Structure-based design, synthesis, and profiling of a β-tryptase inhibitor with a spiro-piperidineamide scaffold, benzylamine P1 group, and a substituted indole P4 group†
Guyan
Liang
*a,
Yong Mi
Choi-Sledeski
b,
Gregory B.
Poli
b,
Xin
Chen
c,
Anne
Minnich
d,
Qingping
Wang
e,
Joseph
Tsay
d,
Keith
Sides
d and
Roy J.
Vaz
a
aDrug Design, Lead Generation and Candidate Realization, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA. E-mail: guyan.liang@sanofi-aventis.com; Tel: +1-908-231-4573
bMolecular Therapeutics, Lead Generation and Candidate Realization, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
cStructural Biology, Lead Generation and Candidate Realization, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
dIn Vitro Pharmacology, Internal Medicine, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
eDrug Metabolism and Pharmacokinetics, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
First published on 30th June 2011
Abstract
A β-tryptase inhibitor with a basic benzylamine P1 group, a substituted indole P4 group, and a spiro-piperidineamide scaffold linker was designed, synthesized, and tested for its β-tryptase potency and ADMET properties. Comparing to its non-spiro analogs, the inhibitor with a spiro-piperidineamide scaffold demonstrated superior metabolic stability in human liver microsome and against semicarbazide-sensitive amine oxidase (SSAO).
The therapeutic utility of human β-tryptase for inflammatory and allergic disorders, like asthma and chronic obstructive pulmonary disease (COPD), has been well established.1–4 Human mast cells, residing in connective tissues and epithelial surfaces, express both α- and β-tryptases. While the physiological and enzymatic function of α-tryptase is controversial,5–7 the present study mainly focused on β-tryptase. Human mast cell β-tryptase, activated intracellularly and stored in secretory granules, can be secreted upon mast cell activation, which in turn triggers inflammatory and allergic responses. It is believed that inhibition of β-tryptase can effectively mitigate the inflammatory and allergic symptoms.
A wide variety of β-tryptase inhibitors, ranging from macromolecule to small synthetic inhibitors, has been investigated for their potential therapeutic functions. Early studies discovered some macromolecular inhibitors, like leech-derived tryptase inhibitors8 and myeloperoxidase which interrupts the tetramerization of β-tryptase thus renders it inactive.9 The majority of research in the field reported in the last two decades has been largely focused on small synthetic organic inhibitors.10–20Inhibitors for trypsin-like serine proteases, of which β-tryptase is a member, can be generally divided into three categories: mechanism-based inhibitors, basic-P1 inhibitors, and non-basic P1 inhibitors. While the discovery for β-tryptase inhibitors has progressed beyond the least desirable mechanism-based category, the hunt for a non-basic P1 inhibitor has been fruitless so far. Variety of basic P1 inhibitors have been synthesized and evaluated by our team at Sanofi-aventis with one of the advanced compounds shown in Fig. 1, compound 1. However, the clinical study of this compound was stopped due to unanticipated observations, among which is that the compound was rapidly oxidized by semicarbazide sensitive amine oxidase (SSAO). The new challenge for the project team was to progress another compound satisfying all previous requirements plus the stability toward SSAO which does not correlate well with either in vitro stability of the compound in human plasma or in vivo animal PK data. Hence, an SSAO stability assay was established using membrane-bound full-length SSAO expressed in Chinese hamster ovary (CHO) cells.
 | ||
| Fig. 1 β-Tryptase inhibitors with a benzylamine P1 group. | ||
Several types of inhibitors were explored in the course of the project. As shown in Fig. 1, relative to β-tryptase binding, compound 1 can be divided into the benzylamine P1, the piperidineamide linker, and the aromatic P4 groups. While benzylamine is the group being oxidized by SSAO and is a target of the optimization, numerous P4 groups and scaffold linkers were also explored including substituted indole rings as P4 and spiro-piperidines as scaffold linkers. Costanzo and co-workers published their discovery of compound 2 based on our disclosure of compound 1.21 However, at that time, our search had already progressed beyond inhibitors with an aromatic alkynyl P4 group to new chemotypes including molecules with a substituted indole P4 group.
