Synthesis and biological evaluation of 2,3-bis(het)aryl-4-azaindole derivatives as protein kinase inhibitors†
Frédéric
Pin
a,
Frédéric
Buron
a,
Fabienne
Saab
a,
Lionel
Colliandre
a,
Stéphane
Bourg
b,
Françoise
Schoentgen
c,
Remy
Le Guevel
d,
Christiane
Guillouzo
d and
Sylvain
Routier
*a
aICOA, University of Orleans, UMR CNRS 6005, Rue de Chartres, BP 6759, 45067, Orleans Cedex 2, France. E-mail: sylvain.routier@univ-orleans.fr; Fax: +33-238-417-354; Tel: +33-238-417-354
bFédération de Recherche “Physique et Chimie du Vivant”, FR CNRS 2708, Rue de Charles Sadron, F-45071, Orleans Cedex 2, France
cIMPMC UMR 7590 CNRS, University of Pierre et Marie Curie-Paris 6, Campus Jussieu, boîte courrier 115, 4 Place Jussieu, 75252, Paris Cedex 05, France
dHôpital de Pontchaillou, INSERM U-522, 65033, Rennes Cedex, France
First published on 3rd August 2011
Abstract
The synthesis of several novel 4-azaindoles was carried out by novel Fischer reaction which offers as a main advantage, the synthesis of the bisfunctionalized 4-azaindolic building block in one step. The final compounds were evaluated on a panel of 5 kinases in order to evaluate their selectivity and on 7 cancer cell lines to determine their cytotoxic effects. RAF-1 and DYRK1A inhibitions were found, docking studies explain fully the results.
Protein kinases constitute a large family of structurally related enzymes that are responsible for the control of a wide variety of signal transduction processes within the cell. Deregulation of kinase activities is frequently observed during the development of human diseases such as cancer, diabetes, Alzheimer's disease, inflammation, etc.1 Consequently, they have been used as targets to identify pharmacological inhibitors of potential therapeutic interest. Among all the kinases, our laboratory is currently dedicated to the synthesis of several disease-relevant kinase inhibitors, and focused their efforts on cyclin-dependent kinases (CDKs), glycogen synthase kinase-3 (GSK3), casein kinase 1 (CK1), dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) and Ras Activating Factor kinase (RAF).
Enzymes were chosen for their implication in regulation processes. CDKs control the eukaryotic cell division apoptosis, numerous neuronal functions, insulin release, and transcription.2GSK3β is involved in embryonic development, protein synthesis, cell proliferation and differentiation, microtubule dynamics, cell motility and apoptosis.3CK1 isoforms are involved in the regulation of many different cellular processes such as canonical Wnt signaling, DNA damage response, and cell cycle progression.4 RAF, a serine/threonine protein kinase, is an integral part of the MAP kinase signaling pathway regulating cellular growth and proliferation.5DYRK1A encodes a member of the dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) family. It may play a significant role in a signaling pathway regulating cell proliferation and may be involved in brain development.6 To our knowledge, four derivatives are currently in clinical trials as RAF inhibitors: RAF265 (Chiron/Novartis), XL281/BMS-908662 (Exelixis/BMS), GSK2118436 (GlaxoSmithKline), and PLX4032 (Plexxikon/Roche), with many others in preclinical development.7 Roscovitine8 was among the first CDK inhibitors that entered clinical trials and interacts with several other targets (DYRK1A and CK1) and NP031112 as GSK3β inhibitor and GSK606616 as DYRK1A inhibitor were studied in clinical trials.7
Many indoles or azaindoles containing derivatives showed an interesting inhibition of various kinases such as PI3K, JAK, JAK2, etc., nevertheless, the 4-azaindole core is scarcely reported as kinase inhibitors.9 These facts prompted us to design and synthesize novel 4-azaindole derivatives as potential kinase inhibitors.
