3D-QSAR studies on purine-carbonitriles as cruzain inhibitors: comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA)
Oscar
Méndez-Lucio
a,
Jaime
Pérez-Villanueva
b,
Antonio
Romo-Mancillas
a and
Rafael
Castillo
*a
aFacultad de Química, Departamento de Farmacia, UNAM, México, DF 04510, Mexico. E-mail: rafaelc@servidor.unam.mx; Fax: +525 56 22 53 29; Tel: +525 56 22 52 87
bDepartamento de Sistemas Biológicos, División de Ciencias Biológicas y de la Salud, UAM-X, México, DF 04960, Mexico
First published on 5th September 2011
Abstract
Cruzain has been identified as the major cysteine protease of Trypanosoma cruzi, the etiological cause of Chaga's disease. For this reason, many efforts have been undertaken to design new inhibitors against this enzyme. Recently, molecules having a purine or triazine nucleus have been reported as potent non-peptidic inhibitors of cruzain. In order to gain an insight into the structural requirements that can lead to the improvement of the activity of these molecules, in this paper we report the CoMFA and CoMSIA studies of a series of purine-carbonitriles as cruzain inhibitors. Quantum semi-empirical calculations of the inhibitors inside the active site of cruzain were used as an approach to obtain reliable conformations for molecular alignment. Two different molecular alignments were used, resulting in 3 CoMFA models and 31 CoMSIA models. These models correspond to all of the possible combinations among five fields: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor. Highly predictive models were obtained. Based on the q2 values, the best CoMFA model had an r2 = 0.98 and a q2 = 0.73, whereas the best CoMSIA model retrieved an r2 = 0.88 and a q2 = 0.62. All models were validated with a rigorous procedure using an external test set. Contour maps obtained from these models show a preference toward the purine ring and indicate that bulky groups with a negative potential at the 3- and 5-positions of the phenyl ring are important structural requirements for inhibitory activity against cruzain.
Introduction
American trypanosomiasis is an infection caused by Trypanosoma cruzi, a flagellated protozoan parasite which infects around 10–12 million people, kills more than 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 each year and has become a world health problem.1 This parasite is transmitted to humans mainly by insects from the genera Triatoma, Panstrongylus and Rhodnius.2 Once a human has been infected, there appears an acute phase characterized by mild symptoms. After this phase, an asymptomatic period starts and can last between 10 and 20 years. Finally, the patient presents a chronic phase which could lead to sudden death caused by the enlarging of hollow viscera and/or heart problems.3Benznidazole and nifurtimox have been used to treat patients; nevertheless, serious side effects and the inefficacy of these drugs in chronic phase patients have stimulated the interest of several research groups to search for new therapeutic agents.3,4
000 each year and has become a world health problem.1 This parasite is transmitted to humans mainly by insects from the genera Triatoma, Panstrongylus and Rhodnius.2 Once a human has been infected, there appears an acute phase characterized by mild symptoms. After this phase, an asymptomatic period starts and can last between 10 and 20 years. Finally, the patient presents a chronic phase which could lead to sudden death caused by the enlarging of hollow viscera and/or heart problems.3Benznidazole and nifurtimox have been used to treat patients; nevertheless, serious side effects and the inefficacy of these drugs in chronic phase patients have stimulated the interest of several research groups to search for new therapeutic agents.3,4
The life cycle of this parasite consists of four stages: the epimastigote and metacyclic trypomastigote, which take place in the insect vector, and the amastigote and bloodstream trypomastigote, which appear only in the infected organism.3 An essential enzyme for the differentiation between these life cycle stages is cruzain, the major cysteine protease of Trypanosoma cruzi. Among other functions, cruzain plays an essential role in the penetration into the host cell, the defense against the immune system of the infected organism and in the parasite's nutrition.3 Based on the vital functions of cruzain, it has been proposed as a promising target in the design of new drugs against this parasite. Some examples of reversible and irreversible inhibitors are the previously reported molecules with a sulfone, a fluoro methyl ketone or a nitrile as a susceptible group to nucleophilic attack by the enzyme.5–8
Recently, a set of 33 purine nitriles as reversible covalent inhibitors of cruzain have been synthesized and tested.8 Although some of these compounds show good activity, an optimization of these molecules can be carried out. For this purpose, computational methodologies are an important tool in the study of the three-dimensional quantitative structure–activity relationships (3D-QSAR). Particularly, Comparative Molecular Field Analysis (CoMFA)9 and Comparative Molecular Similarity Indices Analysis (CoMSIA)10 are two powerful methodologies widely used. These two methodologies allow us to study a set of molecules, their putative active conformation, and binding mode. A great advantage of CoMFA and CoMSIA is the contour maps that highlight the most important structural features that could be useful in the optimization or design of new active compounds. When doing this kind of study, it is important to consider that molecules in the dataset have the same binding mode, act via the same mechanism and have a common pharmacophore (not necessarily the same molecular skeleton).11 In particular, molecules with two different scaffolds (purine and triazine) were analyzed during this study; nevertheless it is important to notice that the triazine is a substructure of the purine nucleus.
