Peptidomimetic nitriles as selective inhibitors for the malarial cysteine protease falcipain-2†
Veronika
Ehmke
a,
Falco
Kilchmann
a,
Cornelia
Heindl
b,
Kunqiang
Cui
c,
Jin
Huang
c,
Tanja
Schirmeister
b and
François
Diederich
*a
aLaboratory of Organic Chemistry, ETH Zürich, Wolfgang-Pauli-Strasse 10, 8093, Zürich, Switzerland. E-mail: diederich@org.chem.ethz.ch; Fax: (+41)44-632-1109
bInstitute of Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074, Würzburg, Germany
cSchool of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China
First published on 5th July 2011
Abstract
Proteases of protozoan parasites have emerged as promising targets in drug design and discovery due to their indispensable roles in the life cycles of the parasites. For the development of new therapeutic agents against the malarial parasite Plasmodium falciparum, attention has turned to a family of cysteine proteases taking part in the degradation of human haemoglobin. The falcipains have key functions in this metabolic process, making them attractive targets for the development of novel antimalarials. To inhibit the cathepsin L-like cysteine protease falcipain-2, we designed peptidomimetic nitriles through rational structure-based molecular modelling focusing on the optimal occupancy of the selectivity-determining subpockets. A series of compounds was efficiently prepared and their biological activity assessed to explore the binding site properties of the target enzyme. Inhibitory affinities down to the single-digit micromolar range were obtained for this first generation of covalent, reversible cysteine protease inhibitors. High selectivity against human cathepsin B and L, as well as against the serine protease α-chymotrypsin was observed for the majority of the synthesised ligands. The ideal occupation of the selectivity-determining S2 pocket and the balanced electrophilicity of the nitrile group seem to be crucial to achieve both potency and selectivity.
Protozoan parasites are an extremely diverse group of organisms that cause many of the most devastating infectious diseases leading to profound health problems in the world.1 Despite the advances in combating these infectious diseases, they continue to cause tremendous social and economic burden, particularly in tropical and subtropical regions.2
Identification of protease-mediated processes in parasitic protozoa revealed that proteolytic enzymes play key roles in the life cycles of the parasites.3 Proteases from all classes are responsible for diverse vital functions of the pathogens. Among the protease family, cysteine proteases are a distinct class that takes advantage of the high nucleophilicity of an active site thiolate to achieve amide bond cleavage. Enzymes of the papain subfamily of cysteine proteases are present in many eukaryotic organisms, including protozoans, performing essential tasks in metabolism. During peptide hydrolysis, the catalytic thiolate attacks the electrophilic carbon of the scissile bond of the substrate to form a tetrahedral intermediate. Therefore, “classical” cysteine protease inhibitors are characterised by the presence of a peptide bond isostere prone to covalently bind to the active site.4 Design and development of cysteine protease inhibitors were mostly driven by the detailed characterisation of papain-like cysteine proteases as pharmaceutically valid targets.5 Only few inhibitors, however, have advanced to clinical trials, which might be partly due to the lack of adequate electrophilic isosteres of the amide bond for selective covalent inhibition. Functional groups capable of irreversible interaction with the active site cysteine have been successfully incorporated into various compounds, for example diazomethanes, halomethyl ketones, acyloxymethyl ketones, Michael systems and epoxides.4 However, their progress and application as therapeutic agents are limited due to observed cross-reactivity with other nucleophiles present in the cell. In contrast to irreversible ligands, only few reversible binding inhibitors have been used in the development of new inhibitors. So far, the best-studied classes of reversible cysteine protease inhibitors, used as transition-state analogues, comprise mainly peptidic