Thus we were interested in finding simple and flexible methods to easily access 2,3-bis(het)aryl-4-azaindoles I. Most of the methods used to form azaindoles have been inspired by various synthetic strategies developed for the indole ring formation. These methods include Madelung-type cyclization,10 Reissert-type procedure,11 Leimgruber–Batcho reaction,12 Lorenz-type cyclization,13palladium-catalyzed heteroannulations14 and Bartoli sequence.15
Among these classical approaches, the most well-known and versatile Fischer indole cyclization has rarely been applied to azaindole chemistry.16 Using this method, Suzenet and co-workers obtained recently a convenient access to 2,3-bisalkyles-4-azaindoles.17 However, they never tried to incorporate simultaneously two (het)aryl moieties in both C-2 and C-3 positions.
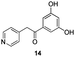
The use of the Fischer reaction required the preparation of several ketones (Scheme 1 and Table 1).18 Anions of 2, 3 or 4-methylpyridines II were obtained with LiHMDS or LDA and condensed with a family of aryl ester III (compounds 1–7) to give ketones IV (8–16). For O-tert-butyldimethylsilyl (TBS) protected derivatives, a supplementary deprotection was carried out by TBAF treatment.
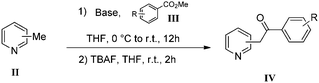 | ||
| Scheme 1 Synthesis of ketones. | ||
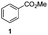
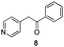
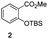
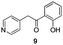
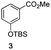
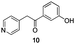
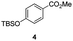
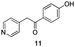
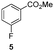
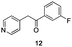
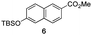
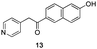
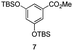
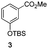
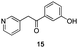
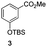
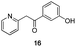
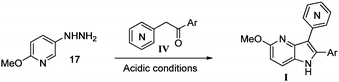
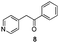
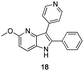
Each ketone was used in the Fischer reaction under Suzenet and co-workers' conditions. 5-Hydrazinyl-2-methoxypyridine 17, which was easily prepared from the corresponding aminopyridine by a diazotation/reduction sequence,19 was first reacted with 1-phenyl-2-(pyridin-4-yl)ethanone 8 under reflux in the presence of a 4% aqueous H2SO4 solution to give 18 in a very disappointing yield (4%).
Similar result was obtained using a 10% aqueous HCl solution (18, 6%). Instability of the hydrazine and salts formation could explain the lack of reactivity of the methylketone in strong aqueous acidic conditions. To solve this problem, we decided to use the organic PTS acid and toluene as solvent (Scheme 2).
 | ||
| Scheme 2 Fischer reaction. | ||
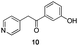
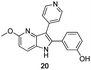
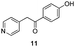
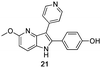
Fortunately, this new procedure allowed the formation of 18 in the encouraging 52% yield (Table 2, entry 1). We next planned to test the Fischer reaction with ketones 9 to 16. The corresponding 4-azaindoles were always formed and isolated after flash chromatography purification (Table 2) except for the azaindoles 23 and 24 (starting from ketones 13 and 14) that showed a strong instability under our conditions (entries 6 and 7).
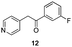
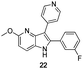
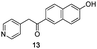
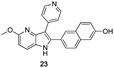
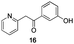
As a last reaction (Scheme 3), we cleaved the methoxy ether group in C-5 of the azaindoles 18 and 22 with in situ formed TMSI (from TMSCl and NaI).20 The two final derivatives 27 and 28 were isolated after only 2 h with respectively 52 and 63% of yields.
