Progress on lamellarins†
Daniel
Pla‡
*a,
Fernando
Albericio
abc and
Mercedes
Álvarez
*abd
aCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona Science Park, Baldiri Reixac 10, 08028, Barcelona, Spain
bInstitute for Research in Biomedicine, Barcelona Science Park-University of Barcelona, Baldiri Reixac 10, E-08028, Barcelona, Spain
cDepartment of Organic Chemistry, University of Barcelona, 08028, Barcelona, Spain
dLaboratory of Organic Chemistry, Faculty of Pharmacy, University of Barcelona, 08028, Barcelona, Spain. E-mail: mercedes.alvarez@irbbarcelona.org; Fax: (+34)934037126; Tel: (+34)934037086
First published on 17th March 2011
Abstract
This review covers recent literature on the lamellarins, a family of marine natural products, and related analogs, encompassing synthetic strategies for total synthesis, structure–activity relationships (SAR), and studies on mechanisms of biological action, namely in the context of anti-tumor activity. It reviews work published from January 2008 to December 2010.
Introduction
Lamellarin D (Lam-D, Fig. 1), which comes from a large family of marine pyrrolealkaloids, is currently in preclinical development as an anti-cancer drug. Comprehensive reviews of this compound were published in 2007 and 2008.1,2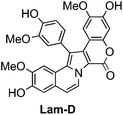 | ||
| Fig. 1 Structure of Lam-D. | ||
Recent advances in the pharmacological development of Lam-D include the identification of new cellular targets of this compound and new insights into its mechanism of action. The malate–aspartate shuttle was recently identified as a new target for Lam-D by proton NMR-based metabolomics,3 and certain protein kinases relevant to cancer have recently been reported as targets of Lam-D, Lam-N and other lamellarins.4 Lam-D exerts its anti-tumor activity through complementary pathways: a nuclear route via topoisomerase I (Topo I) inhibition,5,6 and mitochondrial targeting by induction of mitochondrial permeability transition (MPT).7
Robust synthesis of Lam-D has enabled SAR and mechanism of action studies. Thus, the vast chemical space of the Lam structure has been explored through modified analogs, including 1-dearyl,8 2-dearyl,9lactone-free derivatives,10lactam,11 and polymeric and peptidic nuclear location signal conjugates.12,13
Herein, in addition to the recent synthetic strategies developed for total synthesis of Lam-D and its numerous naturally occurring and synthetic analogs, recent work on SAR studies and on using multi-target mechanisms to induce apoptosis are also discussed.
1. Synthesis of lamellarins
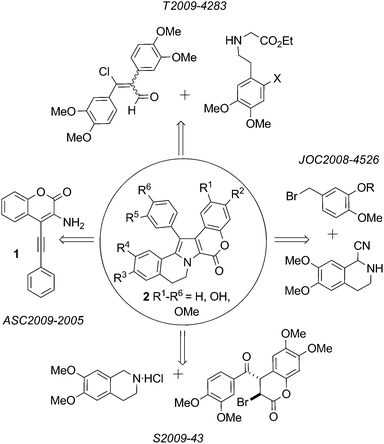 | ||
| Fig. 2 Synthesis of lamellarins based on construction of the pyrrole core. | ||
A new protocol for the construction of the Lamellarin scaffold 2 (Fig. 2 R1–R6 = H) relies on Pd-catalyzed intramolecular hydroamination of the acetylenic aminocoumarin derivative 1.14 This procedure has not yet been exploited in natural product synthesis.
The key step in a recent synthesis of Lam-G trimethyl ether15 is the final step: haloarylation of 3-bromo-4-(3,4-dimethoxybenzoyl)-6,7-dimethoxy-chroman-2-one 3 with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride (Scheme 1).
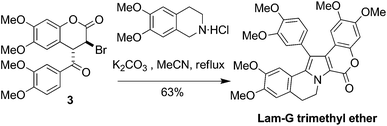 | ||
| Scheme 1 Key step in the synthesis of Lam-G trimethyl ether.15 | ||
1-Benzyl-3,4-dihydroisoquinolines have been transformed to Lam-G trimethyl ether and Lam-U using short reaction sequences. Grob reaction of dihydroisoquinoline 4 with the nitrocinnamate 5 afforded pyrroles6a and 6b.16 Subsequent O-debenzylation and lactonization provided the aforementioned natural product, plus Lam-U (Scheme 2).
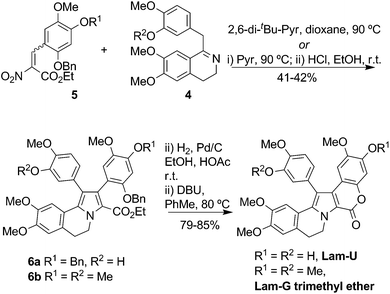 | ||
| Scheme 2 Synthesis of Lam-G trimethyl ether and Lam-U.16 | ||
Gupton et al.17 reported formal syntheses of Lam-G trimethyl ether and ningalin B via formation of the polysubstituted pyrrole derivatives 7a and 7b, respectively, from a vinylogous iminium salt derivative. Subsequent transformations of these highly substituted pyrroles enabled efficient regio-controlled formal syntheses of the targets (Scheme 3).