aldehydes and methyl ketones, in addition to a few nitriles.6,7 Due to similar proteolytic mechanisms and substrate affinities of cysteine and serine proteases, covalently binding compounds are generally prone to interact with both. Apparently, nucleophilic attack is influenced by the electrophilicity of the reactive head group, modulating the reversibility of the substrate–ligand complex formation.8Aldehydes are highly electrophilic and therefore susceptible to off-target effects, whereas nitriles are notably less reactive towards free nucleophiles. In a recent approach, we focused accordingly on aryl nitriles to obtain potent and selective cysteine protease inhibitors.9Nitriles inhibit these enzymes by the formation of a covalent, reversible thioimidate adduct resulting from the nucleophilic attack of the active site cysteine.10 Numerous nitrile-containing pharmaceuticals are in use today and additional candidates entered clinical development, highlighting the relevance of the nitrile pharmacophore in medicinal chemistry.11 The frequent occurrence of this functional group underlines its metabolic resistance and biocompatibility.
The protozoan parasite Plasmodium (P.) falciparum is the clinically most relevant pathogen causing a severe and often fatal form of malaria in humans.12 The emergence of drug-resistant P. falciparum strains and limitations in current chemotherapies demand the development of new therapeutic agents with novel modes of action. Based on the first X-ray crystal structures of the essential cathepsin L-like cysteine protease falcipain-2 from the malarial parasite, available since 2006 (PDB codes: 1YVB, 2GHU, 3BPF),13–15 we became interested in the design, synthesis and biological evaluation of new selective inhibitors. Falcipain-2 is one of the key haemoglobinases of the erythrocytic parasite, playing an essential role in the degradation of human haemoglobin to provide nutrients for the pathogen.5 In order to obtain a new class of falcipain-2 inhibitors, we applied a structure-based molecular design approach that relies on chemical structure intuition and molecular recognition principles, without the need for screening.16 In the present work, we sought to explore the binding site properties and optimal occupancy of the mainly apolar pockets of falcipain-2, in order to obtain potent and particularly selective inhibitors.
The active site of falcipain-2 is relatively shallow and solvent-exposed, typically accommodating short peptidic substrate fragments. Therefore, our design (Fig. 1) focused on peptidomimetic ligands bearing a nitrile group as head group. For peptidic ligands in biological systems, a motif consisting of three amino acids was generally shown to fulfil the optimal length requirement for effective substrate–enzyme interactions.17 Accordingly, 3D modelling using the computer program MOLOC18 revealed a peptidomimetic compound with 1-aminocyclohexanecarboxylic acid as the two amide bond-forming central core as a suitable motif for addressing the active site of falcipain-2. The cyclohexyl residue should provide structural preorganisation to appropriately orientate the vectors towards the S2 subpocket and to occupy the upper part of the shallow S3 pocket. The electrophilic centre is incorporated as “C-terminal” aminoacetonitrile, allowing sufficient conformational freedom for the thiolate attack. According to calculated electrophilicities, aminoacetonitriles should possess a moderately activated nitrile without being too reactive to undergo side-reactions.8 The formed thioimidate intermediate is stabilised through a hydrogen bond with the NH2 of Gln36 (d(N–HGln36⋯N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) = 3.1 Å) which is part of an oxyanion hole. Since the mainly apolar S2 pocket is known to be the key determinant of substrate specificity,13 we particularly focused on its optimal occupancy to gain insight into the molecular recognition properties of this cavity and to establish a structure–activity relationship. An ethylene linker was primarily chosen to direct the preferentially hydrophobic vectors into this pocket lined by Ile85, Phe236, Asp234 and Ser149. The design is in agreement with two co-crystal structures