The synthesis and evaluation of dihydroquinazolin-4-ones and quinazolin-4-ones as thyroid stimulating hormone receptor agonists†
Erika E.
Englund
a,
Susanne
Neumann
b,
Elena
Eliseeva
b,
Joshua G.
McCoy
a,
Steven
Titus
a,
Wei
Zheng
a,
Noel
Southall
a,
Paul
Shinn
a,
William
Leister
a,
Craig J.
Thomas
a,
James
Inglese
a,
Christopher P.
Austin
a,
Marvin C.
Gershengorn
b and
Wenwei
Huang
*a
aNIH Chemical Genomics Center, 9800 Medical Center Drive, Building B, Bethesda, MD 20892-3371, USA
bNational Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Bethesda, MD 20892, USA
First published on 30th August 2011
Abstract
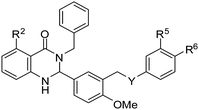
We herein describe the rapid synthesis of a diverse set of dihydroquinazolin-4-ones and quinazolin-4-ones, their biological evaluation as thyroid stimulating hormone receptor (TSHR) agonists, and SAR analysis. Among the compounds screened, 8b was 60-fold more potent than the hit compound 1a, which was identified from a high throughput screen of over 73,000 compounds.
Thyroid-stimulating hormone (TSH) is an α/β heterodimeric glycoprotein hormone that is produced in the anterior pituitary gland and controls function and proliferation of thyroid follicular cells upon interaction with the TSH receptor (TSHR).1–3 TSHR is a G-protein coupled receptor that belongs to the glycoprotein hormone receptor family.4 Two closely related members of this family include LHCGR (Luteinizing Hormone/Chorionic Gonadotropin Receptor)5 and FSHR (Follicle Stimulating Hormone Receptor).6,7 TSHR was first identified in the thyroid cell plasma membrane in 1966,8 and since that time has been found in bone,9 brain,10 kidney,11 testis,12 endometrium13 and the immune system.14 The primary function of TSHR in thyroid follicular cells is the regulation of thyroid hormone synthesis, as well as thyrocyte size and number,7,15 but the roles of TSHR in the other organs and tissues are not fully understood. The identification of a potent and selective small molecule TSHR agonist could provide a valuable tool for researchers interested in elucidating the roles and importance of this receptor in a variety of tissues. A small molecule TSHR agonist could also be clinically valuable. Recombinant TSH is currently used for patients with thyroid cancer receiving thyroid hormone suppression therapy, but it is difficult to produce and must be administered intramuscularly.16 A small molecule agonist could serve as an orally bioavailable alternative to recombinant TSH with advantages for therapeutic applications.
There are several reports of TSHR monoclonal antibody agonists,17–19 but very few of small molecule TSHR agonists. Thienopyrimidine Org 41841 (Fig. 1) was first described in 2002 as a partial LHCGR agonist and was later found to be a partial TSHR agonist with an EC50 of 7700 nM compared to 220 nM for LHCGR.20 This finding was followed with a series of synthesized analogues for SAR studies. Some analogues were LHCGR selective with low μM potency, but none were TSHR selective.21,22 Upon screening a library of 73,180 compounds (PubChem Assay ID: 1401), dihydroquinazolin-4-one 1a (Fig. 1) was identified as a TSHR agonist.23,24 The dihydroquinazolin-4-one chemotype is present in some known drugs and bioactive compounds, such as the diuretics fenquizone25 and metolazone,26 but had not been previously identified as a TSHR modulator. More importantly, 1a was a selective TSHR agonist with no detectable LHR or FSHR efficacy and was therefore selected for further studies. We previously reported that dihydroquinazolin-4-one 1b was a full TSHR agonist with in vivo efficacy in mice.24 In this paper, we describe the synthesis and structure–activity relationship studies that were undertaken to evaluate a library of dihydroquinazolin-4-one 1a analogues.