In order to obtain more information concerning the ligand–protein interactions that can lead to the improvement of purine nitrile cruzain inhibitors, CoMFA and CoMSIA studies were carried out, obtaining high quality models. As far as we know, this is the first time that the inhibition of cruzain by purine nitrile molecules has been studied by 3D-QSAR.
Results and discussion
CoMFA and CoMSIA models
In general, all molecules used in this study have a similar structure as seen in Table 1. A rational procedure to obtain a molecular alignment consists of using a molecular conformation similar to that of the crystallographic protein–ligand complex. Therefore, the first stage of this study was to build CoMFA models derived from two different molecular alignments (Fig. 1) in order to obtain the closer conformation to the one expected in the binding site.|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) | pIC50 |
| a Compounds in the test set. | ||||
| 1 | Ethyl | 0.71 | 6.1487 | |
| 2 | Cyclopentyl | 0.71 | 6.1487 | |
| 3 | Propyl | 0.79 | 6.1024 | |
| 4 | Isopropyl | 0.89 | 6.0506 | |
| 5 a | Methylcyclopropyl | 0.89 | 6.0506 | |
| 6 | Isobutyl | 0.89 | 6.0506 | |
| 7 | Butyl | 0.89 | 6.0506 | |
| 8 | 4-Hydroxybutyl | 0.89 | 6.0506 | |
| 9 | Methyl | 1.26 | 5.8996 | |
| 10 a | Cyclohexyl | 1.41 | 5.8508 | |
| 11 | 2-Hydroxyethyl | 1.58 | 5.8013 | |
| 12 | Cyclopentyl | 3-Nitro | 0.063 | 7.2006 |
| 13 a | Cyclopentyl | 3-Fluoro | 0.079 | 7.1024 |
| 14 | Cyclopentyl | 3-Chloro | 0.079 | 7.1024 |
| 15 | Cyclopentyl | 3-Bromo | 0.1 | 7.0000 |
| 16 | Cyclopentyl | 4-Fluoro | 0.126 | 6.8996 |
| 17 | Cyclopentyl | 3-Methyl | 0.126 | 6.8996 |
| 18 | Cyclopentyl | 4-Bromo | 1.122 | 5.9500 |
| 19 | Cyclopentyl | 3-Phenyl | 2.239 | 5.6499 |
| 20 | Cyclopentyl | 3,5-Difluoro | 0.063 | 7.2007 |
| 21 a | Cyclopentyl | 3,5-Dichloro | 0.398 | 6.4001 |
| 22 | 2,2-Difluoroethyl | 3,5-Difluoro | 0.025 | 7.6021 |
| 23 | Ethyl | 3,5-Difluoro | 0.01 | 8.0000 |
| 24 a | 2,2-Difluoroethyl | 3,5-Difluoro | 0.013 | 7.8860 |
| 25 | Cyclopentyl | 3,5-Difluoro | 0.018 | 7.7447 |
| 26 | Ethyl | 3-Chloro | 0.018 | 7.7447 |
| 27 | Cyclopentyl | 3-Chloro | 0.04 | 7.3979 |
| 28 | 3,5-Difluoro | 2,2-Difluoroethyl | 0.05 | 7.3010 |
| 29 | 3-Chloro | Ethyl | 0.063 | 7.2007 |
| 30 | 3-Chloro | 2,2-Difluoroethyl | 0.071 | 7.1487 |
| 31 a | 3,5-Difluoro | Ethyl | 0.251 | 6.6003 |
| 32 | 3,5-Difluoro | Cyclopentyl | 0.316 | 6.5003 |

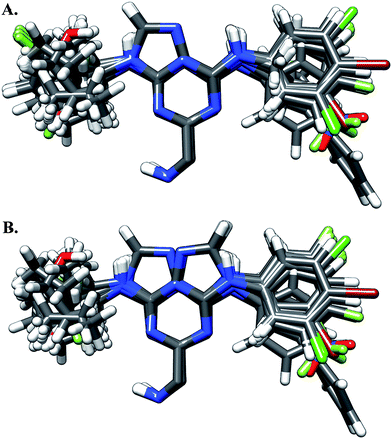 | ||
| Fig. 1 Different alignments used in this study: (A) is based on crystallographic data of compound 23 bound to cruzain; (B) is based on the fact that the S2 pocket has a predilection for hydrophobic residues. | ||
Model 1 was developed using the alignment shown in Fig. 1A, in which the purine scaffold of all molecules is oriented in the same way as the crystallographic structure of compound 23 bound to cruzain. For this alignment, model 1 had a q2 of 0.623 and an r2 of 0.983, which are quite acceptable results for a 3D-QSAR model. The statistical results for model 1 are shown in Table 2.