Amidine-containing groups, in lieu of benzylamine, were explored as possible P1 replacements without much success, which was largely due to their poor oral bioavailability. Direct substitutions at the amino nitrogen or at the carbon atom α to the nitrogen were proven to be detrimental to the β-tryptase activity, which is perfectly reasonable given that the benzylamine group binds in the tight S1 pocket and maintains a critical H-bonding network with surrounding residues, especially Asp 189. Based on β-tryptase X-ray structures, the position at the phenyl ring para to the aminomethyl group is likely the only position accessible to substitutions while one of the meta positions is occupied by the piperidine scaffold. The challenge is to find a suitable substitution at the para position to render the aminomethyl group less susceptible to SSAO-mediated oxidation.
The catalytic center of SSAO contains a critical cofactor, 2,4,5-trihydroxyphenylalanyl quinine (TPQ) which is derived from the tyrosine residue of the highly conserved sequence (SXXNYD) by a post-translational modification.22,23 The oxidized form of TPQ can react with benzylamine to form a Schiff base which can be hydrolyzed to benzaldehyde and further oxidized to benzoic acid, as shown in Scheme 1. The protonated benzylamine must be deprotonated to facilitate the nucleophilic attack on TPQ. While the equilibrium between the protonated and unprotonated species may affect the Schiff base formation, this equilibrium is heavily influence by the local pH environment. Another major factor for the stability of the substrate is the nucleophilicity of the aminomethyl group, which is directly related with the rate of the Schiff base formation.
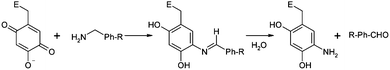 | ||
| Scheme 1 The benzylamine of β-tryptase forms a Schiff base with the oxidized form of TPQ of SSAO before turning into benzaldehyde. | ||
Quantum mechanics calculations using density functional theory were carried out to assess the impact on partial charge distribution by the para-fluoro substitution or the spiro ring scaffold, especially on the aminomethyl group. The Becke-3 parameter hybrid exchange functional24 and the LYP correlation functional25 (B3LYP) were used in the density functional calculation with the uniform basis set of 6-311++G** which includes both polarization functions and defuse functions on all atoms. Using this approach, a molecular electrostatic potential (ESP) map was calculated, from which atomic partial charges26 were derived.
A fluoro substitution of benzylamine at the position para to the aminomethyl group or introducing a spiro ring between benzylamine and piperidine may delocalize the negative charge of the aminomethyl group and render it less nucleophilic. As shown in Fig. 2, the total charge on the aminomethyl group of compounds 6 and 7 are 0.194 and 0.183, respective, which are significantly less negative than −0.406 of compound 5. Arguably, a part of the negative charge is delocalized to the phenyl ring. This charge delocalization renders the amonimethyl group of compounds 6 and 7 less nucleophilic, which consequently decreases the rate of the Schiff base formation and increases the stability of the substrate.
 | ||
| Fig. 2 ESP partial atomic charges of the aminomethyl group calculated using a quantum mechanics software Jaguar® from Schrodinger® with the B3LYP hybrid functional and 6-311++G** basis set. | ||
Another reason which may significantly impact the SSAO oxidation is the flexibility of the potential SSAO substrate. SSAO is a membrane-bound protein with a dimeric fold.27,28 Each monomer has its own catalytic center which is featured by the cofactor, TPQ, and a copper ion coordinated by the side chains of three conserved histidine residues. This catalytic center is deeply buried inside the protein and is accessible only through a narrow channel of 4.5 Å in diameter through which potential substrates have to enter and the oxidized products (aldehyde) have to exit. While the side chain mobility of the residues lining the channel can accommodate the passage of molecules with certain size and flexibility, even molecules with an apparent diameter slightly larger than 4.5 Å. The flexibility of the substrate is another important factor influencing the accessibility and is the focus of the present study.