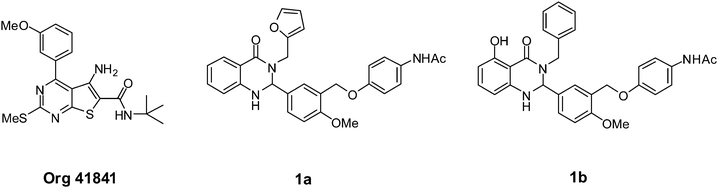 | ||
| Fig. 1 TSHR agonists. | ||
A convergent route was developed for the rapid synthesis of 1a analogues (Scheme 1). The western fragment 3 was synthesized either via coupling of R3NH2 with an acid 2 or ring opening of isatoic anhydride 4. The eastern fragment 7 was synthesized via the benzylation of 6 with 5. This product was combined with fragment 3 in the presence of Yb(OTf)3 to afford dihydroquinazolin-4-ones 1 or 8.27 Quinazolin-4-ones 9 were obtained by oxidation of dihydroquinazolin-4-ones 1 using either DDQ28 or MnO2.29
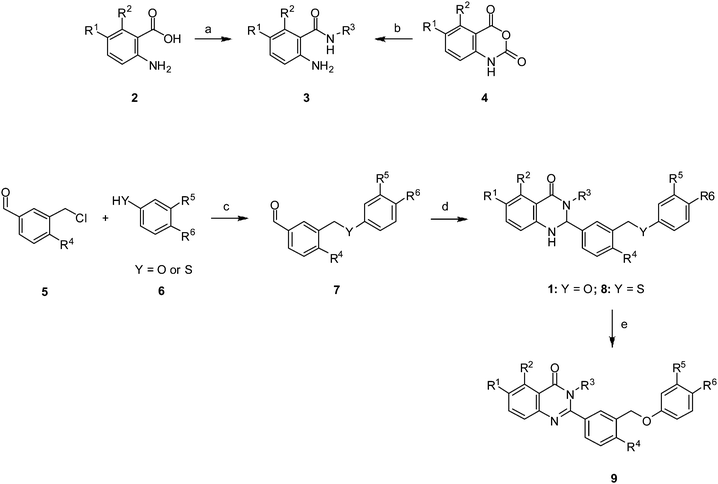 | ||
| Scheme 1 Reagents and conditions: (a) DIPEA, R3NH2, 2-chloro-1,3-dimethylimidazolinium chloride, CH2Cl2; (b) R3NH2, CH3CN (or DMA), 12 h; (c) K2CO3, CH3CN, 150 °C, 30 min; (d) Yb(OTf)3, 3, EtOH, 80 °C, 2–6 h; (e) DDQ, r.t., 1 h or MnO2, DMSO, 80 °C, 12 h. | ||
Our SAR studies first investigated substitution on the amide and several clear trends were observed (R3, Table 1). First, there was little tolerance for hydrophilic groups, such as the amine (1k), alcohol (1l) or pyridine (1i), at this site. Second, while lipophilic groups are generally well tolerated, there appeared to be steric constraints. The benzyl group (1g) performed the best out of all analogues, but either lengthening (phenethyl 1m) or shortening (phenyl 1f) the tether between the aryl ring and amide resulted in a 2 and 10-fold reduction in potency, respectively. Finally, aliphatic groups were tolerated (n-Bu 1h), albeit not especially potent, but a lone hydrogen (H 1j) resulted in a very low potency and decreased efficacy. Among the tested R3 analogues, the benzyl group (1g) was the most potent with a 4-fold higher potency than the hit compound 1a. Based upon these findings, the optimization of other sites was pursued with R3 fixed as benzyl.