| Statistics | CoMFA | CoMSIA | |||||
|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | SED | SEA | SEDA | SEHDA | |
| a Cross-validated correlation coefficient from LOO. b Standard error of predictions derived from the LOO method. c Non-crossvalidated r2. d Standard error estimate. e F-Test value. f Correlation coefficient derived from predictions of test set molecules. g Optimum number of principal components. | |||||||
| Alignment | A | B | B | B | B | B | B |
| Grid spacing/Å | 2.0 | 2.0 | 1.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| q 2 a | 0.623 | 0.736 | 0.692 | 0.627 | 0.618 | 0.565 | 0.460 |
| Pressb | 0.488 | 0.408 | 0.441 | 0.473 | 0.479 | 0.523 | 0.533 |
| r 2 c | 0.983 | 0.980 | 0.977 | 0.888 | 0.874 | 0.901 | 0.712 |
| S d | 0.104 | 0.112 | 0.122 | 0.259 | 0.275 | 0.249 | 0.389 |
| F e | 227.096 | 196.155 | 166.508 | 41.591 | 36.544 | 36.586 | 59.474 |
| R 2 f | 0.856 | 0.763 | 0.753 | 0.806 | 0.833 | 0.815 | 0.758 |
| ONC g | 5 | 5 | 5 | 4 | 4 | 5 | 1 |
| Contribution | |||||||
| Steric | 0.815 | 0.816 | 0.804 | 0.367 | 0.461 | 0.336 | 0.159 |
| Electrostatic | 0.185 | 0.184 | 0.196 | 0.259 | 0.268 | 0.235 | 0.080 |
| Hydrophobic | 0.307 | ||||||
| Hydrogen bond donor | 0.374 | 0.347 | 0.304 | ||||
| Hydrogen bond acceptor | 0.271 | 0.082 | 0.150 | ||||
An alternative alignment was chosen for model 2 (Fig. 1B). In this approach, the phenyl groups of all the molecules were oriented on the same side of the purine scaffolds. This new alignment is based on the fact that the phenyl group of compound 23 is inside a pocket of cruzain which is very similar to the S2 pocket of papain, another cysteine protease. It is important to mention that previous studies on papain have found that the S2 pocket has a predilection for hydrophobic residues, especially for phenylalanine.12 For this CoMFA model, a similar value of r2 (0.980) was obtained, but it had a slightly better q2 of 0.736. These results indicate that both alignments are good enough to derive 3D-QSAR models. Nevertheless, model 2 has a better predictive power and the alignment used to build this model is based on experimental data. For these reasons, the alignment shown in Fig. 1B was used to generate the subsequent models.
Since the calculation of the CoMFA and CoMSIA descriptors takes place in a three-dimensional lattice, another variable that was evaluated in this study is the lattice grid spacing. An exhaustive analysis of several 3D-QSAR models indicates that the smallest grid spacing results in a higher q2.13 To prove this hypothesis in our database, model 3 was built considering the same alignment and parameters as in model 2, but with a grid spacing of 1 Å, whereas the grid spacing of model 2 was 2 Å. Lower values for the statistical parameters (q2 = 0.692 and r2 = 0.977) were obtained from model 3. This unexpected result has been observed before as an indication of the poor effect of more grid points, as compared with a careful choice of parameters at 2 Å.14 Thus, model 2 was selected as the best CoMFA model and the predicted values of activity are plotted and listed in Fig. 2A and Table 3, respectively.
