 (w), 770 (s), 689 (w), 664 (w), 628 (w), 563 (m).
UV-Vis (CH3OH, nm): λ 225 (ε 8846), λ 274 (ε 3056), λ 366 (ε 430).
MS (m/z, QMS-MS/MS): 252.10 [M+].
Micro Analysis Calculated for C11H7N3Cl2 (252.10): Calcd.: C, 52.41%; H, 2.80%; N, 16.67%; Cl, 28.13%; Found: C, 52.31%; H, 2.79%; N, 16.57%; Cl, 28.10%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 9.46 (s, 1H, NCHN), 7.93 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 7.57 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 5.63 (s, 2H, CH2), 3.84 (s, 3H, N–CH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 138.7, 137.5, 133.3, 129.2, 120.5, 118.8, 118.6, 112.0 (NCN + CN + CCl + CCyanobenzyl), 51.0 (CH2), 35.6 (CH3).
IR absorptions (KBr, cm−1): 3430 (m), 3125 (w), 3046 (s), 2926 (m), 2855 (w), 2233 (s), 1580 (s), 1560 (s), 1509 (m), 1454 (m), 1417 (w), 1346 (s), 1197 (s), 1153 (s), 1029 (w), 877 (m), 820 (m), 603 (m), 553 (s).
UV-Vis (CH3OH, nm): λ 224 (ε 14989), λ 269 (ε 5345), λ 361 (ε 1665).
MS (m/z, QMS-MS/MS): 267.96 [M+–I].
Micro Analysis Calculated for C12H10N3Cl2 I (394.04): Calcd.: C, 36.58%; H, 2.56%; N, 10.66%; Cl, 17.99%; I, 32.21%; Found: C, 36.47%; H, 2.48%; N, 10.57%; Cl, 17.98%; I, 32.18%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 9.88 (s, 1H, NCHN), 8.03 (d, J = 8.2 Hz, 1H, CHBenzimid), 7.88 (dd, J = 8.1, 3.8 Hz, 3H, CHBenzimid), 7.70–7.59 (m, 4H, CHCyanobenzyl), 5.89 (s, 2H, CH2), 4.09 (s, 3H, N–CH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 143.7, 139.9, 133.2, 132.4, 131.0, 129.4, 127.1, 127.0, 118.8, 114.2, 113.9, 111.8 (NCN + CN + CBenzimid + CCyanobenzyl), 49.5 (CH2), 33.9 (CH3).
R absorptions (KBr, cm−1): 3575 (s), 3358 (s), 3077 (m), 3023 (m), 2233 (s), 1608 (m), 1568 (s), 1460 (m), 1346 (w), 1280 (w), 1226 (w), 1020 (m), 863 (m), 783 (m), 757 (s), 657 (m), 598 (w), 554 (w), 478 (w), 424 (m).
UV-Vis (CH3OH, nm): λ 229 (ε 9655), λ 269 (ε 6138), λ 373 (ε 1472).
MS (m/z, QMS-MS/MS): 248.39 [M+–H2O–Br].
Micro Analysis Calculated for C16H16N3OBr (346.23): Calcd.: C, 55.51%; H, 4.66%; N, 12.14%; Br, 23.08%; Found: C, 55.23%; H, 4.51%; N, 12.12%; Br, 23.11%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 9.93 (s, 1H, NCHN), 7.90 (d, J = 8.0 Hz, 4H, CHCyanobenzyl), 7.72 (s, 2H, CHBenzimid), 7.66 (d, J = 8.0 Hz, 4H, CHCyanobenzyl), 5.84 (s, 4H, CH2), 2.32 (s, 6H, CH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 142.6, 139.9, 137.3, 133.3, 129.9, 129.3, 118.8, 113.7, 111.8 (NCN + CN + CBenzimid + CCyanobenzyl), 49.8 (CH2), 20.4 (CH3).
IR absorptions (KBr, cm−1): 3430 (w), 3031 (w), 2951 (s), 2791 (w), 1608 (m), 1567 (s), 1488 (w), 1435 (s), 1413 (m), 1346 (w), 1309 (w), 1233 (w), 1186 (m), 1017 (w), 972 (w), 812 (s), 610 (w), 548 (s), 429 (w).
UV-Vis (CH3OH, nm): λ 225 (ε 14115), λ 268 (ε 7126), λ 290(ε 5625), λ 362 (ε 1992).
MS (m/z, QMS-MS/MS): 377.14 [M+–Br].
Micro Analysis Calculated for C25H21N4Br (457.37): Calcd.: C, 65.65%; H, 4.63%; N, 12.25%; Br, 17.47%; Found: C, 65.65%; H, 4.62%; N, 12.11%; Br, 17.77%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 10.09 (s, 1H, NCHN), 7.79 (s, 2H, CHBenzimid), 7.51 (d, J = 7.1 Hz, 4H, CHBenzyl), 7.44–7.32 (m, 6H, CHBenzyl), 5.76 (s, 4H, CH2), 2.32 (s, 6H, CH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 141.8, 137.0, 134.6, 129.9, 129.4, 129.1, 128.6, 113.8 (NCN + CBenzimid + Cbenzyl), 50.2 (CH2), 20.4 (CH3).
IR absorptions (KBr, cm−1): 3435 (s), 3115 (w), 3028 (w), 2965 (s), 1558 (s), 1497 (w), 1454 (s), 1427 (w), 1354 (w), 1225 (w), 1082 (w), 853 (w), 765 (m), 705 (s), 669 (m), 616 (w), 430 (m).
UV-Vis (CH3OH, nm): λ 229 (ε 4622), λ 280 (ε 3341), λ 352 (ε 904).
MS (m/z, QMS-MS/MS): 327.17 [M+–H2O–Br].
Micro Analysis Calculated for C23H25N2OBr (425.36): Calcd.: C, 64.94%; H, 5.92%; N, 6.59%; Br, 18.78%; Found: C, 64.82%; H, 5.73%; N, 6.48%; Br, 18.67%.
1H NMR (δ ppm CDCl3, 400 MHz): 11.05 (s, 1H, NCHN), 7.56–6.89 (m, 20H, CHImid + CHBenzyl), 5.44 (s, 4H, CH2).
13C NMR (δ ppm CDCl3, 100 MHz, proton decoupled): 137.2, 133.2, 132.1, 130.7, 130.4, 129.0, 129.0, 128.8, 128.4, 124.5 (NCN + CImid + CBenzyl), 51.4 (CH2).
IR absorptions (KBr, cm−1): 3421 (m), 3030 (m), 2926 (m), 2855 (w), 1558 (m), 1497 (w), 1452 (s), 1352 (w), 1211 (w), 1178 (m), 1071 (w), 1024 (m), 755 (m), 698 (s), 645 (w), 514 (w), 462 (w).
UV-Vis (CH3OH, nm): λ 237 (ε 17199), λ 255 (ε 11660), λ 299 (ε 3678).