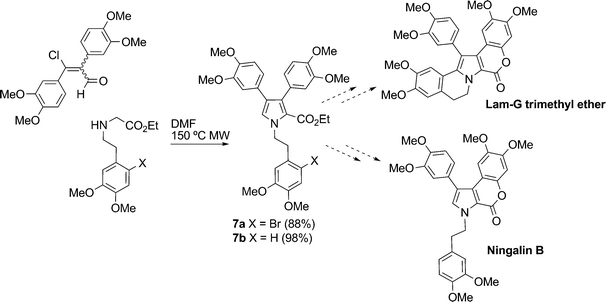 | ||
| Scheme 3 Formal syntheses of Lam-G trimethyl ether and ningalin B.17 | ||
In late 2010 Li et al. reported a new, concise synthesis of Lam-D and Lam-H by condensation of 3-isopropoxy-4-methoxyphenethylamine with two units of the corresponding phenethylaldehyde derivative, followed by AgOAc-mediated oxidative coupling to construct the pyrrole core. Their synthesis featured two additional induced oxidative cyclizations to form the lactone and the pyrrole-arene C–C bond. Thus, Lam-D and Lam-H were prepared in seven steps and in overall yields of 17% and 16%, respectively (Scheme 4).18
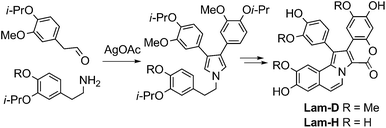 | ||
| Scheme 4 Synthesis of Lam-D and Lam-H.18 | ||
Total syntheses of Lam-O, P, Q, and R have been achieved by using as key synthetic steps the Pd-catalyzed Suzuki–Miyaura cross-coupling reaction of N-benzenesulfonyl-3,4-dibromopyrrole (8) with an arylboronic acid followed by directed α-lithiation.19N-Deprotection and substitution gave the natural products (Scheme 5).
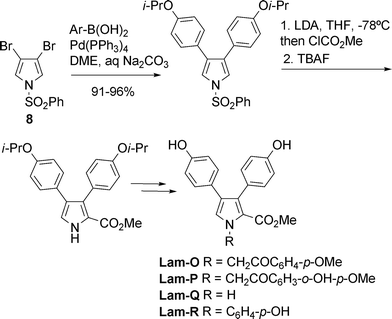 | ||
| Scheme 5 Synthesis of lamellarins O, P, Q, and R19 | ||
Sequential Suzuki-Miyaura coupling of 3,4-dihydroxypyrrole bistriflate (9) as a key reaction20,21 was harnessed to prepare three Lam-α sulfate derivatives: Lam-α 13-sulfate, 20-sulfate, and 13,20-disulfate. The common intermediate 10, in which 13–OH and 20–OH are protected with orthogonal protecting groups, was the shared precursor (Scheme 6).
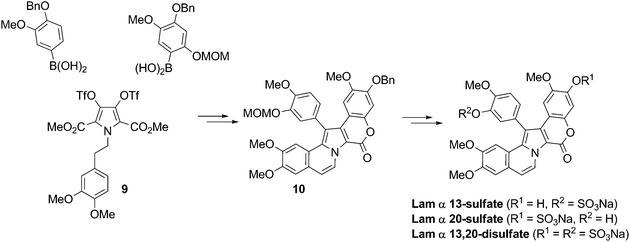 | ||
| Scheme 6 Synthesis of Lam-α 13-, 20-sulfate, and 13,20-disulfate20,21 | ||
A general method for the synthesis of N-unsubstituted 3,4-diarylpyrrole-2,5-dicarboxylates also has been developed.22 The principal reactions comprised a Hinsberg-type synthesis of dimethyl N-benzyl-3,4-dihydroxypyrrole-2,5-dicarboxylate followed by palladium-catalyzed Suzuki-Miyaura coupling of its bis-triflate derivative. The N-benzyl protecting group of the resulting 3,4-diarylpyrrole-2,5-dicarboxylates was cleanly removed under hydrogenolysis or solvolysis. The authors observed that the regioselectivity of the Suzuki cross-coupling of dihalopyrrole esters was influenced by the reaction solvent.23
Banwell et al.24 recently published the first total synthesis of Lam-S and a new synthesis for Lam-G trimethyl ether, based on the same strategy. Their route included two sequential Suzuki–Miyaura cross-coupling reactions to insert the two polyalcoxyaryl groups at positions 3 and 4 of the pyrrole derivative 11. The resulting compound was then subjected to decarboxylative Heck cyclization and, in the case of Lam-S, subsequent O-iPr ether deprotection. (Scheme 7).