 | ||
| Fig. 2 Plots of experimental vs. predicted values of (A) CoMFA model 2 and (B) CoMSIA including SED fields. | ||
| Compound | pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Prediction | Residual | Prediction | Residual | ||
| a Compounds in the test set. | |||||
| 1 | 6.1487 | 6.1119 | 0.0368 | 6.107 | 0.0417 |
| 2 | 6.1487 | 5.9593 | 0.1894 | 6.0697 | 0.0791 |
| 3 | 6.1024 | 6.0296 | 0.0728 | 6.0662 | 0.0362 |
| 4 | 6.0506 | 6.0463 | 0.0043 | 5.9688 | 0.0818 |
| 5 a | 6.0506 | 6.3420 | −0.2914 | 6.3777 | −0.3271 |
| 6 | 6.0506 | 6.0562 | −0.0056 | 6.1253 | −0.0747 |
| 7 | 6.0506 | 6.1141 | −0.0635 | 6.1739 | −0.1233 |
| 8 | 6.0506 | 6.125 | −0.0744 | 5.9938 | 0.0568 |
| 9 | 5.8996 | 5.9918 | −0.0922 | 6.2667 | −0.3671 |
| 10 a | 5.8508 | 5.8323 | 0.0184 | 6.0114 | −0.1606 |
| 11 | 5.8013 | 5.8808 | −0.0795 | 5.7081 | 0.0933 |
| 12 | 7.2006 | 7.3393 | −0.1387 | 7.1142 | 0.0864 |
| 13 a | 7.1024 | 6.9560 | 0.1464 | 6.9091 | 0.1932 |
| 14 | 7.1024 | 7.0989 | 0.0035 | 6.8515 | 0.2509 |
| 15 | 7.0000 | 6.9979 | 0.0021 | 6.7704 | 0.2296 |
| 16 | 6.8996 | 6.8324 | 0.0672 | 6.6669 | 0.2327 |
| 17 | 6.8996 | 6.8793 | 0.0204 | 6.6588 | 0.2408 |
| 18 | 5.9500 | 5.9941 | −0.0441 | 6.6577 | −0.7077 |
| 19 | 5.6499 | 5.6839 | −0.0339 | 5.6500 | −0.0001 |
| 20 | 7.2007 | 7.0986 | 0.1021 | 7.2680 | −0.0674 |
| 21 a | 6.4001 | 6.8935 | −0.4934 | 7.0180 | −0.6179 |
| 22 | 7.6021 | 7.6803 | −0.0782 | 7.5783 | 0.0238 |
| 23 | 8.0000 | 8.1405 | −0.1405 | 7.9463 | 0.0537 |
| 24 a | 7.8860 | 7.6108 | 0.2752 | 7.7680 | 0.1180 |
| 25 | 7.7447 | 7.6108 | 0.1339 | 7.768 | −0.0233 |
| 26 | 7.7447 | 7.7499 | −0.0051 | 7.4673 | 0.2775 |
| 27 | 7.3979 | 7.3678 | 0.0302 | 7.5408 | −0.1429 |
| 28 | 7.3010 | 7.2295 | 0.0716 | 7.4335 | −0.1325 |
| 29 | 7.2007 | 6.9939 | 0.2067 | 6.8395 | 0.3611 |
| 30 | 7.1487 | 7.1043 | 0.0444 | 7.1660 | −0.0173 |
| 31 a | 6.6003 | 7.2303 | −0.6300 | 7.1365 | −0.5361 |
| 32 | 6.5003 | 6.73 | −0.2297 | 6.9896 | −0.4893 |
The CoMSIA studies were carried out using the 1B alignment and a grid spacing of 2 Å. Five fields were used: steric (S), electrostatic (E), hydrophobic (H), hydrogen bond donor (D) and hydrogen bond acceptor (A), and the 31 possible combinations among these were evaluated (Fig. 3). Interestingly, the higher values of q2 were obtained for the combinations SED, SEA, and SEDA (0.627, 0.618 and 0.565, respectively) and not for the one that includes the five fields. A detail that stands out is that almost all the combinations with a q2 > 0.5 include the steric and/or the electrostatic fields, the same kind of descriptors used in the CoMFA models. These results suggest that the structure–activity relationship for this database cannot be explained based on hydrophobicity; however, the similarities of the cruzain pocket with the S2 pocket of papain suggest that it would be interesting to generate inhibitors with a higher molecular diversity (including hydrophobic modifications) at this position.
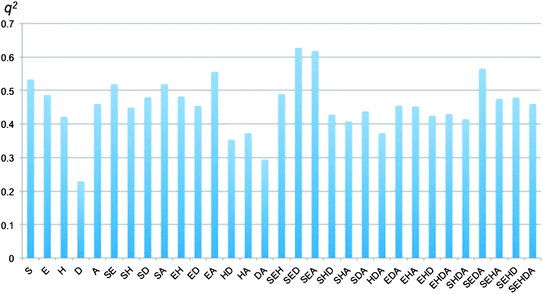 | ||
| Fig. 3 Histogram presenting the q2 value of the 31 possible CoMSIA field combinations. | ||
Validation of CoMFA and CoMSIA models
Although the q2 value is used in several papers to define the predictive ability of a QSAR model, it has been observed that the high value (q2 > 0.5) is a necessary condition, but insufficient to assure the predictive power and model robustness.15 For this reason, Golbraikh and Tropsha15 developed a methodology to validate QSAR models. This methodology consists of evaluating the difference between test set predictions obtained by the derived QSAR model and those determined by a hypothetical ideal model.Hence, this validation methodology was applied to the activity predictions of compounds in the test set of all models reported in this paper. The results of the validation procedure are shown in Table 4 and plotted in Fig. 4.