of falcipain-2 (PDB codes: 1YVB with cystatin, 3BPF with protease inhibitor E64 = trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane). Both substrates found in the X-ray crystal structures bear lipophilic isoleucine vectors occupying the S2 pocket. In lead compound 1a, a pyridin-2-yl moiety was chosen to address the S2 pocket with the nitrogen orientated towards the backbone N–H of Ile85. The peptidomimetic ligand is additionally anchored in the active site through hydrogen bonds with the backbone N–H (d(N–HGly83⋯OC) = 3.0 Å) and CO of Gly83 (d(COGly83⋯H–N) = 2.8 Å) resembling the hydrogen bonding pattern of E64 in the co-crystal structure (Fig. 1 ESI†).
C) = 3.1 Å) which is part of an oxyanion hole. Since the mainly apolar S2 pocket is known to be the key determinant of substrate specificity,13 we particularly focused on its optimal occupancy to gain insight into the molecular recognition properties of this cavity and to establish a structure–activity relationship. An ethylene linker was primarily chosen to direct the preferentially hydrophobic vectors into this pocket lined by Ile85, Phe236, Asp234 and Ser149. The design is in agreement with two co-crystal structures of falcipain-2 (PDB codes: 1YVB with cystatin, 3BPF with protease inhibitor E64 = trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane). Both substrates found in the X-ray crystal structures bear lipophilic isoleucine vectors occupying the S2 pocket. In lead compound 1a, a pyridin-2-yl moiety was chosen to address the S2 pocket with the nitrogen orientated towards the backbone N–H of Ile85. The peptidomimetic ligand is additionally anchored in the active site through hydrogen bonds with the backbone N–H (d(N–HGly83⋯OC) = 3.0 Å) and CO of Gly83 (d(COGly83⋯H–N) = 2.8 Å) resembling the hydrogen bonding pattern of E64 in the co-crystal structure (Fig. 1 ESI†).
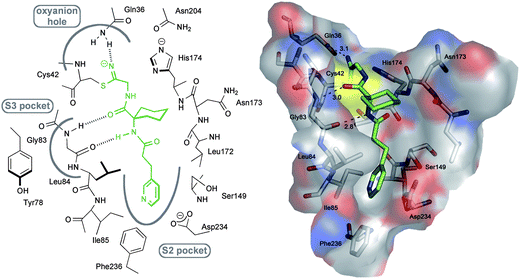 | ||
| Fig. 1 Left: Schematic representation of the active site of falcipain-2 and the positioning of 1a. Right: Proposed binding mode of 1a in falcipain-2 (PDB code: 2GHU)13 as generated with MOLOC showing the occupation of the different pockets. Color code: Cenzyme gray, Cligand green, O red, N blue and S yellow. Hydrogen bond distances between heavy atoms are shown as dotted lines and given in Å. | ||
A series of peptidomimetic aminoacetonitriles was prepared as depicted in Scheme 1. 1-Aminocyclohexanecarboxylic acid (2) was converted to the corresponding methyl ester 3 by treatment with thionylchloride in methanol. Subsequently, ester 3 was coupled with various carboxylic acids using a one-pot peptide coupling procedure with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), 1-hydroxy-1H-benzotriazole (HOBt) and N-ethyldiisopropylamine in DMF. Amides 4a–i were obtained in good to excellent yields. Subsequent saponification using LiOH·H2O gave carboxylic acids 5a–i, which were transformed into the targeted ligands 1a–i in a second amide coupling reaction with aminoacetonitrile bisulfate using the aforementioned conditions. The syntheses of ligands 6–9 (for the structures, see Table 1) followed slightly different routes which are described in detail in the ESI (Schemes 1–4 ESI)†.
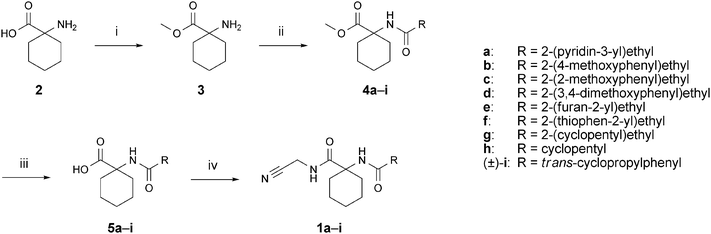 | ||
Scheme 1
Reagents and conditions: (i) SOCl2, MeOH, 0–25 °C, 60 h, 94%; (ii) RCO2H, TBTU, HOBt, iPr2NEt, DMF, 0–25 °C, 15–18 h, 65–95%; (iii) LiOH·H2O, THF/MeOH/H2O 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, 40 °C, 4–6 h, directly used for the next step; (iv) aminoacetonitrile bisulfate, TBTU, HOBt, iPr2NEt, DMF, 0–25 °C, 15–18 h, 33–87%. TBTU = 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate, HOBt = 1-hydroxy-1H-benzotriazole. :2:1, 40 °C, 4–6 h, directly used for the next step; (iv) aminoacetonitrile bisulfate, TBTU, HOBt, iPr2NEt, DMF, 0–25 °C, 15–18 h, 33–87%. TBTU = 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate, HOBt = 1-hydroxy-1H-benzotriazole. | ||
| Compd | Structure | Falcipain-2 | Cathepsin B | Cathepsin L |
|---|---|---|---|---|
| a All results are the average of at least two independent measurements, each performed in duplicate. b n.d.: not determined; inhibition in an initial screen at 20 μM less than 35%. | ||||
| 1a |