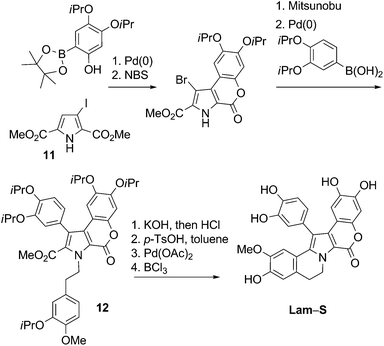 | ||
| Scheme 7 Synthesis of Lam-S24 | ||
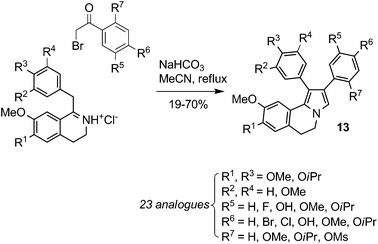 | ||
| Scheme 8 Synthesis of combretastatin A4 and Lam-D hybrid-library10 | ||
Thasana et al. obtained a library of seventeen azalamellarins using microwave-assisted CuI-mediated C–N bond formation.11
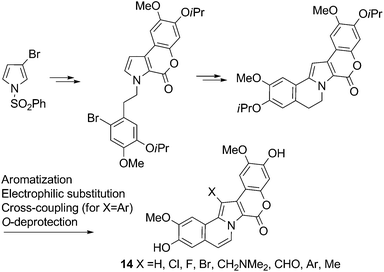 | ||
| Scheme 9 Synthesis of 1-dearyl-Lam-D analogs8 | ||
Given the results of SAR studies on similar 2-dearyl analogs,9 the aforementioned compounds created by the Iwao group should prove interesting for biological evaluation.
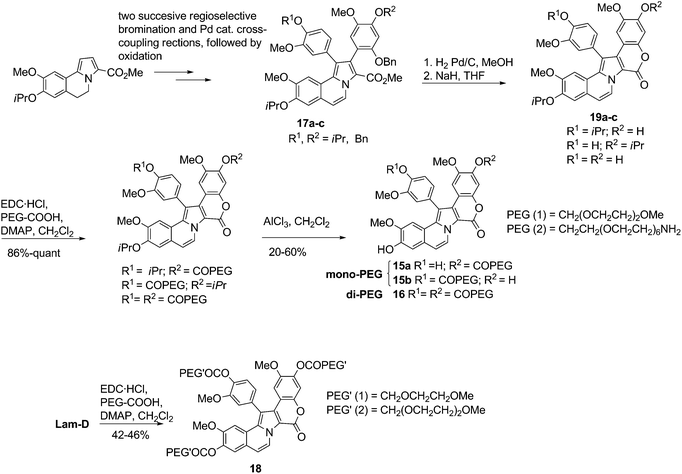 | ||
| Scheme 10 Synthesis of mono-, di- and tri-PEG-bioconjugates of Lam-D.12 | ||
In the synthesis of the dendrimeric- and NLS-Lam-D bioconjugates20 and 21, the amino and guanidino groups of the oligomeric building blocks were protected with N-Boc.13 The PEG-based dendrimer 22 was condensed with protected Lam-D 23 using EDC·HCl and DMAP as coupling agents. Simultaneous removal of the Boc and TBDPS-protecting groups with HF at low temperature afforded 20 (Scheme 11).
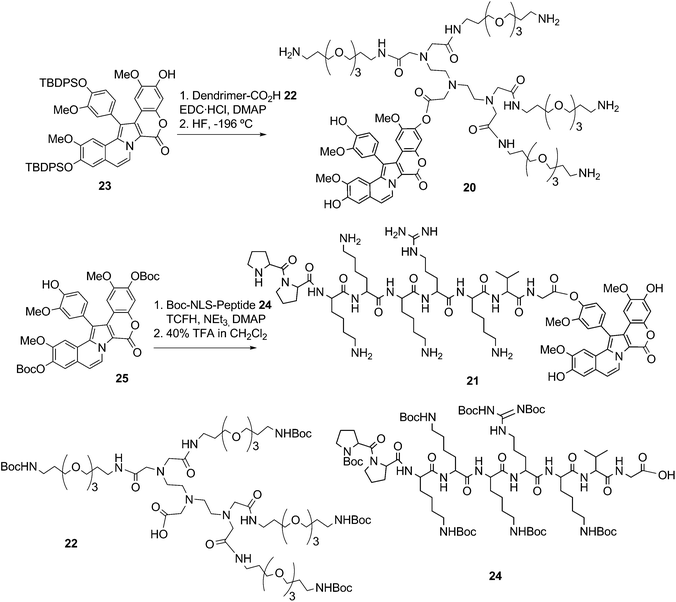 | ||
| Scheme 11 Preparation of dendrimeric- and NLS-bioconjugates of Lam-D13 | ||
Pre-activation of the Boc-protected NLS-peptide24 with N,N,N′,N′- tetramethylchloroformamidinium hexafluorophos-phate (TCFH) and NEt3, followed by the addition of a solution the Boc-protected Lam-D 25 and DMAP, were required to form the bioconjugates, which was obtained in 45% yield. Elimination of the nine Boc protecting groups with 40% TFA in CH2Cl2 gave compound 21 in 17% yield (Scheme 11).