Comparing to compound 3, the one with a benzylamine-spiro-piperidine scaffold, e.g., compound 4, has a significantly decreased flexibility by completely locking the torsion angle between the benzylamine ring and the piperidine ring. A molecular dynamics (MD) simulation was carried out to demonstrate this decreased scaffold flexibility. Both compound 4 and its counterpart without the spiro ring were subjected to a 10 nanosecond (ns) MD simulation at 300 K following a heating period and a 1 ns equilibration period. As shown in Fig. 3, the torsion angle defined by C1–N2–C4–C5 was monitored during the simulation to reflect the rotational flexibility of the benzylamine ring in reference to the piperidine ring, while the torsion angle defined by C1–N2–C3–C4 was traced to reflect the flexibility of the piperidine ring itself. Obviously, with a spiro ring between the benzylamine and the piperidine ring, the torsion angle between the two rings is totally locked with a RMS deviation of 6.0° compared to 10.9° for the non-spiro counterpart, as shown in Fig. 3, (c) and (a). The spiro scaffold even rendered the piperidine ring itself more rigid with the RMS deviation of the torsion angle defined by C1–N2–C3–C4 decreased from 9.5° to 8.4°, as shown in Fig. 3, (b) and (d). This decreased flexibility presumably lowers the probability for the compound to snake through the channel accessing the SSAO catalytic center and, therefore, enhances the stability against the SSAO-mediated oxidation.
 | ||
| Fig. 3 Torsion angle trajectories during a 10 ns MD simulation. Curves (a) and (b) are for the compound 3 and curves (c) and (d) are for compound 4. | ||
Compounds 3 and 4 are hypothesized to bind in the substrate binding pocket of β-tryptase in an almost identical binding mode. As shown in Fig. 4, benzylamine of both compounds bind in the S1 pocket with the amino group H-bonding with the carboxylate side chain of D189. The carbonyl oxygen of the piperidine-amide forms another H-bond with the backbone NH of G219, which allows the scaffold to span the S1 and S4 binding pockets without interacting with the catalytic triad formed by H57, S195, and D102. The substituted indole group in the S4 pocket π-stacks with the amide side chain of Q98 and interacts hydrophobically with the side chain of Y95.
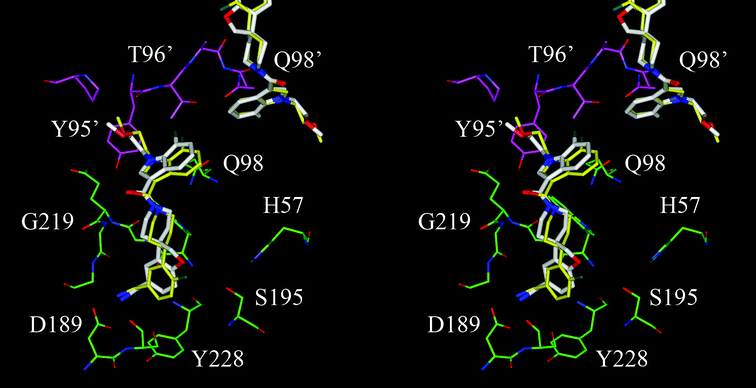 | ||
| Fig. 4 A stereo view of compound 3 and 4 modeled in the β-tryptase substrate binding pocket located between two adjacent monomers of which the carbon atoms are colored in green and magenta, respectively. The carbon atoms of compound 3 are colored in yellow and those of compound 4 are in white. The compounds bound in the binding pocket of the adjacent monomer are partially shown. | ||
The spiro ring constrains the torsion angle between the benzylamine ring and the piperidine ring to about 75° which is well within the acceptable range observed in X-rays structures and in-silico models for similar β-tryptase inhibitors with a piperidineamide scaffold. While most X-ray structures have the torsion angle between the two rings measured between 65° and 80°, some X-ray structures exhibit a torsion angle as large as 90°.29 Although the two ends of the molecule are well anchored, with the benzylamine group binding in the well-defined S1 pocket and the substituted indole ring π-stacking on Q98, the piperidine amide scaffold can still accommodate some changes in orientation. Any change in exiting vectors of the scaffold linker caused by rotation of the piperidine ring can be offset by a counter rotation of the amide group. Based on in-silico models and X-ray structures, the torsion angle between the benzylamine and the piperidine rings can vary within a range of 25° without causing any significant change in either binding mode or binding affinity. Therefore, the risk that the spiro-piperidine scaffold may constrain the torsion angle to a non-preferred conformation is minimal. Considering the additional entropic benefit, a net gain in binding free energy may result.