 | ||
| Scheme 3 Cleavage of methoxy groups. | ||
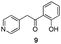
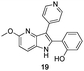
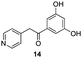
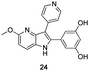
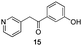
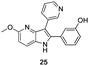
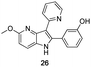
The final products were tested on 5 different in vitrokinase assays to evaluate their inhibition potency against RAF-1, DYRK1A, CDK5, CK1 and GSK3β. The 2,3-bis(het)aryl-4-azaindole derivatives have shown good inhibition of the two kinases RAF-1 and DYRK1A and no activity towards the others (CDK5, CK1 and GSK3β, Table 3). The compounds 19, 20, 21 and 25 gave the most promising results with submicromolar IC50 values against DYRK1A (entries 2–4 and 6). Only the compound 20 showed an interesting submicromolar inhibition of RAF-1 (entry 3, IC50 = 286 nM). Micromolar to submicromolar IC50 values were obtained for the 5-methoxy-4-pyridinyl derivatives 18, 19, 20, 21, and 22 towards RAF-1 and DYRK1A (entries 1–5). The comparison of the IC50 values observed for 27 (entry 8) and 20 (entry 3) shows that the deprotection of the methoxy group in C-5 leads to a strong decrease in the inhibition of the two kinases RAF-1 and DYRK1A. The same conclusion was observed with the compounds 22 and 28 (entries 5 and 9). Interestingly, a beginning of SAR emerges from these results. First, the presence of a pyridinyl group in C-3 as well as a hydroxy group on the phenyl ring in C-2 appears to be essential to have the best activities against DYRK1A and RAF-1. Second, the position of the hydroxyl group on the phenyl ring seems not to be essential for the DYRK1A activity whereas it is important for RAF activity. Third, the position of the nitrogen atom on the pyridinyl group appears to play a crucial role in the selectivity. The comparison of the derivatives 20, 25 and 26, differing only by the position of their nitrogen atom, shows that the meta position is favourable to gain a selectivity against DYRK1Aversus RAF-1 whereas the ortho position leads to the loss of the activity against both.
| Entry | Compounds | RAF-1 | DYRK1A | CDK5 | CK1 | GSK3β |
|---|---|---|---|---|---|---|
| a IC50 (μM) values are presented as means of duplicate experiments. | ||||||
| 1 | 18 | 3.2 | 1.3 | >10 | >10 | >10 |
| 2 | 19 | 2.5 | 0.23 | >10 | >10 | >10 |
| 3 | 20 | 0.286 | 0.7 | >10 | >10 | >10 |
| 4 | 21 | 1.3 | 0.47 | >10 | >10 | >10 |
| 5 | 22 | 4.3 | 1.8 | >10 | >10 | >10 |
| 6 | 25 | >50 | 0.84 | >10 | >10 | >10 |
| 7 | 26 | >50 | >10 | >10 | >10 | >10 |
| 8 | 27 | 16.5 | >10 | >10 | >10 | >10 |
| 9 | 28 | 9.2 | >10 | >10 | >10 | >10 |
In order to investigate the potential inhibition of RAF-1 by our synthesized compounds, docking studies were carried out. A high resolution crystal structure of RAF-1 in complex with (1E)-5-(1-piperidin-4-yl-3-pyridin-4-yl-1H-pyrazol-4-yl)-2,3-dihydro-1H-inden-1-one oxime (S5M) positioned in the binding site was used as the template structure (PDB code 3OMV).21
First of all, S5M was redocked to reproduce the co-crystal complex. Superimposition of the best docked pose, i.e. the top score conformer, to the crystal structure fits well between the two structures: this initial step of rigid docking gave an RMSD deviation, based on heavy atoms, of 0.47 Å.
Docked poses of the reference compound L779,450, a RAF inhibitor, and the most active compound 20 on RAF-1, are superimposed in Fig. 1. They show the same binding mode, more particularly the same positioning of common chemical groups (pyridine ring, central pyrrole ring and functionalized aromatic ring). This binding mode is in agreement with the binding mode of the crystallographic ligand S5M. Specific interactions are seen with RAF-1via common H-bond interactions between the C-3 pyridine nitrogen atom of ligand and Cys424, one of the kinase hinge residues, and in the opposite side between the meta-aryl hydroxy group and Asp486. These results validate again the choice of 2,3-bis(het)aryl-4-azaindole derivatives as RAF-1 inhibitors.
