Novel indolizine compounds as potent inhibitors of phosphodiesterase IV (PDE4): structure–activity relationship
Shoujun
Chen
,
Zhiqiang
Xia
,
Masazumi
Nagai
,
Rongzhen
Lu
,
Elena
Kostik
,
Teresa
Przewloka
,
Minghu
Song
,
Dinesh
Chimmanamada
,
David
James
,
Shijie
Zhang
,
Jun
Jiang
,
Mitsunori
Ono
,
Keizo
Koya
and
Lijun
Sun
*
Synta Pharmaceuticals Corp., 45 Hartwell Ave., Lexington, MA 02421, USA. E-mail: lsun@syntapharma.com; Fax: +1 781 274 8228; Tel: +1 781 274 8220
First published on 22nd December 2010
Abstract
A series of novel indolizine 2-oxoacetamides were designed and synthesized as PDE4 inhibitors. Preliminary SAR of this new class of compounds revealed key structural features required for high potency. Compounds 1ab and 2a are among the most potent inhibitors of PDE4 with low single nM IC50. Cellular activity was demonstrated by the inhibition of TNFα production from human PBMC with IC50 ranging from 14 to 72 nM. Docking analyses suggest the OH group in 1ab enhance the binding via an H-bond interaction with the PDE4 enzyme.
Phosphodiesterases (PDEs) are a family of intracellular enzymes that regulate cellular levels of pivotal second messengers cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) via catalyzing their degradation to the corresponding 5′-monophosphate AMP and GMP, respectively.1,2 The cAMP specific PDE4 is a key isozyme expressed by immune cells, such as mast cells, monocytes, macrophages, neutrophils, and eosinophils. Inhibition of PDE4 leads to the suppression of key inflammatory cytokines including TNFα, IL-2, and GM-CSF. The elevated level of cellular cAMP resulting from inhibition of PDE4 also contributes to the relaxation of airway smooth muscle. Hence, selective inhibition of PDE4 has therapeutic potentials for the treatment of respiratory disorders such as asthma and COPD, and inflammation disorders such as psoriasis.3–6 Recent research has suggested a role of PDE4 in neurological and psychiatric disorders such as depression, Parkinson's disease and Alzheimer's disease.7,8 A number of PDE4 inhibitors, such as Cilomilast,9Roflumilast,10 and AWD12-28111 have entered into clinical development for the treatment of COPD and asthma; and Roflumilst has gained regulatory market approval in 2010. The discovery of next generation PDE4 inhibitors still remains an attractive research area.12,13
We have been interested in developing novel indolizine compounds as therapeutic agents.14Indolizines and their partially or fully hydrogenated derivatives have been widely found in nature as alkaloids with interesting biological activities.15Indolizines are also viewed as bioisosteres of indoles and have been applied, among many others, as anti-tuberculosis agents,16 as MPtpA/B phosphatases inhibitors,17 and as 15-lipoxygenase inhibitors.18 Herein we disclose a novel family of indolizine 2-oxoacetamides (1) and their corresponding 1,3-regioisomers (2) as potent PDE4 inhibitors.
Preparation of the indolizine compounds 1 utilized a novel DMF-Me2SO4 mediated intramolecular [4 + 1] cyclocondensation reaction of picolinium salt developed in our laboratory.19 As illustrated in Scheme 1, α-bromination of acetophenones (3) was followed by the N-alkylation of picolines to give the picolinium bromide (4) in excellent yields. Reaction of 4 with DMF-Me2SO4 in the presence of TEA proceeded smoothly to give regioselectively the 3-mono substituted indolizines (5) in good to excellent yields (58–91%). Reduction of the carbonyl group in 5 was achieved with BH3-THF in acetonenitrile, resulting in the key indolizine intermediates 6 in good yield (50–80%). Compounds 6 were then treated with oxalyl chloride in THF, followed by one-pot amidation in the presence of TEA and DMAP to give the desired products 1. For sterically hindered amines, such as 4-amino-3,5-dichloro-pyridine and its corresponding N-oxide, an alternative method was applied in order to achieve improvement in the isolated yields. Thus, after reaction of 6 with oxalyl chloride, the reaction mixture was treated with 4-nitrophenol. The 4-nitrophenyl esters 7 were isolated and then reacted with 4-amino-3,5-dichloro-pyridine in the presence of NaH resulting in the desired products 1 in moderate yields. The hydroxyl derivatives 1e, 1x, and 1ab were prepared by BBr3 mediated de-methylation of their corresponding methoxy analogues 1d, 1w, and 1ac (BBr3, CH2Cl2, −78 °C to rt, 2-3 h; 50–70%). 1k was synthesized by hydrogenation of its benzylated precursor (H2, 10% Pd/C, EtOH, rt; 91%). The amino derivative 1g was synthesized via the reduction of the nitro group of 1f (SnCl2, EtOH–THF [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2], reflux, 2 h; 62%).
:2], reflux, 2 h; 62%).