The synthesis of spiro-piperidine derivative 4 is described in Scheme 2. Quaternization of 4-hydroxymethylpyridine 8 with benzyl chloride, followed by reduction with NaBH4 in methanol regioselectively gave allyl alcohol 9. Mitsunobu reaction with diisopropylazodicarboxylate and Boc-protected 3-bromo-4-hydroxybenzylamine afforded ether 10. The radical based cyclization of 10 to 11 was effected in high yield by using tributyltin hydride and 2,2′azobis(isobutyronitrile). As shown in the scheme, subsequent hydrogenation of the benzyl protecting group provided the key amine intermediate 12 in high yield. Coupling of amine 12 with 7-fluoro-1-(2-methoxy-ethyl)-1H-indole-3-carboxylic acid 1319 in the presence of EDCI, followed by deprotection with 2N HCl in dioxane provided the desired product 4.
 | ||
| Scheme 2 Reaction scheme for synthesizing compound 4. | ||
Compound inhibition of human β-tryptase was quantified in a chromogenic assay in which enzymatic activities were measured with 50 ng mL−1β-tryptase and 500 μM substrate S-2366 (Diapharma), L-pyroglutamyl-L-prolyl-L-arginine-p-nitroaniline hydrochloride at various compound concentrations. The IC50 of the inhibitor was determined by fitting the activity-concentration data to a sigmoid curve, based on which the corresponding Ki value of the inhibitor was derived using the competitive inhibition equation, Ki = IC50/(1 + [S]/Km).
As shown in Table 1, compounds 3 and 4 have approximately the same potency as compound 1. Based on this observation, one can assume that the newly introduced substituted indole P4 groups are able to contribute a similar level of binding affinity as the aromatic alkynyl P4 group from compound 1. Compound 3 and 4 have an identical structure except that compound 4 has an additional spiro ring between the benzylamine P1 and the piperidine scaffold. The fact that these two compounds have similar activities against β-tryptase indicates that the conformation of the spiro scaffold, while perhaps not the most preferred one, is very close to the unconstrained bioactive conformation exhibited by the binding mode of compound 3.
| β-Tryptase, Ki | SSAO stability | |
|---|---|---|
| Compound 1 | 16 | 8% |
| Compound 3 | 10 | 78% |
| Compound 4 | 15 | 89% |
More importantly, this decreased conformational flexibility lowers the probability of this compound to interact with anti-targets, e.g., SSAO and cytochrome P450s. The SSAO stability was measured by incubating the compound for 20 min with full-length SSAO expressed in CHO cells before stopping the reaction and quantifying the remaining compound through LC/MS/MS.30,31 As shown in Table 1, 78% and 89% of the compounds remained unoxidized after 20 min with SSAO for compounds 3 and 4, respectively, which represent a significant improvement over the 8% remaining for compound 1. This enhanced SSAO stability is presumably attributable to both the electronic effect of the para substitution and the conformational effect of the spiro ring.
Compound 4 is more stable than compound 3 in liver microsomal stability assay. The subcellular microsomal fraction of liver tissues from multiple donors were pooled and purified for any given species. Compound 3 and 4 were incubated with liver microsomes for 20 min before the reaction was stopped and the amount of the remaining compound was measured, from which the microsomal lability of the compound was derived.30 As shown in Table 2, compound 4 is more stable than compound 3 in liver microsomal assay for all three species tested. For example, 71% of compound 3 was metabolized in human liver microsome within 20 min, and only 17% of compound 4 is labile under the same condition. While a MitaSite® calculation indicates that the most susceptible metabolic site is the methoxyethyl group of the indole ring which is conserved between compound 3 and 4, it is a plausible explanation that the less flexible spiro-piperidine scaffold renders the compound more difficult to bind well in the CYP catalytic pocket and is attributable for the enhanced microsomal stability.
| Compound | 3 | 4 |
|---|---|---|
| Liver microsome metabolism (human) | 71% | 17% |
| Liver microsome metabolism (g. pig) | 38% | 12% |
| Liver microsome metabolism (rat) | 21% | 10% |
| hERG inhibittion at 1.0 μM | 11% | |
| hERG inhibittion at 10.0 μM | 16% |
As a part of our routine safety profiling, compound 4 was also tested for hERG channel blockade which is correlated with cardiac QT interval prolongation and may consequently cause sudden cardiac death.32–34 The hERG channel blockade was measured using a well-established patch-clamp electrophysiology assay with hERG-expressing CHO cells from which hERG current and effect of compounds were recorded. As shown in Table 2, compound 4 is completely clean of hERG blockade with only 11% decrease in the hERG current at 1 μM and 16% at 10.0 μM.