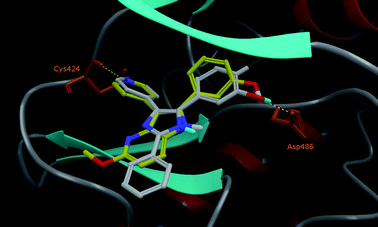 | ||
| Fig. 1 Docking poses of L779,450 (C atoms represented in light grey and polar H in cyan) and 20 (C atoms in yellow) are superimposed in the active site of RAF-1. | ||
The high resolution crystal structure (PDB code 3ANR) of DYRK1A in complex with 7-methoxy-1-methyl-9H-pyrido[3,4-b]indole (HRM)22 was chosen to study the binding mode of our active compounds. In the same way, our docking protocol was validated by the redocking of the co-crystallized ligand HRM. The best pose gave an RMSD deviation, based on heavy atoms, of 0.12 Å.
The most active compound 19 on DYRK1A has been docked with this protocol (Fig. 2). The same binding mode as for 20 in RAF-1 could not be obtained but an alternative binding mode has been found. This last one is in good agreement with the biological data and the crystallographic position of HRM. One representative pose of 19 is superimposed to the crystallographic ligand HRM in Fig. 2. This binding mode is characterized by the formation of two H-bond interactions between the methoxy group of the ligand (4-azaindole substitution) and Leu241, and between the pyridine nitrogen atom of the ligand and the lateral chain of Lys188.
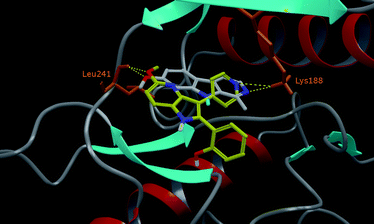 | ||
| Fig. 2 Crystallographic binding mode of HRM and docking pose of 19 (C atoms in yellow) are superimposed in the active site of DYRK1A. Only polar hydrogen atoms are represented for clarity. H bonds are represented by dashed yellow lines. | ||
These results show that the 3-pyridin-4-methoxy-azaindole moieties define a new scaffold to inhibit DYRK1A by strong interactions in the ATP active site.
The anti-proliferative effects on 6 different cancer cell lines were next investigated (Table 4). Positively, each compound exhibited no toxic effect against the fibroblasts (normal cells). Interestingly, the most potent RAF-1 inhibitor 20 gave also the best cytostatic activity on the Caco cell line (entry 3, IC50 = 600 nM) and the NCI (entry 3, IC50 = 1 μM).
| Entry | Compounds | Huh7 (liver) | Caco (colon) | MDA-MB 231 (breast) | HCT-116 (colon) | PC3 (prostate) | NCI (lung) | Fibroblast |
|---|---|---|---|---|---|---|---|---|
| a IC50 values are presented as means of duplicate experiments. | ||||||||
| 1 | 18 | 6 | 15 | 12 | 10 | 15 | 10 | >25 |
| 2 | 19 | 15 | 10 | 20 | 3 | 15 | 4 | 25 |
| 3 | 20 | 8 | 0.6 | 8 | 3 | 10 | 1 | >25 |
| 4 | 21 | 12 | 10 | 8 | 7 | 10 | 15 | 20 |
| 5 | 22 | 5 | >25 | 12 | 6 | 3 | 20 | >25 |
| 6 | 25 | 15 | >25 | >25 | >25 | 20 | >25 | >25 |
| 7 | 26 | 15 | 15 | 10 | 10 | >25 | 20 | 25 |
| 8 | 27 | >25 | >25 | 25 | 25 | >25 | >25 | >25 |
| 9 | 28 | >25 | 20 | >25 | 20 | >25 | >25 | >25 |
Conclusion
In conclusion, a new class of 2,3-bis(het)aryl-4-azaindoles has been prepared by a Fischer type reaction. Our new library led to two promising derivatives which exhibited an IC50 of 286 nM on RAF-1 and IC50 of 230 nM on DYRK1A. The compound 20 presents an inhibition of 600 nM on the Caco cancer cell line without any effect on normal cells. Several modifications are in progress in order to increase the efficiency of such 4-azaindolic drugs as DYRK1A inhibitors.Acknowledgements
We thank the Ligue contre le Cancer (comités du Loiret, Ile-et-Villaine et Loire Atlantique), the Cancéropôle Grand Ouest, the Region Centre (CrikMapAkt program) for their financial support and the Institut National du Cancer (PhD fellowship for F. Saab). Additionally, we thank Dr F. Suzenet for helpful discussions and Dr L. Meijer for CDK, DYRK1A, CK1 and GSK3 kinase evaluations.Notes and references
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309 CrossRef CAS; P. M. Fischer, Curr. Med. Chem., 2004, 11, 1563 Search PubMed; H. Weinmann and R. Metternich, ChemBioChem, 2005, 6, 455 CrossRef.