MS (m/z, QMS-MS/MS): 401.53 [M+–Br].
Micro Analysis Calculated for C29H25N2Br (481.43): Calcd.: C, 72.35%; H, 5.23%; N, 5.82%; Br, 16.60%; Found: C, 72.26%; H, 5.29%; N, 5.79%; Br, 16.58%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 7.81 (d, J = 8.4 Hz, 2H, CHCyanobenzyl), 7.54–7.44 (m, 4H, CHImid + CHCyanobenzyl), 5.40 (s, 2H, CH2), 3.75 (s, 3H, N–CH3), 1.81 (s, 3H, COCH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 179.2 (NCN), 174.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 143.2, 133.1, 128.9, 123.9, 122.6, 119.0, 111.1 (CN + CImid + CCyanobenzyl), 53.9 (CH2), 38.6 (N–CH3), 23.0 (COCH3).
O), 143.2, 133.1, 128.9, 123.9, 122.6, 119.0, 111.1 (CN + CImid + CCyanobenzyl), 53.9 (CH2), 38.6 (N–CH3), 23.0 (COCH3).
IR absorptions (KBr, cm−1): 3401 (w), 3094 (w), 2926 (w), 2228 (s), 1579 (s), 1406 (s), 1232 (m), 1161 (w), 1019 (w), 920 (w), 824 (w), 771 (m), 739 (m), 647 (m), 550 (m).
UV-Vis (CH3OH, nm): λ 230 (ε 8032), λ 273 (ε 4699), λ 368 (ε 1689).
MS (m/z, QMS-MS/MS): 305.90 [M+–O2CCH3].
Micro Analysis Calculated for C14H14N3O2Ag (364.15): Calcd.: C, 46.17%; H, 3.87%; N, 11.53%; Ag, 29.62%; Found: C, 46.37%; H, 3.92%; N, 11.41%; Ag, 29.59%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 7.80 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 7.41 (d, J = 8.0 Hz, 2H, CHCyanobenzyl), 5.55 (s, 2H, CH2), 3.80 (s, 3H, N–CH3), 1.71 (s, 3H, COCH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 181.4 (NCN), 175.0 (CO), 141.4, 133.1, 128.3, 118.8, 118.5, 116.9, 111.3 (CCl + CN + CCyanobenzyl), 53.1 (CH2), 38.2 (N–CH3), 24.1(COCH3).
IR absorptions (KBr, cm−1): 3435 (w), 2925 (s), 2854 (w), 2229 (s), 1579 (s), 1435 (w), 1415 (w), 1385 (m), 1202 (w), 1134 (w), 1083 (w), 1020 (w), 876 (w), 814 (m), 466 (w).
UV-Vis (CH3OH, nm): λ 234 (ε 10753), λ 271 (ε 5756), λ 366 (ε 2078).
MS (m/z, QMS-MS/MS): 374.89 [M+–O2CCH3].
Micro Analysis Calculated for C14H12N3O2Cl2Ag (433.04): Calcd.: C, 38.83%; H, 2.79%; N, 9.70%; Cl, 16.37%; Ag, 24.90%; Found: Calcd.: C, 38.37%; H, 2.91%; N, 9.69%; Cl, 16.53%; Ag, 24.61%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 7.77 (dd, J = 14.9, 8.1 Hz, 3H, CHBenzimid), 7.67 (d, J = 7.8 Hz, 1H, CHBenzimid), 7.50–7.37 (m, 4H, CHCyanobenzyl), 5.84 (s, 2H, CH2), 4.07 (s, 3H, N–CH3), 1.74 (s, 3H, COCH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 181.4(NCN), 174.4(CO), 142.5, 134.6, 133.5, 133.0, 128.5, 124.6, 124.6, 118.8, 112.7, 112.4, 111.1(CN + CBenzimid + CCyanobenzyl), 51.4(CH2), 36.1(N–CH3), 23.8(COCH3).
IR absorptions (KBr, cm−1): 3435 (w), 3060 (w), 2229 (s), 1570 (s), 1410 (s), 1346 (w), 1194 (w), 1093 (w), 1020 (w), 797 (w), 750 (s), 654 (w), 552 (w), 464 (w).
UV-Vis (CH3OH, nm): λ 223 (ε 14835), λ 237 (ε 11269), λ 275 (ε 7612), λ 285 (ε 6123).
MS (m/z, QMS-MS/MS): 355.93 [M+–O2CCH3].
Micro Analysis Calculated for C18H16N3O2Ag (414.21): Calcd.: C, 52.19%; H, 3.89%; N, 10.14%; Ag, 26.04%; Found: Calcd.: C, 52.27%; H, 4.01%; N, 10.29%; Ag, 25.98%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 7.71 (d, J = 8.0 Hz, 4H, CHCyanobenzyl), 7.40 (d, J = 8.0 Hz, 4H, CHCyanobenzyl), 7.38 (s, 2H, CHBenzimid), 5.72 (s, 4H, CH2), 2.21 (s, 6H, CH3), 2.05 (s, 3H, COCH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 181.7(NCN), 176.3(CO), 142.2, 134.3, 133.1, 132.3, 128.3, 118.9, 112.5, 111.1(CN + CBenzimid + CCyanobenzyl), 51.6(CH2), 31.0(CH3), 20.1(COCH3).
IR absorptions (KBr, cm−1): 3436 (s), 3056 (w), 2924 (m), 2855 (m), 2230 (s), 1609 (m), 1577 (s), 1402 (s), 1184 (w), 1019 (m), 848 (w), 814 (w), 678 (w), 548 (m).
UV-Vis (CH3OH, nm): λ 229 (ε 9120), λ 288 (ε 4110), λ 362 (ε 1546).
MS (m/z, QMS-MS/MS): 484.03 [M+–O2CCH3].
Micro Analysis Calculated for C27H23N4O2Ag (543.37): Calcd.: C, 59.68%; H, 4.26%; N, 10.31%; Ag, 19.85%; Found: C, 59.44%; H, 4.18%; N, 10.09%; Ag, 20.08%.
1H NMR (δ ppm DMSO-d6, 400 MHz): 7.45 (s, 2H, CHBenzimid), 7.39–7.24 (m, 10H, CHBenzyl), 5.65 (s, 4H, CH2), 2.25 (s, 6H, CH3), 1.77 (s, 3H, COCH3).
13C NMR (δ ppm DMSO-d6, 100 MHz, proton decoupled): 185.9 (NCN), 176.1 (CO), 136.8, 133.6, 132.4, 129.2, 128.3, 127.7, 112.7 (CBenzimid + Cbenzyl), 52.2 (CH2), 23.8(COCH3), 20.2 (CH3).
IR absorptions (KBr, cm−1): 3437 (s), 2923 (m), 1627 (w), 1577 (s), 1486 (w), 1448 (w), 1403 (s), 1337 (w), 1184 (w), 1024 (w), 845 (w), 707 (s).