2. Molecular structure–activity determinants, and SAR studies
Natural products remain an underutilized source of leads for the pharmaceutical industry.26 Preparing analogs of these products through medicinal chemistry approaches to explore maximum chemical space is a valuable method for improving and/or modulating the parent biological activity. Given their diverse anti-tumor activities, lamellarins have been the subject of several SAR studies in recent years.Hu et al. prepared a library of structurally simplified lactone-free lamellarin (LF-Lam) analogs having a 1,2-diphenyl-5,6-dihydropyrrolo[2,1-a]isoquinoline core structure. This library was partially inspired by the close structural resemblance between Lam-D and combretastatin A4, which contains a conserved cis-stilbene motif. They evaluated twenty-two novel, diversely-substituted analogs, which they then screened for anti-proliferative activities against five cancer cell lines. The compounds generally showed poor activity (sub-millimolar IC50 values); the most active of them are shown in Fig. 3 and listed in Table 1.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| A549 | K-562 | SMM-7721 | SGC-7901 | HCT-116 | |
| a Cell lines: A549, human non-small-cell lung carcinoma; HCT-116, human colon carcinoma; K-562, human erythromyeloblastoid leukemia; SGC-7901, human gastric adenocarcinoma; SMM-7721, human hepatocarcinoma. | |||||
| LF-Lam 1 | >94.1 | 4 | >94.1 | >94.1 | 1.4 |
| LF-Lam 2 | 6.7 | 2.7 | 10.9 | 11.4 | 0.7 |
| LF-Lam 3 | 23.3 | 5.3 | 15 | 22.7 | 13.8 |
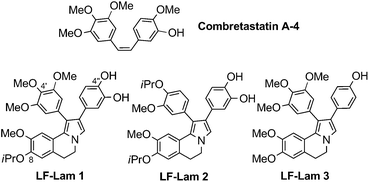 | ||
| Fig. 3 Structures of combretastatin A4 and LF-Lam analogs.10 | ||
Nevertheless, we believe these results reflect a partial but promising structural simplification. Selective removal of the O-iPr groups would easily afford the corresponding free phenolic groups at positions C8 and C4′, which are crucial for the activity of open-chain methyl ester lamellarin analogs.9 The Hu group's results are in agreement with previous findings related to the presence of the OH at C4′′ of the aryl at position 2 of the pyrrolo[2,1-a]isoquinoline, which are also crucial for activity.
Ruchirawat et al. extensively assessed the cytotoxic activities of 25 lamellarins against multiple cancer cell lines derived from six different cancer types (oral, lung, breast, liver, cervix, and blood cell).27 The compounds comprised 22 naturally occurring lamellarins and three synthetic derivatives containing either a saturated or an unsaturated C8–C9 bond, and bearing different OMe, OH and H substitution patterns on the aryl rings. Their SAR studies were the first to reveal the importance of having an OH group at C10 for biological activity and also confirmed prior findings1,2 that the C8![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C9 double bond increases cytotoxicity; that the OH groups at C3 and C11 are crucial to activity; and that methylation of the OH groups at C12, C3′ and C4′ induces only subtle changes in the GI50 values.27
C9 double bond increases cytotoxicity; that the OH groups at C3 and C11 are crucial to activity; and that methylation of the OH groups at C12, C3′ and C4′ induces only subtle changes in the GI50 values.27
Lamellarins-D, -X, -ε, -M, -N, and dehydro-Lam-J were the most potent anti-tumor compounds of the family (Fig. 4): they had IC50 values ranging from sub-nanomolar (0.08 nM) to micromolar (3.2 μM).27 Furthermore, they showed selectivity against cancer cells: these lamellarins were roughly 3 to 1,000,000 times more toxic to cancer cells from all eleven cell lines tested than to the control (normal) cells, MRC-5 human embryonic lung fibroblasts (Table 2).27 In related work, Lam-D and its bioconjugates were similarly selective for cancer cells over BJ skin fibroblasts.12,13
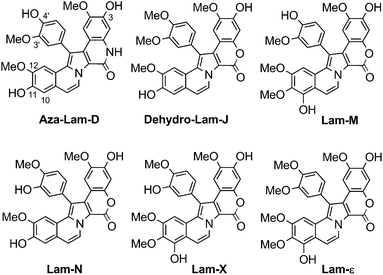 | ||
| Fig. 4 Structures of aza-Lam-D; dehydro-Lam-J; and Lam-X, -ε, -M and -N27,11 | ||
| Comp. | IC50 (μM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oral | Lung | Breast | Liver | Cervix | Blood cell | Leukemia | Fibroblast | ||||||
| KB | A549 | H69AR | T47D | MDA-MB-231 | HuCCA-1 | HepG2 | S102 | HeLa | P388 | HL-60 | MOLT-3 | MRC-5 | |