| Statistics | CoMFA | CoMSIA | |||||
|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | SED | SEA | SEDA | SEHDA | |
| a Correlation coefficient derived from predictions of test set molecules. b Correlation coefficients for the regression through origin for experimental vs. predicted and predicted vs. experimental activity, respectively. c Slopes for regression through origin for experimental vs. predicted and predicted vs. experimental, respectively. | |||||||
| R 2 a | 0.856 | 0.763 | 0.753 | 0.806 | 0.833 | 0.815 | 0.758 |
| R 20 b | 0.705 | 0.654 | 0.540 | 0.676 | 0.695 | 0.725 | 0.553 |
| k c | 1.028 | 1.021 | 1.030 | 1.030 | 1.026 | 1.024 | 1.026 |
| (R2 − R20)/R2 | 0.176 | 0.142 | 0.282 | 0.161 | 0.166 | 0.110 | 0.269 |
| R 0′2b | 0.829 | 0.761 | 0.739 | 0.795 | 0.815 | 0.810 | 0.744 |
| k'c | 0.971 | 0.976 | 0.968 | 0.968 | 0.972 | 0.975 | 0.972 |
| (R′2 − R0′2)/R2 | 0.032 | 0.002 | 0.018 | 0.013 | 0.022 | 0.006 | 0.017 |
 | ||
| Fig. 4 Validation using the test set for (A) CoMFA model 2 and (B) CoMSIA including SED fields. | ||
It can be seen that all CoMFA and CoMSIA models predicted, in a good manner, the activities of the compounds in the test set (this is represented by values of R2 > 0.75). Examining the results for the CoMFA models, it can also be seen that the R2 value of model 1 is higher than the one presented by model 2 (0.856 and 0.763, respectively); this is indicative that model 1 has better predictive power than model 2. Nevertheless, all the models fulfil the criteria for [(R2 − R20)/R2] and k (see Methods); in other words, all the models have an acceptable predictive power. By analyzing the CoMFA validation results in a detailed manner, it can be seen that model 2 has the better results; that is to say, a value of [(R2 − R20)/R2] near zero and the k value closest to 1. Since the only difference between these two models is the molecular alignment, model 2 is still selected as the best CoMFA model because the alignment used for this model is supported by experimental evidence.
Now focusing on the CoMSIA validation results, a similar situation was found. The model based on SEDA fields fulfils the validation criteria better; even so it does not have the highest value of R2, but this can be explained in terms of the molecular diversity in the test set. It would be more useful for the future to use numerous test groups with greater molecular diversity to observe if the value of R2 correlates with the one of q2. An interesting aspect of these results is that in all the three CoMSIA models with the highest value of q2, the R2 value is higher than the one presented in model 2 of the CoMFA study. This difference suggests that the CoMSIA models, in this study, have a better predictive ability than the CoMFA models.
CoMFA contour maps
The most important information that a CoMFA model can give, in addition to the prediction of pIC50, is the contour maps, which can guide to a more rationalized lead optimization. The 3D contour maps obtained from model 2 give information about the structural features to assess active compounds. In order to obtain better-defined maps, a column filtering of 2 kcal was included in model 2; it is important to mention that the predictive ability of model 2 was not affected by this modification. The steric (green and yellow) and electrostatic (red and blue) contour maps are shown in Fig. 5. The most potent inhibitor (23) is shown in balls and sticks, while the rest of the compounds are represented by lines. | ||
| Fig. 5 CoMFA contour maps obtained from model 2 (with a column filtering of 2 kcal). Green contours denote sterically favored regions and yellow contours sterically unfavored regions. Blue contours indicate areas where a negative potential increases activity, whereas red contour regions indicate areas where a negative potential decreases it. | ||
By analyzing contour maps in Fig. 5, it can be seen that a large green contour (sterically favored) and a smaller one are found near positions 3 and 5 of the phenyl ring. This could be indicative that halogens at these positions on the phenyl could increase the biological activity. Indeed, the inhibition presented by molecules 12–32, where most of them are substituted in those positions by bulky groups, mainly halogens, has a pIC50 > 6.4. On the other hand, compounds 1–11, which have no substitution on the phenyl ring, presented considerably low biological activity (<6.2). One exception to the above mentioned is compound 19, which has a 3-phenyl substituent close to a yellow contour (sterically unfavored). This contour is in good agreement with the extremely low pIC50 presented by this compound (5.6499), and gives information about the size of the S2 pocket where this group is placed. Based on the results obtained with the current database we conclude that the S2 pocket is large enough to hold bulky groups such as 3,5-difluorophenyl or 3,5-dichlorophenyl, but it is not big enough to contain groups such as a 3-biphenyl.