UV-Vis (CH3OH, nm): λ 284 (ε 7734), λ 360 (ε 2778), λ 438 (ε 2190).
MS (m/z, QMS-MS/MS): 434.72 [M+–O2CCH3].
Micro Analysis Calculated for C25H25N2O2Ag (493.35): Calcd.: C, 60.86%; H, 5.11%; N, 5.68%; Ag, 21.86%; Found: C, 60.72%; H, 5.15%; N, 5.77%; Ag, 21.73%.
1H NMR (δ ppm CDCl3, 400 MHz): 7.34–7.17 (m, 12H, CHImid + CHBenzyl), 7.04–6.93 (m, 8H, CHBenzyl), 5.33 (s, 4H, CH2), 2.08 (s, 3H, COCH3).
13C NMR (δ ppm CDCl3, 100 MHz, proton decoupled): 178.9(NCN), 176.3(CO), 136.1, 132.6, 130.6, 129.1, 128.6, 128.5, 127.9, 127.7, 127.3(CImid + CBenzyl), 53.7(CH2), 22.8 (COCH3).
IR absorptions (KBr, cm−1): 3437 (s), 2925 (m), 1630 (w), 1570 (s), 1497 (w), 1445 (m), 1405 (m), 1178 (w), 1021 (m), 923 (w), 875 (w), 759 (m), 732 (m), 700 (s).
UV-Vis (CH3OH, nm): λ 214 (ε 16958), λ 263 (ε 9939), λ 364 (ε 2919).
MS (m/z, QMS-MS/MS): 508.73 [M+–O2CCH3].
Micro Analysis Calculated for C31H27N2O2Ag (567.43): Calcd.: C, 65.62%; H, 4.80%; N, 4.94%; Ag, 19.01%; Found: C, 65.54%; H, 4.75%; N, 4.63%; Ag, 18.91%.
2.3 Antibacterial studies
Preliminary in vitro antibacterial activity of non-symmetrically substituted and symmetrically substituted N-heterocyclic carbenes and their corresponding silver complexes were screened against two bacterial strains. The test organisms included Staphylococcus aureus (SA) (NCTC 7447) as a Gram-positive bacteria and Escherichia coli (E. coli) as Gram-negative bacteria.To assess the biological activity of compounds 3a–f and 4a–f, the qualitative Kirby–Bauer disk-diffusion method was applied.26 All bacteria were individually cultured from a single colony in sterile LB medium27 overnight at 37 °C (orbital shaker incubator). All the work carried out was performed under sterile conditions.
For each strain, 70 μL of culture were spread evenly on agar-LB medium. Four 5 mm diameter Whatman paper discs were placed evenly separated on each plate. Two stock solutions (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 DMSO:H2O) of every compound were prepared at 2.2 μM and 4.4 μM to be able to test the effect of different concentrations. Each plate was then tested with 5 μL and 7 μL of 2.2 μM solution and 5 μl and 10 μL for the 4.4 μM solution. The plates were covered and placed in an incubator at 37 °C for 24 h. The plates were then removed and the area of clearance (defined as the distance between the edge of the filter paper disc and the beginning of the bacterial growth) for each sample was measured in millimetres.
:10 DMSO:H2O) of every compound were prepared at 2.2 μM and 4.4 μM to be able to test the effect of different concentrations. Each plate was then tested with 5 μL and 7 μL of 2.2 μM solution and 5 μl and 10 μL for the 4.4 μM solution. The plates were covered and placed in an incubator at 37 °C for 24 h. The plates were then removed and the area of clearance (defined as the distance between the edge of the filter paper disc and the beginning of the bacterial growth) for each sample was measured in millimetres.
2.4 Cytotoxicity studies
Preliminary in vitrocell tests were performed on the human cancerous renal cell line Caki-1 in order to compare the cytotoxicity of the compounds presented in this paper. These cell lines were chosen based on their regular and long-lasting growth behaviour, which is similar to the one shown in kidney carcinoma cells. They were obtained from the ATCC (American Tissue Cell Culture Collection) and maintained in Dulbecco's Modified Eagle Medium containing 10% (v/v) FCS (fetal calf serum), 1% (v/v) penicillin streptomycin and 1% (v/v) L-glutamine. Cells were seeded in 96-well plates containing 200 μL microtitre wells at a density of 5000-cells/200 μL of medium and were incubated at 37 °C for 24 h to allow for exponential growth. Then the compounds used for the testing were dissolved in the minimal amount of DMSO (dimethylsulfoxide) possible and diluted with medium to obtain stock solutions of 5 × 10−4 M in concentration and less than 0.7% of DMSO. The cells were then treated with varying concentrations of the compounds and incubated for 48 h at 37 °C. Then, the solutions were removed from the wells and the cells were washed with PBS (phosphate buffer solution) and fresh medium was added to the wells. Following a recovery period of 24 h incubation at 37 °C, individual wells were treated with a 200 μL of a solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) in medium. The solution consisted of 30 mg of MTT in 30 mL of medium. The cells were incubated for 3 h at 37 °C. The medium was then removed and the purple formazan crystals were dissolved in 200 μL DMSO per well. A Wallac Victor (Multilabel HTS Counter) Plate Reader was used to measure absorbance at 540 nm. Cell viability was expressed as a percentage of the absorbance recorded for control wells. The values used for the dose response curves represent the values obtained from four consistent MTT-based assays for each compound tested.3. Results and discussion