| a NR: not reported; Cell lines: A549, human non-small-cell lung carcinoma; H69AR, human multi-drug-resistant small-cell lung carcinoma; HeLa, human cervical adenocarcinoma; HepG2, human hepatocellular carcinoma; HL-60, human promyelocytic leukemia; HuCCA-1, human cholangiocarcinoma; KB, human oral epidermoid carcinoma; MDA-MB-231, human hormone-independent breast cancer 231; MOLT-3, T-lymphoblast (acute lymphoblastic leukemia); MRC-5, human fetal/embryonic lung fibroblast; P388, mouse lymphoid neoplasm; S102, human hepatocellular carcinoma; T47D, human hormone-dependent breast cancer. | |||||||||||||
| Lam-D | 0.04 | 0.06 | 0.4 | 0.00008 | 0.4 | 0.08 | 0.02 | 3.2 | 0.06 | 0.1 | 0.04 | 0.049 | 9.2 |
| Aza-Lam-D | NR | 0.74 | NR | NR | NR | 0.12 | 0.13 | NR | NR | NR | NR | 0.03 | NR |
| Dehydro-Lam-J | 0.08 | 0.04 | 0.3 | 0.0001 | 0.4 | 0.006 | 0.01 | 2.1 | 0.08 | 0.08 | 0.04 | NR | >97.4 |
| Lam-M | 0.2 | 0.04 | 0.3 | 0.009 | 0.1 | 0.06 | 0.02 | 1.9 | 0.3 | 0.1 | 0.06 | NR | 13.4 |
| Lam-N | 0.06 | 0.04 | 0.06 | 0.0006 | 0.6 | 0.008 | 0.02 | 2.3 | 0.04 | 0.08 | 0.04 | NR | >100.1 |
| Lam-X | 0.08 | 0.3 | 0.3 | 0.006 | 0.08 | 0.04 | 0.2 | 1.6 | 0.09 | 0.3 | 0.2 | NR | 10.1 |
| Lam-ε | 0.3 | 0.3 | 2.3 | 0.006 | 0.3 | 0.07 | 0.1 | 2.1 | 0.3 | 0.3 | 0.1 | NR | 25.8 |
Hannongbua et al. have studied lamellarins using receptor-independent (RI) 4D quantitative SAR (QSAR) models.28,29 They obtained valuable 3D pharmacophore information from a set of 25 structurally complex lamellarins screened in silico against human hormone dependent T47D breast cancer cells. Overall, they identified formation of an intermolecular hydrogen bond, and the hydrophobic interactions of substituents at C10, C11 and C12, as the most important features for cytotoxicity against the cancer cells.
They also suggested that hydrophobic substitutions at C3′ and C4′ could enhance cytotoxicity.
Ruchirawat et al. prepared Lam-D analogs containing a lactam rather than a lactone. Their work was the first report of a SAR studies done for a change at this position.11 The resulting azalamellarins were screened against four cancer cell lines (HuCCA-1, A-549, HepG2, and MOLT-3). Aza-Lam-D showed good activity (IC50 values in the micromolar range), comparable to that of its parent, Lam-D (Table 2). The amideN–H confers the aza-analogs with greater solubility in biological media compared to that of the parent compound. Interestingly, N-alkylated aza-analogs were not as potent as the parent compound.
For pharmacological enhancement through optimized use of chemical space, the Ruchirawat group suggested introducing additional polar functionalities (e.g. alternative H-bond donors, acceptors, and ionizable groups) into this family of compounds.30
Biopolymeric mono-, di- and tri-ester conjugates of Lam-D (at its phenolic sites) are more soluble than the parent compound and can be readily hydrolyzed in physiologic conditions.12 For example, the monoPEG 15a is up to 80-times more soluble in water than Lam-D. Moreover, a release of up to 97% of Lam-D has been achieved by incubation of 15a for 6 h at 37 °C in Dubelcco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and 100 units/mL penicillin and streptomycin.13 The cytotoxicity of the bioconjugates15, 16, 18, 20 and 21 was assessed against three human tumor cell lines (MDA-MB-231 breast, A-549 lung and HT-29 colon). Several compounds exhibited greater cellular internalization than did Lam-D, and more than 85% of the bioconjugates showed a GI50 at lower concentration than did Lam-D.12,13 Cellular internalization was analyzed, and a nuclear distribution pattern was observed for bioconjugate 21, which contains an NLS.13 The mechanisms of action for Lam-D and these derivatives have been indentified as cell cycle arrest at G2 phase and apoptotic cell-death pathways.12
In summary, the various SAR studies and virtual screening models for lamellarins and related analogs discussed above have demonstrated the efficacy of targeted medicinal chemistry. The findings from this work may contribute to future drug development of synthetic analogs and to the selection of synthetic scaffolds.