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | EC50 (μM) | Emax (%) |
| a Activities (EC50) in μM were obtained from the Elisa assay. Emax is expressed as % of the maximal response of 1b, set at 100%.30 n.d. = not determined. | ||||||
| 1a | H | H |
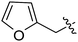
|
OMe | 1.16 | 101 |
| 1b | H | OH | Bn | OMe | 0.09 | 100 |
| 1c | H | OMe | Bn | OMe | 4.48 | 49.1 |
| 1d | OH | H | Bn | OMe | 0.33 | 93.3 |
| 1e | H | F | Bn | OMe | 0.28 | 94.7 |
| 1f | H | H | Ph | OMe | 5.09 | 75.1 |
| 1g | H | H | Bn | OMe | 0.29 | 99.8 |
| 1h | H | H | n-Bu | OMe | 0.83 | 112 |
| 1i | H | H |
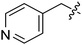
|
OMe | 3.98 | 70.3 |
| 1j | H | H | H | OMe | n.d. | 39.5 |
| 1k | H | H |
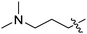
|
OMe | n.d. | 10.1 |
| 1l | H | H |
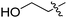
|
OMe | n.d. | 19.7 |
| 1m | H | H |
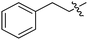
|
OMe | 0.43 | 102.5 |
| 1n | H | H | Bn | H | 1.86 | 36.8 |
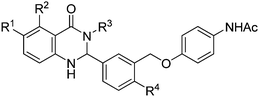
Aryl ring substitution on the western fragment of the molecule was next explored with a focus on the site ortho to the carbonyl (R2, Table 1). Analogues with H (1g) or F (1e) were equipotent, but the OH (1b) analogue resulted in a 3-fold improvement in potency. The free phenol was found to be critical for activity because when it was replaced with a methyl ether (1c) there was a sharp drop in potency. The orientation of the phenol was also critical for activity because when it was moved meta to the carbonyl (1d) the potency returned to the same level as the unsubstituted analogue (1g). This can be rationalized with a model. In our previously published modeling study, we proposed that 1b binds to the transmembrane helical bundle of TSHR. Both the R2OH and the carbonyl group of the dihyroquinazolin-4-one were predicted to participate in hydrogen bonding with Asparagine N5.47. This prediction was supported by a point mutation study where 1b lost significant activity with the N5.47A mutation.24 In our analogues, the OH at R1 was not appropriately situated to engage in hydrogen bonding with Asparagine N5.47 and the R2OMe analogue (1c) had no hydrogen available for hydrogen bonding. The phenol analogue (1b) was the most potent analogue identified from this series. Removal of the R4methyl ether (1n) resulted in significant losses of potency and efficacy and hence the methyl ether was maintained in all subsequent analogues.
Substitution on the eastern fragment was next explored (Table 2). There was little tolerance for change on the eastern most ring. The lead compound substituted the R6 site with NHAc. When this site was instead substituted with a OMe (1o), CN (1r) or H (1p) there were losses in potency and efficacy. When NHAc was moved to the R5 position (1q) there were also sharp drops in potency and efficacy. Among the tested analogues, the R6 as NHAc motif of the original hit could not be altered without detrimental effects on the activity. Y was also explored in this series. All of the previous analogues maintained Y as oxygen, but when the oxygen was replaced with a sulfur group there was up to a 5-fold increase in potency (8bvs.1b, 8avs.1g). In fact, 8b was the most potent compound evaluated from our library and was also found to maintain selectivity for TSHR over LHR and FSHR.