3.1 Synthesis
The synthetic route for non-symmetrically substituted and symmetrically substituted N-heterocyclic carbenes as ligand precursors and their corresponding silver complexes described in this work is given in Schemes 1, 2, 3 and 4. The non-symmetrically substituted NHC precursors 1-methyl-3-(4-cyanobenzyl)imidazolium bromide (3a) and 1-methyl-3-(4-cyanobenzyl)benzimidazolium bromide (3c) were prepared by stirring 1-methylimidazole (1a) and 1-methylbenzimidazole (1c) with p-cyanobenzyl bromide (2a) in toluene at room temperature for 3-4 d with 81% and 76% yields respectively. 4,5-Dichloro-1-(4-cyanobenzyl)imidazole (1bII) is formed in 81% yield from the deprotonation of 4,5-dichloro-1H-imidazole (1b) with K2CO3 and subsequent alkylation with p-cyanobenzyl bromide (2a) in acetonitrile. 4,5-Dichloro-1-(4-cyanobenzyl)-3-methylimidazolium iodide (3b) was prepared by heating of 4,5-dichloro-1-(4-cyanobenzyl)imidazole (1bII) with iodomethane in acetonitrile for 1d with a yield of 75%. The symmetrically substituted NHCs precursors 1,3-bis(4-cyanobenzyl)-5,6-dimethylbenzimidazolium bromide (3d), 1,3-dibenzyl-5,6-dimethylbenzimidazolium bromide (3e) and 1,3-dibenzyl-4,5-diphenylimidazolium bromide (3f) were prepared by stirring 5,6-dimethyl-1H-benzimidazole (1d) and 4,5-diphenyl-1H-imidazole (1e) with 2 equivalents of p-cyanobenzyl bromide (2a) and benzyl bromide (2b) in the presence of K2CO3 as a base in CH3CN at room temperature for 4 d with 76%, 82% and 68% yields respectively. | ||
| Scheme 1 General reaction scheme for the synthesis of non-symmetrically substituted N-heterocyclic carbenes (3a and 3c) and their corresponding N-heterocyclic carbene–silver complexes (4a and 4c). | ||
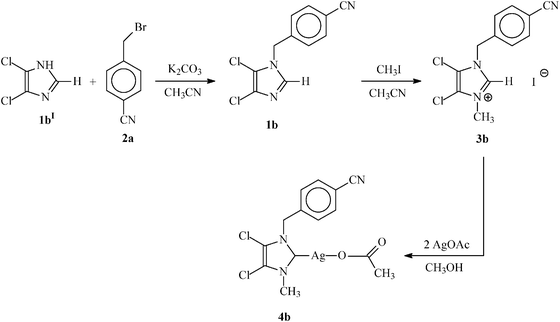 | ||
| Scheme 2 General reaction scheme for the synthesis of non-symmetrically substituted N-heterocyclic carbene (3b) and its N-heterocyclic carbene–silver complex (4b). | ||
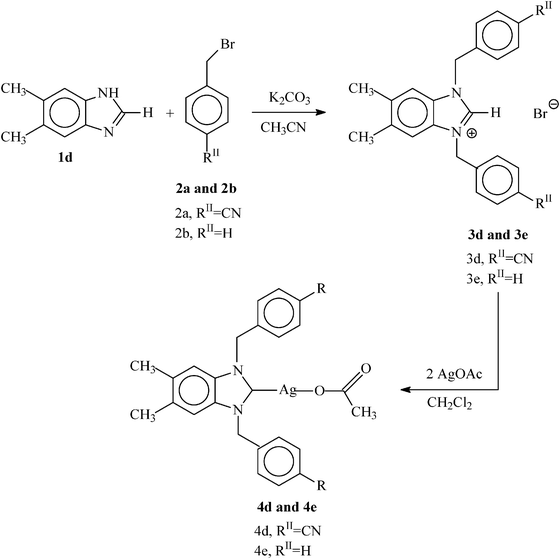 | ||
| Scheme 3 General reaction scheme for the synthesis of symmetrically substituted N-heterocyclic carbenes (3d and 3e) and their corresponding N-heterocyclic carbene–silver complexes (4d and 4e). | ||
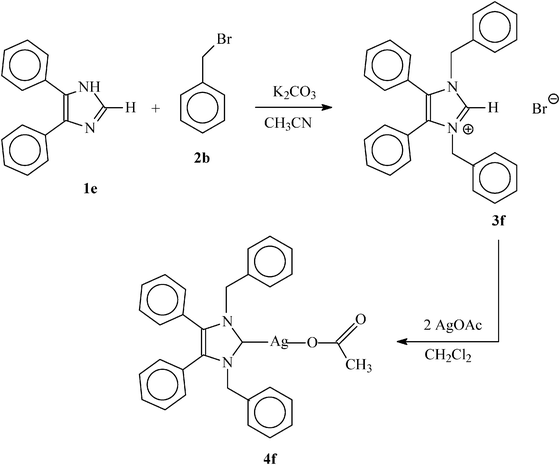 | ||
| Scheme 4 General reaction scheme for the synthesis of symmetrically substituted N-heterocyclic carbene (3f) and its N-heterocyclic carbene–silver complex (4f). | ||
The NHC precursors were characterized by spectral (1H, 13C NMR, IR, UV-visible and mass) and elemental analysis studies. Additionally, the solid state structure of the NHC precursors 3c–e were determined by single crystal X-ray diffraction. The 1H NMR spectra of all NHC precursors 3a–f show a characteristic downfield shift in the range δ = 9.27–11.05 ppm for the NCHN proton attributable to the positive charge of the molecule.28–30 In addition, their identities have also been confirmed by a base peak for the [M+–Br] fragments in their positive mode ESI mass spectra.
The NHC–silver complexes (1-methyl-3-(4-cyanobenzyl)imidazole-2-ylidene) silver(I) acetate (4a), (4,5-dichloro-1-(4-cyanobenzyl)-3-methyl)imidazole-2 ylidene) silver(I) acetate (4b), (1-methyl-3-(4-cyanobenzyl)benzimidazole-2-ylidene) silver(I) acetate (4c), (1,3-bis(4-cyanobenzyl)5,6-dimethylbenzimidazole-2-ylidene) silver(I) acetate (4d), (1,3-dibenzyl-5,6-dimethylbenzimidazole-2-ylidene) silver(I) acetate (4e) and (1,3-dibenzyl-4,5-diphenylimidazol-2-ylidene) silver(I) acetate (4f) were synthesized by the reaction of 3a–f with 2 equivalents of silver acetate in dichloromethane. The reaction mixture was stirred for 1–3 d at room temperature or refluxed for 2–4 d to afford the NHC–silver acetate complexes as off white solid in 23% to 79% yield. The complexes were characterized by spectral (1H, 13C NMR, IR, UV-visible and mass) and elemental analysis studies. Furthermore, the solid state structures of 4b and 4d–f were analysed by single crystal X-ray diffraction. The absence of a downfield NCHN signal and presence of new signals at 2.08–1.70 ppm for the acetate protons in all the 1H NMR spectra for 4a–f however, indicates a successful complex formation. The 13C NMR resonances of the carbene carbon atoms in complexes 4a–f occur in the range δ 181.7–178.9 ppm respectively. These signals are shifted downfield compared to the corresponding precursors of 3a–fcarbene carbons resonance at the range 141.8–137.2 ppm respectively which further demonstrates the formation of expected NHC–silver acetate complexes. Also the appearance of the 13C NMR resonances for the carbonyl and methyl carbons of the acetate group of complexes 4a–f in the range 176.3–173.6 and 24.1–20.1 ppm respectively showed the formation of the NHC–silver complexes.6,8 Furthermore, positive mode ESI mass spectra of all six NHC–silver complexes (4a–f) are dominated by [M+–O2CCH3] fragment peaks arising from the loss of one acetate ligand.