- M. Malumbres and M. Barbacid, Trends Biochem. Sci., 2005, 30, 630 CrossRef CAS; A. Camins, E. Verdaguer, J. Folch, A. M. Canudas and M. Pallas, Drug News Perspect., 2006, 19, 453 Search PubMed; T. K. Pareek, J. Keller, S. Kesavapany, H. C. Pant, M. J. Iadarola, R. O. Brady and A. B. Kulkarni, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 791 CrossRef; A. Borgne, I. Versteege, M. Mahé, A. Studeny, S. Léonce, I. Naime, M. Rodriguez, J. A. Hickman, L. Meijer and R. M. Golsteyn, Oncogene, 2006, 25, 7361 CrossRef; J. Garriga and X. Grana, Gene, 2004, 337, 15 CrossRef; P. Loyer, J. H. Trembley, R. Katona, V. J. Kidd and J. M. Lahti, Cell. Signalling, 2005, 17, 1033 CrossRef; F. Y. Wei, K. Nagashima, T. Ohshima, Y. Saheki, Y. F. Lu, M. Matsushita, Y. Yamada, K. Mikoshiba, Y. Seino, H. Matsui and K. Tomizawa, Nat. Med., 2005, 11, 1104 CrossRef.
- K. Ishiguro, A. Shiratsuchi, S. Sato, A. Omori, M. Arioka, S. Kobayashi, T. Uchida and K. Imahori, FEBS Lett., 1993, 325, 167 CrossRef CAS; S. Lovestone, C. H. Reynolds, D. Latimer, D. R. Davis, B. H. Anderton, J. M. Gallo, D. Hanger, S. Mulot and B. Marquardt, Curr. Biol., 1994, 4, 1077 CrossRef; P. S. Klein and D. A. Melton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8455 CrossRef; M. Pap and G. M. Cooper, J. Biol. Chem., 1998, 273, 19929 CrossRef; G. I. Welsh and C. G. Proud, Biochem. J., 1993, 294, 625 Search PubMed; A. S. Wagman, K. W. Johnson and D. E. Bussiere, Curr. Pharm. Des., 2004, 10, 1105 CrossRef; X. Li, F. Lu, Q. Tian, Y. Yang, Q. Wang and J. Z. Wang, J. Neural Transm., 2006, 113, 93–102 CrossRef; J. T. Neary and Y. Kang, J. Neurosci. Res., 2006, 84, 515 CrossRef; A. Martinez, A. Castro, I. Dorronsoro and M. Alonso, Med. Res. Rev., 2002, 22, 373 CrossRef.
- S. D. Gross and R. A. Anderson, Cell. Signalling, 1998, 10, 699 CrossRef CAS; U. Knippschild, A. Gocht, S. Wolff, N. Huber, J. Lohler and M. Stoter, Cell. Signalling, 2005, 17, 675 CrossRef.
- C. Peyssonnaux and A. Eychene, Biol. Cell, 2001, 93, 53 CrossRef CAS; H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. C. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949 CrossRef; P. M. Pollock, U. L. Harper, K. S. Hansen, L. M. Yudt, M. Stark, C. M. Robbins, T. Y. Moses, G. Hostetter, U. Wagner, J. Kakereka, G. Salem, T. Pohida, P. Heenean, P. Duray, O. Kallioniemi, N. K. Hayward, J. M. Trent and P. S. Meltzer, Nat. Genet., 2003, 33, 19 CrossRef.