Another yellow contour appears near the cyclopentyl substituent at position 9 of the purine ring in molecules 12–21, 25, 27 and 32. In this area, bulky groups would decrease activity. In fact, this observation is exemplified by comparing compounds 25, 27 and 32 with molecules 23, 26 and 31, respectively. In all cases, molecules with an ethyl group instead of the cyclopentyl are more active. This effect could be related to the fact that these groups are exposed to the solvent; the less lipophilic these groups are, the more interactive they are with water molecules in the solvent, nevertheless more specialized calculations are needed to support this idea.
The electrostatic contours are in red when a negative potential is favorable for the enzyme inhibition, whereas blue contours are close to areas where a negative potential is unfavorable. A blue contour appears near position 3 of the phenyl group and it is related to the 3-phenyl substituent of compound 19, the compound with lowest biological activity of the entire database. As it can be seen in Fig. 5, the blue contour crosses through the center of a phenyl group, exactly where a high electron density is concentrated due to the π electrons.
CoMSIA contour maps
One of the advantages of the CoMSIA over CoMFA methodology is that the former produces better-defined contour maps. This circumstance arises from the fact that the use of molecular similarity indices avoids the arbitrary definitions of cutoff values, and the descriptors can be calculated in all grid points.10 In this section, contour maps obtained from the CoMSIA model in which SEDA fields were included (Fig. 6) will be discussed. In Fig. 6A the contour maps corresponding to steric and electrostatic fields are displayed; compound 23 is shown in a ball and stick representation while the rest of the compounds are just in lines. Interestingly, the contour maps for steric interactions are very similar to those of CoMFA; that is, a green contour (sterically favored) upon position 3 of the phenyl group, while a yellow one (sterically disfavored) is above the cyclopentyl substituent. Nevertheless, some differences are found in electrostatic contours as compared with CoMFA. In CoMSIA, red contours (areas where a negative potential is favorable) are near positions 3 and 5 of the phenyl group, very close to the green contours discussed before. These red contours are related to the halogens in compounds 20–25, 28, 31, 32, and to the 3-nitro substituent of compound 12. It is important to remark that within these compounds they are the most active molecules in this database. Blue contours related to areas where negative potentials are unfavorable almost disappeared. This is a good example of how CoMSIA and CoMFA complement each other. In this case CoMFA was able to detect that the π electrons decrease the activity of compound 19, whereas CoMSIA detected the importance of halogens for biological activity. | ||
| Fig. 6 CoMSIA contour maps including SEDA fields. (A) Depicts steric and electrostatic contour maps, the ones that have the same color code as CoMFA. (B) Orange contours depict regions in which hydrogen bond acceptors are favorable, whereas those near yellow contours are unfavorable. In the same way cyan contours indicate regions where hydrogen bond donors increase activity, whereas those near purple contours decrease activity. | ||
Fig. 6B shows the contour maps related to the hydrogen bond donor and acceptor moieties. Hydrogen bond acceptor moieties are favorable for biological activity in regions near orange contours and unfavorable near yellow contours. In the case of hydrogen bond donors, these favor the activity if they are close to cyan contours and disfavor when near purple contours. Fig. 6B shows that a yellow contour surrounds the triazine nitrogen that is overlapped with the bridgehead carbon of purine. This contour might be explained as the preference of a carbon instead of nitrogen at this position in molecules with a triazine ring; in terms of the compounds used in this study, this result reflects a preference for the purine over the triazine ring. Comparing compounds 14 with 27 and 22 with 24, it is clearly seen that compounds with a purine scaffold are better cruzain inhibitors than those with a triazine ring. Interestingly, no interaction was found between cruzain and the purine scaffold in the crystal structure of compound 23. There exists the possibility that the purine scaffold could increase the electrophilicity of the nitrile group making it more susceptible to a nucleophilic attack from the thiolate of Cys25 as studied by Oballa et al.16 For this reason, a possible explanation for the differences in biological activity among the triazine and the purine inhibitors could be the different reactivity toward the enzyme, but it would be necessary to have a deeper study to confirm this hypothesis.