3. Activities and mechanism of action of Lam-D
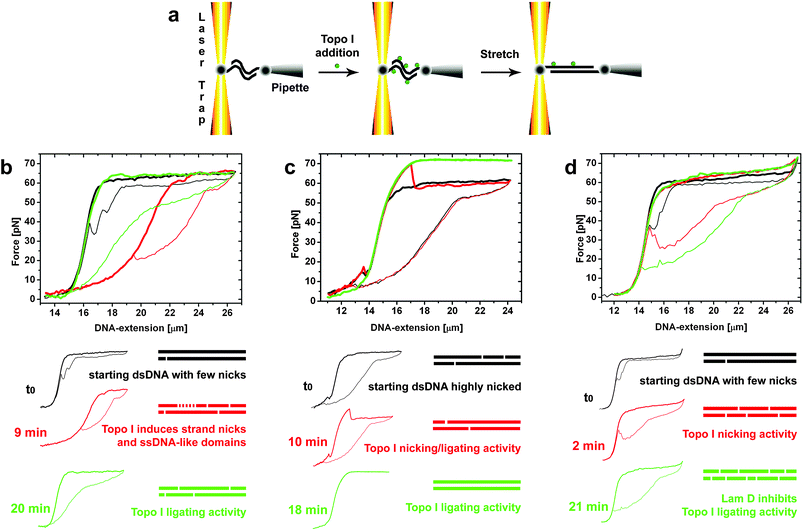 | ||
| Fig. 5 Optical tweezers (OT) force assay of Topoisomerase I (Topo I) activity. (a) Schematic of the experiments, in which each 3′ biotinylated end of the dsDNA is attached to one streptavidin-coated bead. (b-d) OT Force-DNA extension measurements. b) Force-stretching experiment starting from a dsλDNA molecule with a few nicks, as indicated by the small hysteresis area (black curve). Stretching-relaxing cycles are indicated by half-bold half-thin plots. The cleavage activity of Topo I after 9 min of reaction generated an ssDNA-like domain, a heavily nicked dsDNA which exhibited the mechanical properties of ssDNA (red curve). After a further 11 min the ligating activity of Topo I removed the ssDNA-like domain (green curve). c) Force-stretching experiment starting with highly-nicked dsDNA, as indicated by the large hysteresis area (black curve). Topo I activity is reflected by a higher-level plateau, which indicates total religation of the initial DNA nicks, and by complete disappearance of the overstretching hysteresis (green curve, 18 min reaction time). The arrow indicates a drop in the stretching curve resulting from Topo I cleavage during the cycle. d) Force assay of Topo I activity in the presence of Lam-D. Starting λDNA exhibited a small hysteresis area (black curve) corresponding to a dsDNA molecule with only a few nicks. The cleavage activity of Topo I induced a large increase in hysteresis in the presence of Lam-D after the first two minutes of reaction (red curve). Stabilization by Lam-D of this intermediate complex resulted in inhibition of the religating activity of Topo I (green curve, acquired at 21 min reaction).6,31 | ||
Inhibition of Topo I by Lam-D has also been demonstrated by atomic force microscopy (AFM) imaging of plectonemic DNA molecules with Topo I enzyme attachments that were treated with a combination of Lam-D and a reaction buffer used to relax supercoiled DNA (see Fig. 6).6
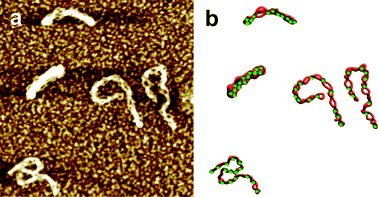 | ||
| Fig. 6 AFM imaging of Topo I-treated plasmid DNA. a) Supercoiled circular DNA treated by Topo I in the presence of Lam-D. Horizontal scale is 0.75 μm, vertical scale is 2 nm for all images. b) Interpretive illustration. The green dots represent Topo molecules.6,31 | ||
Nuclear penetration remains a challenge in the pharmacological development of Lam-D and related compounds. Albeit the aforementioned work elegantly demonstrates how Lam-D blocks the nuclear enzyme Topo I, if the drug cannot reach the nucleus, then this activity is useless. Confocal microscopy experiments on internalization of Lam-D and its PEG conjugates 15, 16 and 18 in three different cell lines show that these compounds chiefly end up in the cytoplasm,12 although Lam-D and the PEG-NH2 compound 15a were able to weakly reach the nucleus in one of the tested cell cultures. Chemical bioconjugation of Lam-D to a nuclear localization signal (NLS) peptide to give the Lam-D NLS bioconjugate21 was explored as a strategy to enable active transport of the drug through the nuclear membrane. Experiments on the co-localization and cellular distribution of Lam-D NLS bioconjugate in three green fluorescent protein-Topo I (GFP-Topo I) transfected cell cultures (HT-29, A-549, and MDA-MD-231 cell lines), indicated that it localized to the nucleus in HT-29 and A-549 cells, while GFP-Topo I was located in the nucleus for all three cell types. Thus, the NLS peptidic sequence is at least partly responsible for nuclear import of 21 in those cell lines. The authors concluded that greater cytotoxicity correlated with greater nuclear localization.13
In 2009 Bailly, Marchetti et al. reported further and more extensive studies using various tumor cell lines, including several leukemia and carcinoma lines.5 Their results suggest that for all cell types tested, Lam-D exerts its cytotoxicity primarily by inducing mitochondrial apoptosisindependently of nuclear signaling.5 This work not only contributes to deciphering the apoptotic pathways activated by Lam-D but also provides evidence that Lam-D is still active even if the classical apoptotic pathways are blocked in cancer cells (i.e. when p53 is null/mutated, Topo I is mutated, and/or Bcl-2 is overexpressed).