- K. H. Baek, A. Zaslavsky, R. C. Lynch, C. Britt, Y. Okada, R. J. Siarey, M. W. Lensch, I. H. Park, S. S. Yoon, T. Minami, J. R. Korenberg, J. Folkman, G. Q. Daley, W. C. Aird, Z. Galdzicki and S. Ryeom, Nature, 2009, 459, 1126 CrossRef CAS.
- ClinicalTrials.gov web site, http://www.clinicaltrials.gov/ CrossRef CAS; G. L. Schwartz, S. Robertson, A. Shen, E. Wang, L. Pace, H. Dials, D. Mendelson, P. Shannon and M. J. Gordon, J. Clin. Oncol., 2009, 27, 3513 CrossRef CAS; R. Kefford, H. Arkenau, M. P. Brown, M. Millward, J. R. Infante, G. V. Long, D. Ouellet, M. Curtis, P. F. Lebowitz and G. S. Falchook, J. Clin. Oncol., 2010, 28, 8503 Search PubMed; K. T. Flaherty, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. J. O'Dwyer, R. J. Lee, J. F. Grippo, K. Nolop and P. B. Chapman, N. Engl. J. Med., 2010, 363, 809 CrossRef; J. Tang, T. Hamajima, M. Nakano, H. Sato, S. H. Dickerson and K. E. Lackey, Bioorg. Med. Chem. Lett., 2008, 18, 4610 CrossRef; A. L. Smith, F. F. DeMorin, M. A. Paras, Q. Huang, J. K. Petkus, E. M. Doherty, T. Nixey, J. L. Kim, D. A. Whittington, L. F. Epstein, M. R. Lee, M. J. Rose, C. Babij, M. Fernando, K. Hess, Q. Le, P. Beltran and J. Carnahan, J. Med. Chem., 2009, 52, 6289 Search PubMed.
- L. Meijer and E. Raymond, Acc. Chem. Res., 2003, 36, 417 CrossRef CAS; L. Meijer, K. Bettayeb and H. Galons, (R)-Roscovitine (CYC202, Seliciclib), in Inhibitors of Cyclin-Dependent Kinases as Anti-tumor Agents, Enzyme Inhibitors Series, ed. P. J. Smith and E. W. Yue, CRC Press, Boca Raton, FL, 2006, vol. 3, ch. 9, pp. 187–226 Search PubMed.
- Q. Ye, G. Xu, D. Lv, Z. Cheng, J. Li and Y. Hu, Bioorg. Med. Chem., 2009, 17, 4302 CrossRef CAS; J. Porter, S. Lumb, R. J. Franklin, J. M. Gascon-Simorte, M. Calmiano, K. L. Riche, B. Lallemand, J. Keyaerts, H. Edwards, A. Maloney, J. Delgado, L. King, A. Foley, F. Lecomte, J. Reuberson, C. Meier and M. Batchelor, Bioorg. Med. Chem. Lett., 2009, 19, 2780 CrossRef; H. Koolman, T. Heinrich, H. Böttcher, W. Rautenberg and M. Reggelin, Bioorg. Med. Chem. Lett., 2009, 19, 1879 CrossRef; A. Trejo, H. Arzeno, M. Browner, S. Chanda, S. Cheng, D. D. Comer, S. A. Dalrymple, P. Dunten, J. Lafargue, B. Lovejoy, J. Freire-Moar, J. Lim, J. Mcintosh, J. Miller, E. Papp, D. Reuter, R. Roberts, F. Sanpablo, J. Saunders, K. Song, A. Villasenor, S. D. Warren, M. Welch, P. Weller, P. E. Whiteley, L. Zeng and D. M. Goldstein, J. Med. Chem., 2003, 46, 4702 CrossRef.
- D. Hands, B. Bishop, M. Cameron, J. S. Edwards, I. F. Conttrell and S. H. B. A. Wright, Synthesis, 1996, 877 CrossRef CAS.