In conclusion, a benzylamine-containing β-tryptase inhibitor with a spiro-piperidineamide scaffold and a substituted indole P4 group was designed, synthesized, and tested. The spiro-piperidineamide scaffold and its unique combination with the P1 and P4 groups constrain the molecule to the bioactive bound conformation which is important for preserving the β-tryptase activity. More importantly, this decreased conformational flexibility mitigates the risk for the molecule to be recognized by anti-targets, e.g., SSAO and hERG, which is attributable to the improved SSAO stability, lower liver microsomal metabolism, and almost non-existent hERG channel blockade.
References
- A. Norris, Expert Opin. Invest. Drugs, 2004, 13, 739 CrossRef CAS.
- W. J. Tremaine, A. Brzezinski, J. A. Katz, D. C. Wolf, T. J. Fleming, J. Mordenti, L. C. Strenkoski-Nix, M. C. Kurth, C. Barish, A. Brzezinski, F. C. Fowler, M. Gaspari, J. A. Katz, G. Koval, D. Riff, M. Safdi, B. Salzberg, W. J. Tremaine and D. C. Wolf, Aliment. Pharmacol. Ther., 2002, 16, 407 CrossRef CAS.
- K. D. Rice and P. A. Sprengeler, Curr. Opin. Drug Discovery Dev., 1999, 2, 463 CAS.
- J. M. Clark, W. R. Moore and R. D. Tanaka, Drugs Future, 1996, 21, 811 CAS.
- L. Li, Y. Li, S. W. Reddel, M. Cherrian, D. S. Friend, R. L. Stevens and S. A. Krilis, J. Immunol., 1998, 161, 5079 CAS.
- L. B. Schwartz, K. Sakai, T. R. Bradford, S. Ren, B. Zweiman, A. S. Worobec and D. D. Metcalfe, J. Clin. Invest., 1995, 96, 2702 CrossRef CAS.
- G. H. Caughey, J. Allergy Clin. Immunol., 2006, 117, 1411 CrossRef CAS.
- A. S. Tanaka, M. M. Silva, R. J. S. Torquato, M. A. E. Noguti, C. A. M. Sampaio, H. Fritz and E. A. Auerswald, FEBS Lett., 1999, 458, 11 CrossRef CAS.
- L. Cregar, K. C. Elrod, D. Putnam and W. R. Moore, Arch. Biochem. Biophys., 1999, 366, 125 CrossRef CAS.
- G. H. Caughey, W. W. Raymond, E. Bacci, R. J. Lombardy and R. R. Tidwell, J. Pharmacol. Exp. Ther., 1993, 264, 676 CAS.
- J. M. Clark, W. M. Abraham, C. E. Fishman, R. Forteza, A. Ahmed, A. Cortes, R. L. Warne, W. R. Moore and R. D. Tanaka, Am. J. Respir. Crit. Care Med., 1995, 152, 2076 CAS.
- K. D. Rice, R. D. Tanaka, B. A. Katz, R. P. Numerof and W. R. Moore, Curr. Pharm. Des., 1998, 4, 381 CAS.
- L.-M. Ni and J. C. Powers, Bioorg. Med. Chem., 1998, 6, 1767 CrossRef CAS.
- K. D. Combrink, H. B. Guelgeze, N. A. Meanwell, B. C. Pearce, P. Zulan, G. S. Bisacchi, D. G. M. Roberts, P. Stanley and S. M. Seiler, J. Med. Chem., 1998, 41, 4854 CrossRef CAS.
- L. E. Burgess, B. J. Newhouse, P. Ibrahim, J. Rizzi, M. A. Kashem, A. Hartman, B. J. Brandhuber, C. D. Wright, D. S. Thomson, G. P. A. Vigers and K. Koch, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8348 CrossRef CAS.
- K. D. Rice, A. R. Gangloff, E. Y. L. Kuo, J. M. Dener, V. R. Wang, R. Lum, W. S. Newcomb, C. Havel, D. Putnam, L. Cregar, M. Wong and R. L. Warne, Bioorg. Med. Chem. Lett., 2000, 10, 2357 CrossRef CAS.