In 2010 the same research group ultimately identified direct induction of mitochondrial permeability transition (MPT), which leads to swelling and cytochrome c release, as the mechanism behind the pro-apoptotic function of Lam-D.7
These results indicate that functional mitochondria are required for Lam D-induced apoptosis. Furthermore, inhibition of mitochondrial respiration is responsible for MPT-dependent apoptosis of cancer cells induced by Lam-D.7
The aforementioned findings open the path to new cancer therapy strategies that target mitochondria, thereby reinforcing the potential value of mitochondriophilic drugs for this indication. Given the problems posed by drug resistant cancer cells, targeting mitochondria may prove an effective approach, as long as Lam-D or a related drug is sufficiently selective in affecting exclusively the mitochondria of cancer cells.5,7,32
Conclusions and future trends
The unique structures and biological activities of the lamellarins and related pyrrole-derived alkaloids have attracted significant synthetic efforts in recent years. Thus, elegant synthetic routes to these compounds or their key intermediates have been developed. Since 2008, Lam-G trimethyl ether has been synthesized using four novel strategies, in total yields ranging from 5% to 45%15–17,24 These highly modular approaches can provide access to any member of the family; one need only choose the appropriately substituted and protected building blocks. Additionally, these chemistries have proven valuable for preparation of structurally simplified lamellarin analogs, affording sufficient quantities for SAR and other biological studies.The initial SAR studies on lamellarins have provided the first evidence that an OH group at C10 position is important for biological activity.27 Latter work demonstrated the importance of the C8C9 double bond, and confirmed the importance of the OH groups at C3 and C11. To date, Lam-D, X, ε, M and N, and dehydro-Lam-J are the most potent anti-tumor compounds of this family.
Further SAR studies of the 1-dearyl Lam-D analogs synthesized by Iwao et al.8 could provide useful data and a more thorough overview on structural simplification of the parent compound.
Incorporation of polar groups onto the core to facilitate further H-bonding interactions, as well as ionizable groups to enhance the solubility, can yield improved lamellarin analogs. For example, aza-Lam-D,11 which contains one more H-bond donor than the parent compound; several PEG analogs of Lam-D (15, 16, 18, and 20) analogs;12 and a peptidebioconjugate of Lam-D (21)13 have shown enhanced bioactivity and physicochemical properties relative to the parent compound.
Future medicinal chemistry efforts should be addressed to provide derivatives and bioconjugates with greater specificity for biological targets. Given that Lam-D has been shown to inhibit the nuclear enzyme Topo I, medicinal chemists should prioritize achieving nuclear transport and release of this drug, and creating analogs able to cross the nuclear membrane on their own. Researchers should also explore if and how chemical modifications in the form of new lamellarin analogs could lead to improved or distinct mitochondrial/nuclear pathways of action.
Lastly, direct targeting of mitochondria by Lam-D can circumvent the drug resistance often encountered in tumor cells. Thus, Lam-D constitutes a new pro-apoptotic agent that may bypass certain forms of apoptosis resistance.5
Further studies can contribute to a better understanding of the mechanism of lamellarins and their analogs, and will be beneficial for the ongoing pharmacological optimization of this class of compounds.
Acknowledgements
This review was partially supported by CICYT (Grant CTQ 2009-07758), Generalitat de Catalunya (2009SGR 1024), and the Barcelona Science Park. DP would like to acknowledge the support of the European Commission through a 3-year Marie Curie International Outgoing Fellowship under the 7th Framework Programme (FP7-PEOPLE-2009-IOF-253205).Notes and references
- D. Pla, F. Albericio and M. Álvarez, Anti-Cancer Agents Med. Chem., 2008, 8, 746–760 CAS.
- H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2007, 108, 264–287 Search PubMed.
- M. Bayet-Robert, S. Lim, C. Barthomeuf and D. Morvan, Biochem. Pharmacol., 2010, 80, 1170–1179 Search PubMed.
- D. Baunbæk, N. Trinkler, Y. Ferandin, O. Lozach, P. Ploypradith, S. Ruchirawat, F. Ishibashi, M. Iwao and L. Meijer, Mar. Drugs, 2008, 6, 514–527 CrossRef CAS.
- C. Ballot, J.r. Kluza, A. Martoriati, U. Nyman, P. Formstecher, B. Joseph, C. Bailly and P. Marchetti, Mol. Cancer Ther., 2009, 8, 3307–3317 Search PubMed.