3.2 Structural discussion
Suitable crystals for X-ray crystallography to determine the molecular structure of 3c–e were grown from the slow evaporation of a saturated methanol solution, while crystals of 4b and 4d–f were formed in a saturated dichloromethane solution with slow infusion of pentane. Compounds 3c, 3d, 4b and 4d–f crystallised in the monoclinic space groupP21/n (#14), P21/c (#14), P2/n (#13), P21/c (#14) and C2/c (#15) respectively while compound 3e crystallised in the triclinic space groupP(#2). Compounds 3e and 4e crystallised with 2 and 8 molecules in the unit cell, whilst 3c, 3d, 4b, 4d and 4f crystallised with 4 molecules in the unit cell. Compounds 3c and 3e have a lattice held water molecules in the unit cell of the determined structure. In compounds 3d, 4b and 4d–f there is an absence of any lattice held water molecules or organic solvent molecules in the unit cells of the determined structures. The molecular structures of the compounds 3c–e, 4b and 4d–f are shown in Fig. 1–8. The crystal data and refinement details for all seven compounds are tabulated in Table 1, whereas selected bond lengths and bond angles are compiled in Table 2.
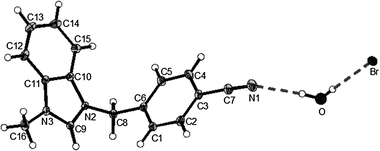 | ||
| Fig. 1 X-Ray diffraction structure of 3c; molecule; thermal ellipsoids are drawn on the 15% probability level. | ||
 | ||
| Fig. 2 X-Ray diffraction structure of 3d; molecule; thermal ellipsoids are drawn on the 15% probability level. | ||
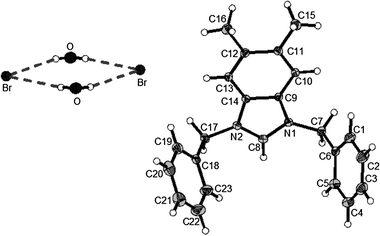 | ||
| Fig. 3 X-Ray diffraction structure of 3e; molecule; thermal ellipsoids are drawn on the 15% probability level. | ||
 | ||
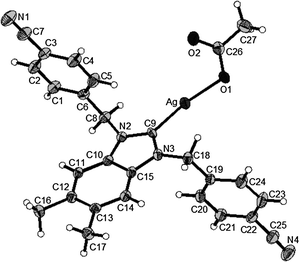
| Fig. 4 X-Ray diffraction structure of 4b; molecule; thermal ellipsoids are drawn on the 50% probability level. | ||
 | ||
| Fig. 5 X-Ray diffraction structure of 4d; molecule; thermal ellipsoids are drawn on the 50% probability level. | ||
 | ||
| Fig. 6 X-Ray diffraction structure of 4e; molecule; thermal ellipsoids are drawn on the 50% probability level. | ||
 | ||
| Fig. 7 X-Ray diffraction structure of 4f; molecule; thermal ellipsoids are drawn on the 50% probability level. | ||
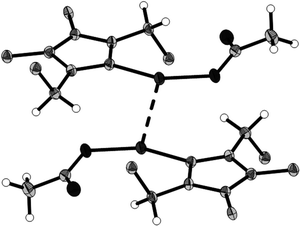 | ||
| Fig. 8 Dimer held together by Ag⋯Ag interaction of 4f; thermal ellipsoids are drawn on the 50% probability level, phenyl groups are represented by their ipso carbons only. | ||
| Bond lengths/Å | 3c | 3d | 3e | Bond lengths/Å | 4b | 4d | 4e | 4f |
|---|---|---|---|---|---|---|---|---|
| N(2)–C(9) | 1.330(3) | 1.3311(18) | N(2)–C(9) | 1.362(5) | 1.352(4) | 1.398(2) | ||
| N(2)–C(10) | 1.392(2) | 1.3935(17) | N(2)–C(10) | 1.383(5) | 1.385(4) | |||
| C(9)–N(3) | 1.323(3) | 1.3325(18) | C(9)–N(3) | 1.356(5) | 1.359(4) | |||
| C(11)–N(3) | 1.389(3) | 1.3950(18) | C(11)–N(3) | 1.382(5) | ||||
| C(10)–C(11) | 1.388(3) | 1.3923(18) | C(10)–C(11) | 1.352(6) | ||||
| N(1)–C(8) | 1.330(2) | C(15)–N(3) | 1.393(4) | |||||
| N(1)–C(9) | 1.391(2) | C(10)–C(15) | 1.387(4) | |||||
| C(8)–N(2) | 1.330(2) | C(10)–Cl(1) | 1.700(4) | |||||
| C(14)–N(2) | 1.394(2) | C(11)–Cl(2) | 1.693(4) | |||||
| C(9)–C(14) | 1.388(2) | Ag–C(9) | 2.069(4) | 2.052(3) | ||||
| Ag–O(1) | 2.140(3) | 2.113(2) | 2.1585(12) | |||||
| O(1)–C(13) | 1.255(5) | |||||||
| O(2)–C(13) | 1.241(5) | |||||||
| C(13)–C(14) | 1.530(6) | |||||||
| O(1)–C(26) | 1.276(4) | |||||||
| O(2)–C(26) | 1.239(4) | |||||||
| C(26)–C(27) | 1.508(5) | |||||||
| N(1)–C(8) | 1.358(5) | 1.351(2) | ||||||
| N(3)–C(33) | 1.359(5) | |||||||
| N(1)–C(10) | 1.394(2) | |||||||
| C(8)–N(2) | 1.354(5) | 1.348(2) | ||||||
| C(33)–N(4) | 1.361(5) | |||||||
| C(9)–C(10) | 1.363(2) | |||||||
| Ag–C(8) | 2.0896(17) | |||||||
| O(1)–C(30) | 1.285(2) | |||||||
| O(2)–C(30) | 1.242(2) | |||||||
| C(30)–C(31) | 1.514(3) | |||||||
| N(1)–C(9) | 1.395(5) | |||||||
| C(14)–N(2) | 1.394(5) | |||||||
| C(9)–C(14) | 1.391(6) | |||||||
| Ag(1)–C(8) | 2.073(4) | |||||||
| Ag(1)–O(1) | 2.171(3) | |||||||
| O(1)–C(24) | 1.271(5) | |||||||
| O(2)–C(24) | 1.235(6) | |||||||
| C(24)–C(25) | 1.497(6) | |||||||
| N(3)–C(34) | 1.398(5) | |||||||
| C(39)–N(4) | 1.391(5) | |||||||
| C(34)–C(39) | 1.390(6) | |||||||
| Ag(2)–C(33) | 2.073(4) | |||||||
| Ag(2)–O(3) | 2.137(3) | |||||||
| O(3)–C(49) | 1.281(5) | |||||||
| O(4)–C(49) | 1.242(6) | |||||||
| C(49)–C(50) | 1.511(6) |
| Bond angles [°] | 3c | 3d | 3e | Bond angles [°] | 4b | 4d | 4e | 4f |
|---|---|---|---|---|---|---|---|---|
| N(2)–C(9)–N(3) | 110.33(17) | 109.81(13) | N(2)–C(9)–N(3) | 104.5(3) | 105.3(2) | |||
| C(9)–N(2)–C(10) | 108.24(16) | 108.78(11) | C(9)–N(2)–C(10) | 110.8(3) | 111.6(2) | |||
| C(9)–N(3)–C(11) | 108.46(16) | 108.62(11) | C(9)–N(3)–C(11) | 111.2(3) | ||||
| C(10)–C(11)–N(3) | 106.60(16) | 106.43(12) | C(10)–C(11)–N(3) | 106.6(4) | ||||
| C(11)–C(10)–N(2) | 106.36(17) | 106.36(11) | C(11)–C(10)–N(2) | 106.9(3) | ||||