|
9.4 ± 1.8 | n.d.b | n.d. |
| 1b |

|
8.9 ± 0.2 | n.d. | n.d. |
| 1c |

|
5.2 ± 0.4 | n.d. | 25.1 ± 0.08 |
| 1d |

|
7.3 ± 0.9 | n.d. | n.d. |
| 1e |
|
17.2 ± 2.1 | n.d. | n.d. |
| 1f |

|
8.8 ± 1.4 | n.d. | n.d. |
| 1g |

|
8.2 ± 0.8 | 25 ± 0.4 | 29.8 ± 0.1 |
| 1h |

|
7.6 ± 0.2 | n.d. | n.d. |
| (±)-1i |

|
2.1 ± 0.4 | 5.0 ± 0.7 | 13.1 ± 0.43 |
| 6 |

|
1.2 ± 0.2 | n.d. | 26.5 ± 1.1 |
| 7 |

|
n.d. | n.d. | n.d. |
| (S)-(−)-8 |

|
n.d. | n.d. | n.d. |
| 9 |
|
0.065 ± 0.005 | 1.35 ± 0.015 | 0.82 ± 0.05 |
All compounds were evaluated for their activity against falcipain-2 from P. falciparum in standard fluorescence-based assays (see ESI†).19,20Lead compound 1a showed a promising inhibitory constant of Ki = 9.4 ± 1.8 μM (Table 1). The in vitro activity of the majority of the tested compounds was comparable to the one of 1a. Ligands 1b–i inhibited the target enzyme with binding affinities in the low micromolar range between Ki = 17.2 ± 2.1 μM and Ki = 2.1 ± 0.4 μM for 1e and (±)-1i, respectively. Consequently, the S2 pocket seems to be tolerant towards various 5- or 6-membered aromatic systems such as pyridin-3-yl (1a), 4-methoxyphenyl (1b), 2-methoxyphenyl (1c), 3,4-dimethoxyphenyl (1d), furan-2-yl (1e) and thiophen-2-yl (1f) moieties connected with an ethylene linker to the cyclohexyl core. Moreover, saturated systems such as cyclopentyl bearing the N-terminal ethylene linker or directly connected to the central amino acid are tolerated, as observed for compounds 1g and 1h. Rigidifying the linker with a cyclopropyl moiety lead to a similar inhibitory constant for nitrile (±)-1i. Interestingly, carbamate analogue 6 also showed a Ki value in the single-digit micromolar range (Ki = 1.2 ± 0.2 μM). The carbamate motif seems to be a possible surrogate for the methyleneamido group (–NH–CO–CH2–) in compounds 1a–g. The S2 pocket is therefore accessible for different substitutents, thus validating our design concept with the cyclohexyl scaffold as a suitable core. The similarity in binding affinity for 1a–i and 6 assumedly might originate from the comparable positioning of the pharmacophores through the cyclohexyl core in the active site of falcipain-2, as shown in Fig. 2.
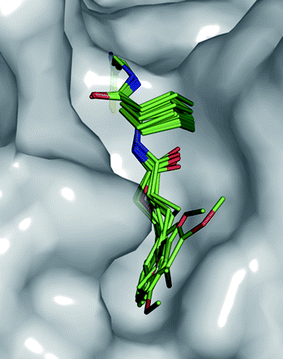 | ||
| Fig. 2 Superimposition of compounds 1a–i and 6 docked into the active site of falcipain-2 (PDB code: 2GHU)13 showing their similar occupancy of the S2 pocket. | ||
Loss of inhibition was observed for compound 7 with a 2-aminoisobutyric residue as the central amino acid. We assume that the cyclohexyl analogue 6 has a better fit between the two peptide fragments lined by Gly82–Gly83 on one side and Leu172–Asn173 on the other side of the S2 pocket than aminoisobutyric derivate 7. This goes along with a more efficient desolvation in the case of compound 6, validating the cyclohexyl moiety as a suitable central motif. Replacement of the aminoacetonitrile moiety with a more rigid (S)-2-cyanopyrrolidine substituent in compound (S)-(−)-8 resulted also in reduced activity. This decrease might be a consequence of a disadvantageous orientation of the nitrile group caused by the pyrrolidine ring, hindering an effective attack of the catalytic thiolate. Aldehyde 9 was the most active ligand in this series with a Ki value of 0.065 ± 0.005 μM, most likely because of the higher reactivity of this functionality compared to the nitrile group as aforementioned. These findings consistently validate the aminoacetonitrile moiety as an appropriate electrophilic head group for the inhibition of falcipain-2 and reveal a suitability of hydrophobic S2 substitutents connected to the cyclohexyl core. Additional compounds synthesised and evaluated in the course of this study (data not shown) further support the structure–activity relationship found for the occupancy of the S2 pocket and the aminoacetonitrile moiety.