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R2 | R5 | R6 | Y | EC50 (μM) | Emax (%) |
| a Activities (EC50) in μM were obtained from the Elisa assay. Emax is expressed as % of the maximal response of 1b, set at 100%.30 n.d.= not determined. | ||||||
| 1b | OH | H | NHAc | O | 0.09 | 100 |
| 1g | H | H | NHAc | O | 0.29 | 99.8 |
| 1o | H | H | OMe | O | 2.16 | 46.3 |
| 1p | H | H | H | O | 2.57 | 25.6 |
| 1q | H | NHAc | H | O | 4.34 | 24.8 |
| 1r | H | H | CN | O | n.d. | 11.7 |
| 1s | H | F | H | O | n.d. | 7.4 |
| 8a | H | H | NHAc | S | 0.08 | 95.7 |
| 8b | OH | H | NHAc | S | 0.018 | 69.6 |
In solution, dihydroquinazolin-4-one analogues were slowly oxidized by air to the corresponding quinazoline-4-ones. The dihydroquinazolin-4-ones suffer from poor stability in acidic aqueous conditions due to hydrolysis. For example, compound 1b degraded at pH = 2 with a half-life of 3 h.24 Quinazoline-4-ones 9a–c (Table 3) were prepared to preemptively address these issues that could limit the application of these analogues. Although there was a modest decrease in potency of all the oxidized analogues, these compounds still maintained potency in the sub-μM range and had the advantage of improved stability over dihydroquinazolin-4-ones (Table 3). The selectivity of 9c was also tested and it was found to be selective for TSHR over LHR and FSHR.
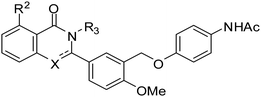
In conclusion, we have successfully synthesized a library of analogues for SAR study. Among them, 1b, 8a, and 8b were at least 10-fold more potent than the hit 1a. In addition, we identified several quinazolin-4-one TSHR agonists (9a–c) with improved stability and sub-μM potency.
Acknowledgements
This work was supported by the Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases and the Molecular Libraries Initiative of the Roadmap for Medical Research, National Institutes of Health. We thank Chris LeClair, Jim Bougie, Danielle VanLeer and Thomas Daniel for compound management and analytical support.References and notes
- Y. Nagayama, D. Russo, H. L. Wadsworth, G. D. Chazenbalk and B. Rapoport, J. Biol. Chem., 1991, 266, 14926–14930 Search PubMed.
- H. Loosfelt, C. Pichon, A. Jolivet, M. Misrahi, B. Caillou, M. Jamous, B. Vannier and E. Milgrom, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3765–3769 Search PubMed.
- G. Vassart, L. Pardo and S. Costagliola, Trends Biochem. Sci., 2004, 29, 119–126 Search PubMed.
- G. Kleinau and G. Krause, Endocr. Rev., 2009, 30, 133 Search PubMed.
- M. Tsampalas, V. Gridelet, S. Berndt, J.-M. Foidart, V. Geenena and S. P. d'Hauterivea, J. Reprod. Immunol., 2010, 85, 93–98 Search PubMed.
- Q. R. Fan and W. A. Hendrickson, Nature, 2005, 433, 269–277 CrossRef CAS.
- T. F. Davies, X. Yin and R. Latif, Thyroid, 2010, 20, 727–736 Search PubMed.
- I. Pastan, J. Roth and V. Macchia, Proc. Natl. Acad. Sci. U. S. A., 1966, 56, 1802–1809 Search PubMed.
- E. Abe, R. C. Marians, W. Yu, X. B. Wu, T. Ando, Y. Li, J. Iqbal, L. Eldeiry, G. Rajendren, H. C. Blair, T. F. Davies and M. Zaidi, Cell, 2003, 115, 151 Search PubMed.
- O. Labudova, N. Cairns, T. Koeck, E. Kitzmuellera, H. Rink and G. Lube, Life Sci., 1999, 64, 1037–1044 Search PubMed.
- C. M. Dutton, W. Joba, C. Spitzweg, A. E. Heufelder and R. S. Bahn, Thyroid, 1997, 7, 879–884 Search PubMed.
- R. Goto-Kazeto, Y. Kazeto and John M. Trant, Gen. Comp. Endocrinol., 2009, 161, 313–319 Search PubMed.
- L. Aghajanova, A. Stavreus-Evers, M. Lindeberg, B. M. Landgren, L. S. Sparre and O. Hovatta, Fertil. Steril., 2011, 95, 230–237 Search PubMed.
- E. U. Bagriacik and J. R. Klein, J. Immunol., 2000, 164, 6158 Search PubMed.