| N(1)–C(8)–N(2) | 110.39(15) | C(9)–Ag–O(1) | 173.86(14) | 170.49(10) | ||||
| C(8)–N(1)–C(9) | 108.20(14) | C(13)–O(1)–Ag | 105.5(3) | |||||
| C(8)–N(2)–C(14) | 108.20(14) | C(26)–O(1)–Ag | 107.55(19) | |||||
| C(9)–C(14)–N(2) | 106.48(14) | C(9)–N(3)–C(15) | 111.1(2) | |||||
| C(14)–C(9)–N(1) | 106.71(13) | N(2)–C(10)–C(15) | 106.1(2) | |||||
| C(10)–C(15)–N(3) | 106.0(2) | |||||||
| N(2)–C(8)–N(1) | 106.0(3) | 104.88(14) | ||||||
| C(8)–N(1)–C(10) | 111.51(14) | |||||||
| C(10)–C(9)–N(2) | 106.27(15) | |||||||
| C(9)–C(10)–N(1) | 106.06(15) | |||||||
| C(8)–N(2)–C(9) | 111.28(14) | |||||||
| C(8)–Ag–O(1) | 163.40(6) | |||||||
| C(30)–O(1)–Ag | 107.04(11) | |||||||
| C(8)–N(2)–C(14) | 111.0(3) | |||||||
| C(9)–C(14)–N(2) | 106.1(3) | |||||||
| C(14)–C(9)–N(1) | 106.1(3) | |||||||
| C(8)–N(1)–C(9) | 110.8(3) | |||||||
| C(8)–Ag(1)–O(1) | 165.38(15) | |||||||
| O(2)–C(24)–O(1) | 122.7(4) | |||||||
| C(33)–N(4)–C(39) | 110.8(3) | |||||||
| C(34)–C(39)–N(4) | 106.4(3) | |||||||
| C(39)–C(34)–N(3) | 106.2(3) | |||||||
| C(33)–N(3)–C(34) | 110.7(3) | |||||||
| C(33)–Ag(2)–O(3) | 166.60(14) | |||||||
| O(4)–C(49)–O(3) | 123.8(4) |
In the five-membered ring (NCNCC) of compounds 3c–e, the bond distances and angles are in good agreement with each other and the bond distances found in the similar compound 1,3-diisopropylbenzimidazolium bromide reported in literature (see Table 2).31 The NHC–silver complexes 4b and 4d–f are mononuclear complexes. In the NHC–silver complexes 4b and 4d–f reported here, the bond lengths and angles in and directly around the NHC core agree very well among each other and with literature data.32–36 Comparing the precursors 3d and 3e with the corresponding complexes 4d and 4e (see Table 2), one finds a slight increase of both the C(9)–N and C(8)–N distances. In the NHC–silver complexes 4b, 4d, 4e and 4fsilver is two-coordinate in an approximately linear fashion. In 4f there is a significant argentophilic interaction with an Ag⋯Ag distance of 2.9107(3) Å (Fig. 7). This is accompanied by a slight increase of the silver–carbene bond length (Ag–C(8) = 2.0896(17) Å), compared to those in the complexes 4b, 4d and 4e (see Table 2).
3.3 Biological evaluation
The in vitro antibacterial activities of NHC-precursors and NHC–silver complexes were tested using the qualitative Kirby-Bauer disk diffusion method. The antibacterial activities of the NHC-precursors and their corresponding NHC–silver complexes are summarized in Fig. 9–12. The results are tabulated in Tables 3 and 4, respectively. The metal salt (silver acetate) used to prepare the complexes and the solvent (DMSO) used to prepare the stock solutions played no role in growth inhibition on the same bacteria as previously reported.22,37 With respect to our previously tested compounds,22 the concentration of the stock solutions is reduced by 4-folds, hence the amount of compound used in each test is significantly less, indicating a remarkably stronger biological activity. Almost no antibacterial activity was observed for compounds 3a–c against the both Gram-positive bacteria Staphylococcus aureus and Gram-negative bacteria Escherichia coli. Medium to high antibacterial activity was observed for compounds 3d–f against Gram-positive bacteria Staphylococcus aureus but no antibacterial activity was observed against Gram-negative bacteria Escherichia coli. The compounds 4a–d have shown medium antibacterial activity against the both Gram-positive bacteria Staphylococcus aureus and Gram-negative bacteria Escherichia coli. The best antibacterial activity was observed for the compounds 4e and 4f against Gram-positive bacteria Staphylococcus aureus with areas of clearance 12 to 15 mm while medium activity was observed towards Gram-negative bacteria Escherichia coli with areas of clearance 6 to 7 mm at the highest amount (0.44 μmol) used (see Tables 3 and 4).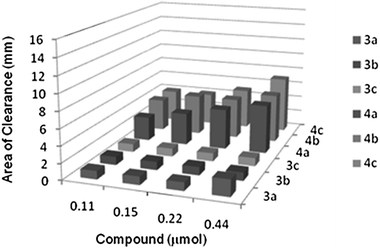 | ||
| Fig. 9 Area of clearance on Staphylococcus aureus (Gram +ve) by 3a–c and 4a–c. | ||
 | ||
| Fig. 10 Area of clearance on Escherichia coli (Gram −ve) by 3a–c and 4a–c. | ||
 | ||
| Fig. 11 Area of clearance on Staphylococcus aureus (Gram +ve) by 3d–f and 4d–f. | ||
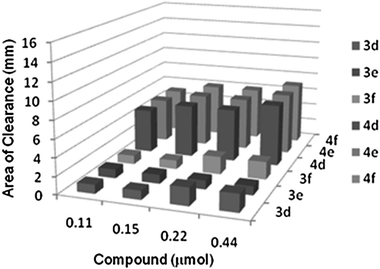 | ||
| Fig. 12 Area of clearance on Escherichia coli (Gram −ve) by 3d–f and 4d–f. | ||
| Compounds | Staphylococcus aureus (Gram +ve) | Escherichia coli (Gram −ve) | |
|---|---|---|---|
| 3a/mm | 0.11 μmol (30.5 μg) | 1 | 1 |
| 0.15 μmol (42.8 μg) | 1 | 1 | |
| 0.22 μmol (61.1 μg) | 1 | 1 | |
| 0.44 μmol (122.3 μg) | 2 | 1 | |
| 3b/mm | 0.11 μmol (43.3 μg) | 1 | 1 |
| 0.15 μmol (60.6 μg) | 1 | 1 | |
| 0.22 μmol (86.6 μg) | 1 | 2 | |
| 0.44 μmol (173.3 μg) | 1 | 2 | |
| 3c/mm | 0.11 μmol (36.1 μg) | 1 | 1 |
| 0.15 μmol (50.5 μg) | 1 | 1 | |
| 0.22 μmol (72.2 μg) | 1 | 1 | |
| 0.44 μmol (144.4 μg) | 1 | 2 | |
| 3d/mm | 0.11 μmol (50.3 μg) | 5 | 1 |
| 0.15 μmol (70.4 μg) | 5 | 1 | |