Another yellow contour appears pointing to the secondary amine that links the purine ring with the R2 substituents in compounds 29–32; in the same way, an orange and a cyan contour point to the amine linking the purine and the phenyl ring in compounds 22–28. It is worth mentioning that these contours are giving information about the purine orientation based on the secondary amine position, instead of showing the interaction of the amine as a hydrogen bond donor or hydrogen bond acceptor. In this set of compounds, these contours can be interpreted as a preference for the phenylamino group at position 6 of the purine ring as it is in compounds 22–28. A direct comparison between compounds 23–26 and 31, 28, 32, 29, respectively, shows that the phenylamino group at position 6 (and not at position 9) of purine confers a major activity on these compounds.
Methods
Dataset
The dataset used in this study contained 32 triazine and purine nitrile molecules which have been previously published.8 These compounds were divided into a training set used to derive the 3D-QSAR models and a test set used for the validation procedure. The test set was selected manually in order to screen the whole range of activities and diversity of structures. All the activities were converted to pIC50 (−log IC50) values shown in Table 1.Preparation of ligands and molecular alignment
All molecules were constructed using as a template the structure of the inhibitor 6-[(3,5-difluorophenyl)amino]-9-ethyl-9H-purine-2-carbonitrile (23) co-crystallized with cruzain obtained from the protein data bank (access number: 3I06).8 Then, the calculations of the ligands' energy minimization were aided by using TRITON,17 a graphic user interface for MOPAC that allowed us to minimize the ligands inside a cavity formed by 20 amino acids in the cruzain's active site (Glu19, Cys22, Gly23, Ser24, Cys25, Trp26, Cys63, Ser64, Gly65, Gly66, Leu67, Met68, Val137, Ala138, Val139, Leu160, Asp161, His162, Gly163, Glu208). Finally, the calculations were carried out with MOPAC2009 employing the PM6 hamiltonian, the localized molecular orbital method MOZYME18–22 and a continuum COSMO model with the water dielectric constant of 78.4.23 The backbone atoms of the amino acids were fixed and all the side chain and ligand atoms were optimized using the BFGS method.24–27After the calculations, the molecules were extracted from the cavity used for the minimization to start the alignment procedure. It is well known that the alignment of molecules plays a fundamental role in 3D-QSAR to obtain high quality models; for this reason, two different alignments were used in this research. The first one consisted of overlapping the imine group, which is a common substructure in all molecules (Fig. 1A). In a second approach, the bond between the purine ring and the nitrile moiety was rotated 180° in compounds 28–32, thus making the phenyl group match with that of compound 23 inside a pocket next to Cys25. After these modifications, the molecules were submitted to a second geometry optimization inside the cavity and finally aligned using the common substructure previously defined (Fig. 1B).
CoMFA and CoMSIA
For the CoMFA calculations, a three-dimensional lattice with a grid spacing of 2.0 Å was created and extended 4 Å away from the molecule for each model using SYBYL8.0.28 As an alternative approach, the same lattice was used with a grid space of 1.0 Å. In each lattice intersection a sp3 atom with a van der Waals radius of 1.52 Å was used as a steric probe and a +1 charge as an electrostatic probe. The energy cut-off default value of 30 kcal mol−1 was used. The CoMSIA models were derived using the same training set and alignment as those of the best CoMFA model. A grid spacing of 2.0 Å was used to calculate five different similarity indices: steric, electrostatic, hydrophobic, hydrogen bond donor and hydrogen bond acceptor. An attenuation factor of 0.3 was used for the Gaussian function.Statistical analysis and validation
In order to derive CoMFA and CoMSIA models, partial least squares analysis was carried out using the descriptors as independent variables and the pIC50 as a dependent variable. Cross-validation analysis was performed using the Leave-One-Out (LOO) method to determine the optimal number of components and to avoid data over fitting. The best model was chosen using as parameters the cross-validation (q2) and the correlation (r2) coefficients.Additional validation was performed predicting the activity of an external test set with 6 compounds using all the CoMFA and CoMSIA models. The predictions were analyzed by regression analysis, applying the criteria proposed by Golbraikh and Tropsha,15 as follows:
(1) q2 > 0.5.
(2) R2 > 0.6.
(3) [(R2 − R20)/R2] < 0.1 or [(R2 − R0′2)/R2] < 0.1.
(4) 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15.
where q2 is the cross-validated correlation coefficient from LOO; R2 is the correlation coefficient for experimental (y) vs. predicted (ỹ) activities for test set molecules; R20 and R0′2 are the correlation coefficients for the regression through origin for y vs. ỹ and ỹ vs. y respectively; k and k′ are the slopes for regression through origin yr0 = kỹ and ỹr0 = k'y.