- M. W. Szkudlinski, V. Fremont, C. Ronin and B. D. Weintraub, Physiol. Rev., 2002, 82, 473 Search PubMed.
- J. K. Fernandes, T. A. Day, M. S. Richardson and A. K. Sharma, Curr. Treat. Options Oncol., 2005, 6, 47–57 Search PubMed.
- J. Sanders, M. Evans, L. D. K. E. Premawardhana, H. Depraetere, J. Jeffreys, T. Richards, J. Furmaniak and B. R. Smith, Lancet, 2003, 362, 126–128 Search PubMed.
- J. Sanders, R. N. Miguel, J. Furmaniak and B. R. Smith, Methods Enzymol., 2010, 485, 393–420 Search PubMed.
- C. R. Chen, S. M. McLachlan and B. Rapoport, Endocrinology, 2008, 149, 3427–3434 Search PubMed.
- N. C. R.v. Straten, G. G. Schoonus-Gerritsma, R. G.v. Someren, J. Draaijer, A. E. P. Adang, C. M. Timmers, R. G. J. M. Hanssen and C. A. A.v. Boeckel, ChemBioChem, 2002, 3, 1023–1026 CrossRef CAS.
- H. Jaschke, S. Neumann, S. Moore, C. J. Thomas, A.-O. Colson, S. Costanzi, G. Kleinau, J.-K. Jiang, R. Paschke, B. M. Raaka, G. Krause and M. C. Gershengorn, J. Biol. Chem., 2006, 281, 9841–9844 Search PubMed.
- S. Moore, H. Jaeschke, G. Kleinau, S. Neumann, S. Costanzi, J. K. Jiang, J. Childress, M. R. B, A. Colson, R. Paschke, G. Krause, J. C. Thomas and M. C. Gershengorn, J. Med. Chem., 2006, 49, 3888–3896 Search PubMed.
- S. Titus, S. Neumann, W. Zheng, N. Southall, S. Michael, C. Klumpp, A. Yasgar, P. Shinn, C. J. Thomas, J. Inglese, M. C. Gershengorn and C. P. Austin, J. Biomol. Screening, 2008, 13, 120–127 Search PubMed.
- S. Neumann, W. Huang, S. Titus, G. Krause, G. Kleinau, A. T. Alberobello, W. Zheng, N. T. Southall, J. Inglese, C. P. Austin, F. S. Celi, O. Gavrilova, C. J. Thomas, B. M. Raaka and M. C. Gershengorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12471–12476 Search PubMed.
- F. V. Costa, C. Borghi, S. Boschi, A. Mussi and E. Ambrosioni, J. Clin. Pharmacol., 1990, 30, 254–261 Search PubMed.
- A. Kiyingi, M. J. Field, C. C. Pawsey, J. Yiannikas, J. R. Lawrence and W. J. Arter, Lancet, 1990, 335, 29–31 Search PubMed.
- General procedure for the synthesis of 1,2-dihydroquinazolin-4-ones 1 and 8. A solution of 7 (400 μmol) in EtOH (6 mL) was treated with benzamide 3 (440 μmol) and ytterbium trifluoromethanesulfonate (200 μmol) and heated to 80 °C for 2–6 h. The reaction mixture was concentrated under reduced pressure and purified viacolumn chromatography on Silica-gel. The isolated product was triturated with Et2O to afford the desired product. Example: 1e1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.98 (s, 3 H), 3.79 (s, 3 H), 3.80 (d, J = 15.6 Hz, 1H), 4.93 (s, 2 H), 5.20 (d, J = 15.3 Hz, 1 H), 5.67 (br.s., 1 H), 6.31–6.51 (m, 2 H), 6.85 (d, J = 8.6 Hz, 2 H), 6.99 (d, J = 8.4 Hz, 1 H), 7.08–7.35 (m, 7 H), 7.38 (s, 1 H), 7.45 (d, J = 8.6 Hz, 2 H), 7.65 (br. s., 1 H), 9.75 (s, 1 H); HPLC: tR = 5.58 min, UV254 = 96%; HRMS (ESI): m/z calcd for C31H28FN3O4 [M + Na]+ 548.1956, found 548.1967.