| 0.22 μmol (100.6 μg) | 6 | 2 | |
| 0.44 μmol (201.2 μg) | 7 | 2 | |
| 3e/mm | 0.11 μmol (44.8 μg) | 6 | 1 |
| 0.15 μmol (62.7 μg) | 6 | 1 | |
| 0.22 μmol (89.6 μg) | 10 | 1 | |
| 0.44 μmol (179.2 μg) | 10 | 1 | |
| 3f/mm | 0.11 μmol (52.9 μg) | 9 | 1 |
| 0.15 μmol (74.1 μg) | 10 | 1 | |
| 0.22 μmol (105.9 μg) | 11 | 2 | |
| 0.44 μmol (211.8 μg) | 13 | 2 | |
| Compounds | Staphylococcus aureus (Gram +ve) | Escherichia coli (Gram −ve) | |
|---|---|---|---|
| 4a/mm | 0.11 μmol (40.0 μg) | 3 | 5 |
| 0.15 μmol (56.0 μg) | 4 | 5 | |
| 0.22 μmol (80.1 μg) | 5 | 6 | |
| 0.44 μmol (160.2 μg) | 6 | 7 | |
| 4b/mm | 0.11 μmol (47.6 μg) | 4 | 5 |
| 0.15 μmol (66.6 μg) | 5 | 6 | |
| 0.22 μmol (95.2 μg) | 5 | 7 | |
| 0.44 μmol (190.5 μg) | 6 | 8 | |
| 4c/mm | 0.11 μmol (45.5 μg) | 4 | 5 |
| 0.15 μmol (63.7 μg) | 4 | 6 | |
| 0.22 μmol (91.1 μg) | 5 | 6 | |
| 0.44 μmol (182.2 μg) | 7 | 7 | |
| 4d/mm | 0.11 μmol (59.7 μg) | 4 | 5 |
| 0.15 μmol (83.6 μg) | 4 | 6 | |
| 0.22 μmol (119.5 μg) | 5 | 6 | |
| 0.44 μmol (239.0 μg) | 5 | 7 | |
| 4e/mm | 0.11 μmol (54.2 μg) | 6 | 5 |
| 0.15 μmol (75.9 μg) | 9 | 6 | |
| 0.22 μmol (108.5 μg) | 10 | 6 | |
| 0.44 μmol (217.0 μg) | 12 | 7 | |
| 4f/mm | 0.11 μmol (62.5 μg) | 10 | 5 |
| 0.15 μmol (87.5 μg) | 11 | 6 | |
| 0.22 μmol (125.0 μg) | 13 | 6 | |
| 0.44 μmol (250.0 μg) | 15 | 7 | |
Based on the above discussions it can be stated that (i) the NHC–silver complexes 4a–f are significantly more active against both Gram-positive bacteria Staphylococcus aureus and Gram-negative bacteria Escherichia coli than the NHC-precursors 3a–f. (ii) It was concluded that, as the NHC-precursors and NHC–silver complexes concentration increases, the antimicrobial activity becomes higher and (iii) It was also observed that, compared to the NHC-precursors the NHC–silver complexes exhibited enhanced antibacterial activity, which is due to the synergistic effect of the increased lipophilicity of the complexes. Chelation decreases the polarity of the metal ion, which further leads to the enhanced lipophilicity of the complex. Since the bacterial cell wall is surrounded by a lipid membrane which favours the passage of lipid soluble materials, increased lipophilicity allows the penetration of complex into and through the membrane and deactivates the active enzyme sites of the microorganisms.38
In comparison with the known reported NHC-precursors and NHC–silver complexes from the literature6,8,22 the NHC-precursors (3a–f) and their corresponding NHC–silver complexes (4a–f) have remarkably higher antibacterial activity.
3.4 Cytotoxicity studies
We are interested in the different types of NHC–silver complexes as possible anti-cancer drugs. Therefore the in vitro cytotoxicity of non-symmetrically substituted NHC–silver acetate complexes 4a–c and symmetrically substituted NHC–silver acetate complexes 4d–f was evaluated by MTT-based assays39 on the human cancerous renal-cell line Caki-1. This test involves a 48 h drug exposure period, followed by a 24 h recovery time. The log dose response curve for NHC–silver acetate complexes 4a–f can be viewed in Fig. 13 and 14, respectively. Non-symmetrically substituted NHC–silver acetate complexes 4a–c, which contains 1-methylimidazole, 4,5-dichloro-1-(4-cyanobenzyl)imidazole and 1-methylbenzimidazole groups, has an IC50 values 6.2 (±1.0), 7.7 (±1.6) and 1.2 (±0.6) μM, respectively. Compound 4c is the most promising candidate in this paper because of lowest IC50 value and has an approximately five-fold and six-fold increase in magnitude when compared with compounds 4a and 4b respectively. In comparison with cisplatin (IC50 value = 3.3 μM), 4c has an approximately three-fold increase in magnitude. Symmetrically substituted NHC–silver acetate complexes 4d–f, which also contains 5,6-dimethyl-1H-benzimidazole and 4,5-diphenyl-1H-imidazole groups, has an IC50 values 10.8 (±1.9), 24.2 (±1.8) and 13.6 (±1.0) μM, respectively. Compounds 4d and 4f show a very similar IC50 value and two-fold increase in magnitude when compared with 4e and in comparison with cisplatin the IC50 values for the three compounds are not impressive.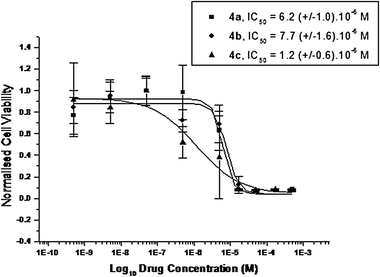 | ||
| Fig. 13 Cytotoxicity curves from typical MTT assays showing the effect of compounds 4a–c on the viability of Caki-1 cells. | ||
 | ||
| Fig. 14 Cytotoxicity curves from typical MTT assays showing the effect of compounds 4d–f on the viability of Caki-1 cells. | ||
Non-symmetrically and symmetrically substituted NHC–silver acetate complexes show almost similar IC50 values; both compound classes are easily soluble in DMSO and all compounds are stable in saline solution with respect to silver chloride precipitation. It was also observed that, compared to known reported NHC–silver complexes from the literature,9,22 the NHC–silver complexes (4a–f) have shown slightly high cytotoxic activity.