- D. Pla, A. Sischka, F. Albericio, M. Álvarez, X. Fernàndez-Busquets and D. Anselmetti, Small, 2009, 5, 1269–1272 Search PubMed.
- C. Ballot, J. Kluza, S. Lancel, A. Martoriati, S. Hassoun, L. Mortier, J.-C. Vienne, G. Briand, P. Formstecher, C. Bailly, R. Nevière and P. Marchetti, Apoptosis, 2010, 15, 769–781 Search PubMed.
- T. Ohta, T. Fukuda, F. Ishibashi and M. Iwao, J. Org. Chem., 2009, 74, 8143–8153 CrossRef CAS.
- D. Pla, A. Marchal, C. A. Olsen, A. Francesch, C. Cuevas, F. Albericio and M. Álvarez, J. Med. Chem., 2006, 49, 3257–3268 CrossRef CAS.
- L. Shen, X. Yang, B. Yang, Q. He and Y. Hu, Eur. J. Med. Chem., 2010, 45, 11–18 CrossRef CAS.
- S. Boonya-udtayan, N. Yotapan, C. Woo, C. J. Bruns, S. Ruchirawat and N. Thasana, Chem.–Asian J., 2010, 5, 2113–2123 Search PubMed.
- D. Pla, A. Francesch, P. Calvo, C. Cuevas, R. Aligue, F. Albericio and M. Álvarez, Bioconjugate Chem., 2009, 20, 1100–1111 Search PubMed.
- D. Pla, M. Marti, J. Farrera-Sinfreu, D. Pulido, A. Francesch, P. Calvo, C. Cuevas, M. Royo, R. Aligue, F. Albericio and M. Álvarez, Bioconjugate Chem., 2009, 20, 1112–1121 Search PubMed.
- L. Chen and M. H. Xu, Adv. Synth. Catal., 2009, 351, 2005–2012 CrossRef CAS.
- J. S. Yadav, K. U. Gayathri, B. V. Reddy, P. Subba and A.R., Synlett, 2009, 43–46 CrossRef CAS.
- J. C. Liermann and T. Opatz, J. Org. Chem., 2008, 73, 4526–4531 CrossRef CAS.
- J. T. Gupton, B. C. Giglio, J. E. Eaton, E. A. Rieck, K. L. Smith, M. J. Keough, P. J. Barelli, L. T. Firich, J. E. Hempel, T. M. Smith and R. P. F. Kanters, Tetrahedron, 2009, 65, 4283–4292 CrossRef CAS.
- Q. Li, J. Jiang, A. Fan, Y. Cui and Y. Jia, Org. Lett., 2011, 13, 312–315 Search PubMed.
- T. Fukuda, E.-i. Sudo, K. Shimokawa and M. Iwao, Tetrahedron, 2008, 64, 328–338 CrossRef CAS.
- T. Fukuda, T. Ohta, S. Saeki and M. Iwao, Heterocycles, 2010, 80, 841–846 Search PubMed.
- The atom numbering shown corresponds to the system used in the original reference, which does not follow IUPAC rules.
- T. Fukuda, Y. Hayashida and M. Iwao, Heterocycles, 2009, 77, 1105–1122 Search PubMed.
- Y. Zhang and S. T. Handy, The Open Organic Chemistry Journal, 2008, 2, 58–64 Search PubMed.
- K. Hasse, A. C. Willis and M. G. Banwell, Eur. J. Org. Chem., 2011, 76, 88–99 Search PubMed.
- D. Pla, A. Marchal, C. A. Olsen, F. Albericio and M. Álvarez, J. Org. Chem., 2005, 70, 8231–8234 CrossRef CAS.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef.
- M. Chittchang, P. Batsomboon, S. Ruchirawat and P. Ploypradith, ChemMedChem, 2009, 4, 457–465 CrossRef CAS.
- P. Thipnate, J. Liu, S. Hannongbua and A. J. Hopfinger, J. Chem. Inf. Model., 2009, 49, 2312–2322 Search PubMed.
- P. Thipnate, M. Chittchang, N. Thasana, P. Saparpakorn, P. Ploypradith and S. Hannongbua, Monatsh. Chem., 2011, 142, 97–109 Search PubMed.
- M. Chittchang, M. Paul Gleeson, P. Ploypradith and S. Ruchirawat, Eur. J. Med. Chem., 2010, 45, 2165–2172 Search PubMed.
- See ref. 6. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
- M.-A. Gallego, C. Ballot, J. Kluza, N. Hajji, A. Martoriati, L. Castera, C. Cuevas, P. Formstecher, B. Joseph, G. Kroemer, C. Bailly and P. Marchetti, Oncogene, 2008, 27, 1981–1992 Search PubMed.
Footnotes |
| † Dedicated to Prof. Rafael Suau a good friend and master |
| ‡ Current address: Memorial Sloan-Kettering Cancer Center, New York NY, 10065, USA; E-mail: mailto:plaquerd@mskcc.org |
| This journal is © The Royal Society of Chemistry 2011 |