In order to study the selectivity against related proteases, the compounds were tested for their ability to inhibit the structurally closely related human cysteine proteases cathepsin B and L and the serine protease α-chymotrypsin (Table 1). Affinities for cathepsin B were only detected for nitriles 1g and (±)-1i. Apart from them, all falcipain-2 inhibitors showed good selectivity against cathepsin B. Similar results were found for the activity against cathepsin L. Compounds 1a, 1b, 1d–f and 1h are highly selective for the target enzyme and do not affect cathepsin L. The selectivity for the majority of compounds against both cathepsins can be explained by the moderate electrophilicity of the nitrile. In agreement with this finding is the capability of aldehyde 9 to bind both cathepsin B and L. An adequate choice of the electrophile is therefore essential to reduce off-target effects. Balanced reactivity of the head group seems to be decisive to control and enhance selectivity of cysteine protease inhibitors. Additionally, close comparison of the S2 pockets of falcipain-2 and cathepsin B and L reveals slight differences in their overall shape and extension, explaining the observed selectivities (Fig. 2 ESI†). In the case of falcipain-2, the S2 vector of the selective inhibitor 1a fully occupies the narrow and well-defined pocket with close contacts to the enzyme (Ile85, Phe236 and Asp234). In contrast to falcipain-2, the S2 pockets of cathepsin B and L are wider and more solvent-exposed and therefore not fully occupied by our ligands. Mecozzi and Rebek showed in model systems that ligands leaving unoccupied space in apolar cavities are not well-accommodated because the intermolecular interactions are not better than those that the ligand experiences in the bulk solvent.21 They concluded that undesirable empty space leads to energetically unfavoured desolvation of the interior surface. More recently it was shown by our group that this rule is also applicable to enzymatic pockets.22 These results are in agreement with the observed selectivities of the peptidomimetic nitriles. The ligands are not able to sufficiently fill the S2 pocket of cathepsin B and L and to undergo attractive van der Waals contacts, which explains the loss of inhibition. Finally, unexceptionally all compounds were inactive against α-chymotrypsin proving also their selectivity for cysteine over serine proteases, which might result again from the moderate reactivity of the nitrile and the higher nucleophilicity of the catalytic thiolate compared to serine.
In conclusion, we developed a series of new reversible inhibitors for the parasitic cysteine protease falcipain-2 through rational structure-based design. Molecular modelling led to the design of peptidomimetic nitriles with various S2 substituents to gain insight into binding site properties and to obtain highly selective compounds. In vitro affinities down to the single-digit micromolar range were obtained for the majority of the synthesised compounds. These results validate the aminoacetonitrile moiety as a suitable electrophilic head group and support the proposed binding mode. Biological assays revealed very good selectivity against closely related human cathepsins taking advantage of the moderate electrophilicity of the nitrile functionality and of the differences in the shape of the individual S2 pockets. Remarkably, all inhibitors showed no cross-reactivity with the serine nucleophile at the active site of α-chymotrypsin making them interesting targets for further optimisation studies towards highly effective inhibitors of the malarial protein.