4. Conclusions and outlook
Six new non-symmetrically substituted and symmetrically substituted N-heterocyclic carbene–silver acetate derivatives (4a–f) were synthesised through the reaction of appropriately non-symmetrically substituted and symmetrically substituted N-heterocyclic carbenes (3a–f) with silver acetate. Almost all the complexes have shown high antibacterial activity compared to the precursors against both Gram-positive bacteria Staphylococcus aureus and Gram-negative bacteria Escherichia coli. While screening this novel series we have noted a marked improvement in antibacterial activity with respect to the compounds previously reported.22 We were able to significantly reduce the substrate concentration in the stock solution (from 18.4 μM previously utilised down to 4.4 μM, four-fold) and achieve a greater area of clearance in many cases (from 7 mm with complexes (4,5-dichloro-1,3-bis(4-methoxybenzyl)imidazole-2-ylidene)silver(I)acetate and (1,3-dibenzylimidazole-2-ylidene)silver(I)acetate22 to 15 mm with compound 4f when tested against Staphylococcus aureus for example). We are currently looking at the possibility of determining MIC50 values for the best compounds. NHC–silver complexes 4a–f yielded antitumor IC50 values of 6.2 (±1.0), 7.7 (±1.6), 1.2 (±0.6) and 10.8 (±1.9), 24.2 (±1.8) and 13.6 (±1.0) μM respectively on the Caki-1 cell line. One of the compounds contained in this paper (1-methyl-3-(4-cyanobenzyl)benzimidazole-2-ylidene)silver(I) acetate (4c) shows the highest cytotoxicity [IC50 value of 1.2 (±0.6) μM] up to now for the N-heterocyclic carbene–silver complexes synthesised in our research group, indicating the high potential as an anti-cancer drug. Further work is currently underway in order to improve these values by performing formulation experiments to improve solubility of these NHC-silver acetate complexes, which should allow for in vivo testing of 4c in the nearby future.Acknowledgements
The authors thank the Irish Research Council for Science Engineering and Technology (IRCSET) for funding through a postdoctoral fellowship for Dr Siddappa Patil.References
- K. Öfele, J. Organomet. Chem., 1968, 12, 42 CrossRef.
- H. W. Wanzlick and H. J. Schönberr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef.
- K. M. Hindi, T. J. Siciliano, S. Durmus, M. J. Panzner, D. A. Medvetz, D. V. Reddy, L. A. Hogue, C. E. Hovis, J. K. Hilliard, R. J. Mallet, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2008, 51, 1577 CrossRef CAS.
- A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884 CrossRef CAS.
- A. Kascatan-Nebioglu, A. Melaiye, K. M. Hindi, S. Durmus, M. J. Panzner, L. A. Hogue, R. J. Mallett, C. E. Hovis, M. Coughenour, S. D. Crosby, A. Milsted, D. L. Ely, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2006, 49, 6811 CrossRef CAS.
- D. A. Medvetz, K. M. Hindi, M. J. Panzner, A. J. Ditto, Y. H. Yun and W. J. Youngs, Metal-Based Drugs, 2008 Search PubMed , Article ID 384010, 7 pages, http://dx.doi.org/10.1155/2008/384010.
- A. Melaiye, R. S. Simons, A. Milsted, F. Pingitore, C. Wesdemiotis, C. A. Tessier and W. J. Youngs, J. Med. Chem., 2004, 47, 973 CrossRef CAS.
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042 CrossRef CAS.
- C. L. Cannon, L. A. Hogue, R. K. Vajravelu, G. H. Capps, A. Ibricevic, K. M. Hindi, A. Kascatan-Nebioglu, M. J. Walter, S. L. Brody and W. J. Youngs, Antimicrob. Agents Chemother., 2009, 53, 3285 CrossRef.
- B Rosenberg, L. V. Van Camp and T. Krigas, Nature, 1965, 205, 698 CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467 CrossRef CAS.
- J. M. Rademaker-Lakhai, D. V. Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717 CAS.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140 CrossRef CAS.
- A. Vessières, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529 RSC.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 3474 RSC.
- B. Thati, A. Noble, B. Creaven, M. Walsh, M. McCann, K. Kavanagh, M. Devereux and D. Egan, Cancer Lett., 2007, 248, 321 CrossRef CAS.
- H. Zhu, X. Zhang, X. Liu, X. Wang, G. Liu, A. Usman and H. Fun, Inorg. Chem. Commun., 2003, 6, 1113 CrossRef CAS.
- J. Liu, P. Galetis, A. Farr, L. Maharaj, H. Samarasinha, A. McGechan, B. Baguley, R. Bowen, S. Berners-Price and M. McKeage, J. Inorg. Biochem., 2008, 102, 303 CrossRef CAS.
- S. Patil, J. Claffey, A. Deally, M. Hogan, B. Gleeson, L. M. M. Méndez, H. Müller- Bunz, F. Paradisi and M. Tacke, Eur. J. Inorg. Chem., 2010, 1020 CrossRef CAS.
- G. M. Sheldrick, SADABS Version 2.03, University of Göttingen, Germany, 2002 Search PubMed.
- Program CrysalisPro Version 1.171.33.55, Oxford Diffraction Limited, 2010 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- A. Bondi, H. E. Spaulding, E. D. Smith and C. C. Dietz, Am. J. Med. Sci., 1947, 213, 221.
- S. E. Luria, Bacteriol Rev., 1947, 11, 1.
- W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS.
- A. J. Arduengo, H. V. Rasika-Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405 CrossRef.
- J. C. Garrison, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2005, 690, 6008 CrossRef CAS.
- H. V. Huynh, Y. Han, J. H. H. Ho and G. K. Tan, Organometallics, 2006, 25, 3267 CrossRef CAS.
- P. De Fremont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301 CrossRef CAS.
- Y. Han, Y. T. Hong and H. V. Huynh, J. Organomet. Chem., 2008, 693, 3159 CrossRef CAS.
- C. P. Newman, G. J. Clarkson and J. P. Rourke, J. Organomet. Chem., 2007, 692, 4962 CrossRef CAS.
- V. Lillo, J. Mata, J. Ramirez, E. Peris and E. Fernandez, Organometallics, 2006, 25, 5829 CrossRef CAS.
- M. Viciano, E. Mas-Marza, M. Sanau and E. Peris, Organometallics, 2006, 25, 3063 CrossRef CAS.
- B. Gleeson, J. Claffey, D. Ertler, M. Hogan, H. Müller-Bunz, F. Paradisi, D. Wallis and M. Tacke, Polyhedron, 2008, 27, 3619 CrossRef CAS.
- R. Karvembu, C. Jayabalakrishnan and K. Natarajan, Transition Met. Chem., 2002, 27, 574 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
Footnote |
| † CCDC reference numbers 777915–777921. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0mt00034e |
| This journal is © The Royal Society of Chemistry 2011 |