Conclusions
A 3D-QSAR (CoMFA and CoMSIA) study was carried out on a set of 32 purine and triazine carbonitriles, which have been identified as inhibitors of cruzain, the most important cysteine protease of Trypanosoma cruzi. Geometry optimization of the ligands inside the active site of the enzyme using semi-empirical calculations yielded a good approach to obtain reliable conformations. The problem of molecular alignment was faced by using valuable experimental data (crystallographic structure of compound 23 (ref. 8) and information of the catalytic site of papain12) that led us to two different alignments, from which CoMFA models 1 and 2 were derived. Model 2 was selected as the best CoMFA model, which yielded quite satisfactory values of q2 (0.736) and r2 (0.980).The 31 possible combinations among the 5 different fields of CoMSIA were studied. The CoMSIA model that included the SEDA fields presented good statistical parameters with a q2 of 0.565, an r2 of 0.901 and a R2 of 0.815 and it was the one that has given us more information about the structural requirements for biological activity. All the CoMFA and CoMSIA models fulfil the validation procedure proposed by Golbraikh and Tropsha.15
Four main structural requirements for biological activity were revealed by this study: a purine scaffold is preferred over the triazine ring; bulky groups with a negative electrostatic potential, such as halogens, seem to be important at positions 3 and 5 of the phenyl group; small groups are preferred at position 9 of the purine scaffold; the phenyl group must be linked to purine at position 6 by a secondary amine.
In conclusion, the models reported in this paper show good predictive power, and the information concerning the structural requirements could be useful for optimization of these molecules or for the design of new compounds.
Acknowledgements
This research was supported by CONACyT project no. 80093. O. M.-L and A. R.-M are very grateful to CONACyT for the fellowships granted (no. 245408 and 173861, respectively).References
- J. Clayton, Nature Outlook, June 24th, 2010, 54–55 Search PubMed.
- J. Rodrigues-Coura and P. Albajar-Viñas, Nature Outlook, June 24th, 2010, 56–57 Search PubMed.
- J. Cazzulo, V. Stoka and V. Turk, Curr. Pharm. Des., 2001, 7, 1143–1156 CrossRef CAS.
- M. McGrath, A. Eakin, J. Engel, J. McKerrow, C. Craik and R. Fletterick, J. Mol. Biol., 1995, 247, 251–259 CrossRef CAS.
- E. Dufour, A. Storer and R. Menard, Biochemistry, 1995, 34, 9136–9143 CrossRef CAS.
- J. Palmer, D. Rasnick, J. Klaus and D. Bromme, J. Med. Chem., 1995, 38, 3193–3196 Search PubMed.
- J. Drenth, K. H. Kalk and H. M. Swen, Biochemistry, 1976, 1–8 Search PubMed.
- B. T. Mott, R. S. Ferreira, A. Simeonov, A. Jadhav, K. K.-H. Ang, W. Leister, M. Shen, J. T. Silveira, P. S. Doyle, M. R. Arkin, J. H. Mckerrow, J. Inglese, C. P. Austin, C. J. Thomas, B. K. Shoichet and D. J. Maloney, J. Med. Chem., 2010, 53, 52–60 Search PubMed.
- R. Cramer, D. Patterson and J. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS.
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS.
- H. Kubinyi, Drug Discovery Today, 1997, 2, 457–467 Search PubMed.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1968, 32, 898–902.
- R. R. Mittal, R. A. McKinnon and M. J. Sorich, J. Mol. Model., 2008, 14, 59–67 Search PubMed.
- J. L. Melville and J. D. Hirst, J. Chem. Inf. Comput. Sci., 2004, 44, 1294–1300 Search PubMed.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS.
- R. M. Oballa, J.-F. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998–1002 CrossRef CAS.
- M. Prokop, J. Adam, Z. Kriz, M. Wimmerova and J. Koca, Bioinformatics, 2008, 24, 1955–1956 Search PubMed.
- J. J. P. Stewart, Mopac 2009 Version Version 10.105L, Fujitsu Limited, Tokyo, Japan, 2006 Search PubMed.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2009, 15, 765–805 CrossRef CAS.
- J. J. P. Stewart, Int. J. Quantum Chem., 1996, 58, 133–146 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Struct., 1997, 401, 195–205 Search PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- C. G. Broyden, J. Inst. Math. Appl., 1970, 6, 222–231 Search PubMed.
- R. Fletcher, Comput. J., 1970, 13, 317–322 Search PubMed.
- D. Goldfarb, Math. Comput., 1970, 24, 23–26.
- D. F. Shanno, Math. Comput., 1970, 24, 647–656.
- SYBYL 8.0, Tripos International, 1699 South Hanley Rd., St Louis, Missouri, 63144, USA.
| This journal is © The Royal Society of Chemistry 2011 |