- L. N. Yakhontov, V. A. Azimov and E. I. Lapan, Tetrahedron Lett., 1969, 1909 CrossRef CAS; M. H. Fischer and A. R. Matzuk, J. Heterocycl. Chem., 1969, 6, 775 CrossRef; R. H. Dodd, X. Doisy and P. Potier, Heterocycles, 1989, 28, 1101 CrossRef; N. R. Curtis, J. J. Kulagowski, P. D. Leeson, M. P. Ridgill, F. Emms, S. B. Freedman and S. Patel, Bioorg. Med. Chem. Lett., 1999, 9, 585 CrossRef; R. H. Dodd, C. Ouannès, M. Robert-Gréco and P. Potier, J. Med. Chem., 1989, 32, 1272 CrossRef.
- A. R. Battersby, E. McDonald, H. K. W. Wurziger and K. J. James, J. Chem. Soc., Chem. Commun., 1975, 493 RSC; I. Mahadevan and M. Rasmussen, J. Heterocycl. Chem., 1992, 29, 359 CrossRef CAS.
- I. Mahadevan and M. Rasmussen, J. Heterocycl. Chem., 1992, 29, 359 CrossRef CAS; R. R. Lorenz, B. F. Tullar, C. F. Koelsch and S. Archer, J. Org. Chem., 2000, 65, 1045 Search PubMed.
- G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS; M. McLaughlin, M. Palucki and I. W. Davies, Org. Lett., 2006, 8, 3307 CrossRef; N. R. Curtis, J. J. Kulagowski, P. D. Leeson, M. P. Ridgill, F. Emms, S. B. Freedman and S. Patel, Bioorg. Med. Chem. Lett., 1999, 9, 585 CrossRef; S. S. Park, J. K. Choi, E. K. Yum and D. C. Ha, Tetrahedron Lett., 1998, 39, 627 CrossRef; L. Xu, I. R. Lewis, S. K. Davidsen and J. B. Summers, Tetrahedron Lett., 1998, 39, 5159 CrossRef; F. Ujjainwalla and D. Warner, Tetrahedron Lett., 1998, 39, 5355 CrossRef; Y. Q. Fang, J. Yuen and M. Lautens, J. Org. Chem., 2007, 72, 5152 CrossRef.
- Z. Zhang, Z. Yang, N. A. Meanwell, J. F. Kadow and T. Wang, J. Org. Chem., 2002, 67, 2345 CrossRef CAS.
- For reviews, see: G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS; D. L. Hughes, Org. Prep. Proced. Int., 1993, 25, 607 CrossRef; B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef.
- M. Jeanty, J. Blu, F. Suzenet and G. Guillaumet, Org. Lett., 2009, 11, 5142 CrossRef CAS.
- M. Fujita, T. Hirayama and N. Ikeda, Bioorg. Med. Chem. Lett., 2002, 10, 3113 CrossRef CAS.
- J. Regan, A. Capolino, P. F. Cirillo, T. Gilmore, A. G. Graham, E. Hickey, R. Kroe, J. Madwed, M. Moriak, R. Nelson, C. A. Pargellis, A. Swinamer, C. Torcellini, M. Tsang and N. Moss, J. Med. Chem., 2003, 46, 4676 CrossRef CAS.
- K. Gesenberg, P. P. Deshpande, A. Pullockaran, X. Feng, W. Dedong, G. Qi, A. C. Pathiran, J. Castoro, N. Soundararajan and A. Staab, Tetrahedron Lett., 2007, 48, 2675 CrossRef CAS.
- G. Hatzivassiliou, K. Song, I. Yen, B. J. Brandhuber, D. J. Anderson, R. Alvarado, M. J. Ludlam, D. Stokoe, S. L. Gloor, G. Vigers, T. Morales, I. Aliagas, B. Liu, S. Sideris, K. P. Hoeflich, B. S. Jaiswal, S. Seshagiri, H. Koeppen, M. Belvin, L. S. Friedman and S. Malek, Nature, 2010, 464, 431–435 CrossRef CAS.
- Y. Ogawa, Y. Nonaka, T. Goto, E. Ohnishi, T. Hiramatsu, I. Kii, M. Yoshida, T. Ikura, H. Onogi, H. Shibuya, T. Hosoya, N. Ito and M. Hagiwara, Nat. Commun., 2010, 1, 1–9 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00141h |
| This journal is © The Royal Society of Chemistry 2011 |