Acknowledgements
V.E. is grateful for a Kekulé fellowship of the German Fonds der Chemischen Industrie. Financial support by the Deutsche Forschungsgemeinschaft (SFB 630, TP A4, for T.S.) and the Swiss National Science Foundation is gratefully acknowledged. We thank Dr Daniel Bur (Actelion Pharmaceuticals Ltd.) for helpful discussions.References
- K. R. Matthews, Science, 2011, 331, 1149–1153 CrossRef CAS.
- M. Klemba and D. E. Goldberg, Annu. Rev. Biochem., 2002, 71, 275–305 CrossRef CAS.
- P. J. Rosenthal, Adv. Parasitol., 1999, 43, 105–159 CrossRef CAS.
- D. Leung, G. Abbenante and D. P. Fairlie, J. Med. Chem., 2000, 43, 305–341 CrossRef CAS.
- F. Lecaille, J. Kaleta and D. Brömme, Chem. Rev., 2002, 102, 4459–4488 CrossRef CAS.
- H.-H. Otto and T. Schirmeister, Chem. Rev., 1997, 97, 133–172 CrossRef CAS.
- R. Löser, M. Frizler, K. Schilling and M. Gütschow, Angew. Chem., Int. Ed., 2008, 47, 4331–4334 CrossRef.
- R. M. Oballa, J.-F. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998–1002 CrossRef CAS.
- V. Ehmke, C. Heindl, M. Rottmann, C. Freymond, W. B. Schweizer, R. Brun, T. Schirmeister and F. Diederich, ChemMedChem, 2011, 6, 273–278 CrossRef CAS.
- J. B. Moon, R. S. Coleman and R. P. Hanzlik, J. Am. Chem. Soc., 1986, 108, 1350–1351 CrossRef CAS.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS.
- R. W. Snow, C. A. Guerra, A. M. Noor, H. Y. Myint and S. I. Hay, Nature, 2005, 434, 214–217 CrossRef CAS.
- T. Hogg, K. Nagarajan, S. Herzberg, L. Chen, X. Shen, H. Jiang, M. Wecke, C. Blohmke, R. Hilgenfeld and C. L. Schmidt, J. Biol. Chem., 2006, 281, 25425–25437 CrossRef CAS.
- I. D. Kerr, J. H. Lee, K. C. Pandey, A. Harrison, M. Sajid, P. J. Rosenthal and L. S. Brinen, J. Med. Chem., 2009, 52, 852–857 CrossRef CAS.
- S. X. Wang, K. C. Pandey, J. R. Somoza, P. S. Sijwali, T. Kortemme, L. S. Brinen, R. J. Fletterick, P. J. Rosenthal and J. H. McKerrow, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11503–11508 CrossRef CAS.
- M. Zürcher and F. Diederich, J. Org. Chem., 2008, 73, 4345–4361 CrossRef.
- P. Ung and D. A. Winkler, J. Med. Chem., 2011, 54, 1111–1125 CrossRef CAS.
- P. R. Gerber and K. Müller, J. Comput.-Aided Mol. Des., 1995, 9, 251–268 CrossRef CAS.
- A. Breuning, B. Degel, F. Schulz, C. Büchold, M. Stempka, U. Machon, S. Heppner, C. Gelhaus, M. Leippe, M. Leyh, C. Kisker, J. Rath, A. Stich, J. Gut, P. J. Rosenthal, C. Schmuck and T. Schirmeister, J. Med. Chem., 2010, 53, 1951–1963 CrossRef CAS.
- F. Schulz, C. Gelhaus, B. Degel, R. Vicik, S. Heppner, A. Breuning, M. Leippe, J. Gut, P. J. Rosenthal and T. Schirmeister, ChemMedChem, 2007, 2, 1214–1224 CrossRef CAS.
- S. Mecozzi and J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- M. Zürcher, T. Gottschalk, S. Meyer, D. Bur and F. Diederich, ChemMedChem, 2008, 3, 237–240 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data for new compounds, biological assays and additional figures. See DOI: 10.1039/c1md00115a. |
| This journal is © The Royal Society of Chemistry 2011 |