- Procedure for the synthesis of quinazolin-4-ones 9a and 9b. To a solution of 1,2-dihydroquinazolin-4-ones 1 (200 μmol) in EtOH (5 mL) was added DDQ (240 μmol in 4 mL of acetonitrile). After stirring at r.t. for 4 h, the mixture was concentrated under reduced pressure and purified viacolumn chromatography on silica gel to give the desired products. 9a: 1H NMR (400 MHz, CHLOROFORM-d) δ 2.15 (s, 3 H), 3.95 (s, 3 H), 5.12 (s, 2 H), 5.19 (s, 2 H), 6.12 (d, J = 2.7 Hz, 1 H), 6.22–6.28 (m, 1 H), 6.93 (d, J = 9.0 Hz, 2 H), 7.00 (d, J = 8.6 Hz, 1 H), 7.12 (br. s., 1 H), 7.24 (s, 1 H), 7.39 (d, J = 9.0 Hz, 2 H), 7.46–7.56 (m, 2 H), 7.64–7.82 (m, 2 H), 8.33 (d, J = 8.2 Hz, 1 H); HPLC: tR = 5.49 min, UV254 = 95%; HRMS (ESI): m/z calcd for C29H25N3O5 [M + 1]+ 496.1872, found 496.1873. 9b: 1H-NMR (400 MHz, CHLOROFORM-d) δ 2.13 (s, 3 H), 3.91 (s, 3 H), 5.04 (s, 2 H), 5.26 (s, 2 H), 6.81–7.04 (m, 5 H), 7.11–7.26 (m, 5 H), 7.38 (d, J = 9.0 Hz, 2 H), 7.46–7.58 (m, 2 H), 7.71–7.83 (m, 2 H), 8.36 (d, J = 7.8 Hz, 1 H); HPLC: tR = 5.73 min, UV254 = 98%; HRMS (ESI): m/z calcd for C31H27N3O4 [M + 1]+ 506.2086, found 506.2082.
- Procedure for the synthesis of quinazolin-4-one 9c. To a solution of 1b (15.0 mg, 29 μmol) in 2 mL of DMSO solution was added MnO2 (87 mg, 0.29 mmol). The mixture was heated at 80 °C for 12 h. The solid was filtered and the solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel (7%–60% ethyl acetate in hexanes) to yield 9c (8.1 mg, 54%). 1H NMR (400 MHz, CHLOROFORM-d) δ (ppm) 2.14 (s, 3 H), 3.90 (s, 3 H), 5.04 (s, 2 H), 5.20 (s, 2 H), 6.79–7.01 (m, 6 H), 7.08 (br.s., 1H), 7.20–7.25 (m, 4 H), 7.27–7.42 (m, 3 H), 7.52 (d, J = 2.0 Hz, 1 H), 7.64 (t, J = 8.1 Hz, 1 H), 11.69 (s, 1 H); HPLC: tR = 6.23 min, UV254 = 98%; HRMS (ESI): m/z calcd for C31H27N3O5 [M + 1]+ 522.2027, found 522.2028.
- ELISA cAMP assay: Transiently transfected HEK-EM293 cells or cells stably expressing TSHR, were seeded into 96-well plates at a density of 50,000 cells/well in DMEM containing 10% FBS. Cells were cultured for 24 h before incubation for 1 h in serum-free DMEM containing 1 mM 3-isobutyl-1-methylxanthine (Sigma) and TSH, or small-molecule ligands in a humidified 5% CO2 incubator at 37 °C. Following aspiration of the media, cells were lysed using lysis buffer of the cAMP-Screen Direct system (Applied Biosystems). The cAMP contents of the cell lysates were determined using the method described in the manufacturer's protocol.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00145k |
| This journal is © The Royal Society of Chemistry 2011 |