- J. M. Dener, V. R. Wang, K. D. Rice, A. R. Gangloff, E. Y. L. Kuo, W. S. Newcomb, D. Putnam and M. Wong, Bioorg. Med. Chem. Lett., 2001, 11, 2325 CrossRef CAS.
- C. Hopkins, K. Neuenschwander, A. Scotese, S. Jackson, T. Nieduzak, H. Pauls, G. Liang, K. Sides, D. Cramer, J. Cairns, S. Maignan and M. Mathieu, Bioorg. Med. Chem. Lett., 2004, 14, 4819 CrossRef CAS.
- C. R. Hopkins, M. Czekaj, S. S. Kaye, Z. Gao, J. Pribish, H. Pauls, G. Liang, K. Sides, D. Cramer, J. Cairns, Y. Luo, H.-K. Lim, R. Vaz, S. Rebello, S. Maignan, A. Dupuy, M. Mathieu and J. Levell, Bioorg. Med. Chem. Lett., 2005, 15, 2734 CrossRef CAS.
- J. Levell, P. Astles, P. Eastwood, J. Cairns, O. Houille, S. Aldous, G. Merriman, B. Whiteley, J. Pribish, M. Czekaj, G. Liang, S. Maignan, J.-P. Guilloteau, A. Dupuy, J. Davidson, T. Harrison, A. Morley, S. Watson, G. Fenton, C. McCarthy, J. Romano, R. Mathew, D. Engers, M. Gardyan, K. Sides, J. Kwong, J. Tsay, S. Rebello, L. Shen, J. Wang, Y. Luo, O. Giardino, H.-K. Lim, K. Smith and H. Pauls, Bioorg. Med. Chem., 2005, 13, 2859 CrossRef CAS.
- M. J. Costanzo, S. C. Yabut, H.-C. Zhang, K. B. White, L. de Garavilla, Y. Wang, L. K. Minor, B. A. Tounge, A. N. Barnakov, F. Lewandowski, C. Milligan, J. C. Spurlino, W. M. Abraham, V. Boswell-Smith, C. P. Page and B. E. Maryanoff, Bioorg. Med. Chem. Lett., 2008, 18, 2114 CrossRef CAS.
- M. Mure, S. A. Mills and J. P. Klinman, Biochemistry, 2002, 41, 9269 CrossRef CAS.
- P. Dunkel, A. Gelain, D. Barlocco, N. Haider, K. Gyires, B. Sperlagh, K. Magyar, E. Maccioni and A. Fadda, Curr. Med. Chem., 2008, 15, 1827 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, B37, 785 Search PubMed.
- S. R. Cox and D. E. Williams, J. Comput. Chem., 2, p. 304 Search PubMed.
- T. T. Airenne, Y. Nymalm, H. Kidron, D. J. Smith, M. Pihlavisto, M. Salmi, S. Jalkanen, M. S. Johnson and T. A. Salminen, Protein Sci., 2005, 14, 1964 CrossRef CAS.
- E. Jakobsson, J. Nilsson, D. Ogg and G. J. Kleywegt, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, D61, 1550 CrossRef CAS.
- Torsion angles were derived from proprietary β-tryptase X-ray structures of soaking or co-crystallization experiments.
- G. Liang, Y. M. Choi-Sledeski, G. Poli, X. Chen, P. Shum, A. Minnich, Q. Wang, J. Tsay, K. Sides, J. Cairns, G. Stoklosa, T. Nieduzak, Z. Zhao, J. Wang and R. J. Vaz, Bioorg. Med. Chem. Lett., 2010, 20, 6721 CrossRef CAS.
- A. Adedoyin, M. R. Angelastro, J. A. Bick, J. Cairns, Y. Huang, G. Liang and H.-K. Lim, WO Patent 2008086325, 2008, 38.
- R. J. Vaz, Y. Li and D. Rampe, Prog. Med. Chem., 2005, 43, 1 CrossRef CAS.
- A. M. Brown, Cell Calcium, 2004, 35, 543 CrossRef CAS.
- R. R. Shah, Br. J. Clin. Pharmacol., 2002, 54, 188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00104c |
| This journal is © The Royal Society of Chemistry 2011 |