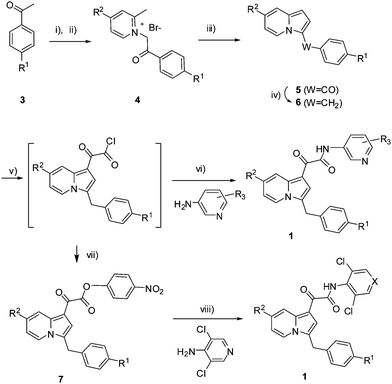 | ||
| Scheme 1 Reagent and conditions: i) Br2, CH2Cl2, rt; ii) Picoline, CH3CN, rt, >80% (2 steps); iii) DMF-Me2SO4, TEA, rt, 58–91%; iv) BH3-THF, CH3CN, 45–50 °C, 50–80%; v) (COCl)2, ether, 0 °C, 10 min; vi) DMAP, THF, 0 °C, 72–85% (2 steps from 6); vii): 4-nitrophenol, TEA, rt; viii) NaH, DMF, 0 °C to rt 42–55% (3 steps from 6). | ||
The indolizine core 1,3-isomeric derivatives (2a and 2b) were synthesized as depicted in Scheme 2. Following the literature procedure, reaction of 8 with 1-ethoxycarbonylmethyl-pyridinium bromide (9) and TEA in reflux EtOH afforded the indolizine intermediate 10 in 42% yield.20 The carboxylic ester moiety was then removed by hydrolysis (KOH, MeOH) followed by decarboxylation (PPA) to afford 11 in 35% overall yield. Reduction of the carbonyl group in 11 with borane produced the key 1-benylated indolizine intermediate 12 (85% yield). The corresponding isomeric products 2 were prepared, following similar procedures for the syntheses of 1, by the reaction of 12 with oxalyl chloride, ester formation with 4-nitrophenol, and finally the amidation of the resulting 4-nitrophenol ester intermediate with 3,5-dichloro-4-aminopyrindine (2a) and 3,5-dichloro-4-aminopyrindine N-oxide (2b).
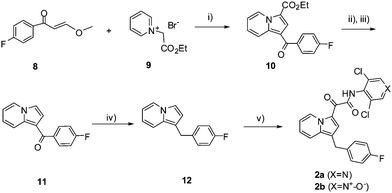 | ||
| Scheme 2 Reagents and conditions: i) TEA, EtOH, reflux, 8 h, 42%; ii) KOH, MeOH, reflux., 3h; iii) Polyphosphoric acid, 100 °C, 1 h; 35% (2 steps); iv) BH3-THF, THF, rt, 45 min, 85%; v) (a) (COCl)2, CH2Cl2, 0 °C, 10 min; (b) 4-nitrophenol, TEA, rt, 15 min; (c) 3,5-dichloro-4-aminopyrindine (2a, X = N) or 1-oxo-3,5-dichloro-4-aminopyrindine (2b, X = N+-O−), NaH, DMF, 0 °C, 30 min, 25–30% from 12. | ||
The PDE4 inhibitory activity of prepared compounds was evaluated by using PDE4 enzyme prepared from U937 human monocytic cells.21 In brief, U937 cells were homogenized and centrifuged. PDE4 activity in the supernatant was assayed with [3H]-cAMP and DMSO solution of a testing compound. The assay mixture was incubated at 37 °C for 30 min followed by the addition of yttrium silicate SPA beads. The beads were spun down, washed twice with 6 mM ZnSO4, suspended in 0.1 N NaOH, and then counted for radioactivity in a liquid scintillation counter. AWD 12-281 and (±)-rolipram were used as positive controls. The IC50 obtained in our assay (14 nM and 880 nM for AWD 12-281 and rolipram, respectively) are consistent with literature reports.11,21b The assay data are reported in Tables 1 and 2 as IC50.
|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (nM) |
| 1a | H | H | H | 348 |
| 1b | F | H | H | 341 |
| 1c | Cl | H | H | 165 |
| 1d | MeO | H | H | 447 |
| 1e | OH | H | H | 327 |
| 1f | NO2 | H | H | 569 |
| 1g | NH2 | H | H | 824 |
| 1h | CN | H | H | 719 |
| 1i | CF3 | H | H | 372 |
| 1j | i-Pr | H | H | 362 |
| 1k | F | OH | H | 414 |
| 1l | F | Cl | H | 239 |
| 1m | Cl | MeO | H | 1,000 |
| 1n | CN | H | 2-Cl | 4,700 |
| 1o | Cl | H | 2,4-(Cl)2 | 84 |
| 1p | CN | H | 2,4-(Cl)2 | 250 |
| 1q | F | H | 2,4-(Br)2 | 10,000 |
| 1r | CN | H | 2,4-(MeO)2 | 10,000 |
| 1s | CN | H | 6-OMe | 1,500 |
| AWD 12-281 | 14 | |||
| Rolipram | 880 | |||
|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | X | IC50 (nM) |
| 1t | CN | H | N | 116 |
| 1u | F | H | N | 13 |
| 1v | Cl | H | N | 16 |
| 1w | MeO | H | N | 40 |
| 1x | OH | H | N | 15 |
| 1y | Cl | Cl | N | 26 |
| 1z | F | Cl | N | 28 |
| 1aa | F | F | N | 13 |
| 1ab | OH | Cl | N | 2 |
| 1ac | MeO | Cl | N | 12 |
| 1ad | CN | Cl | N | 82 |
| 1ae | F | Cl | N+−O− | 54 |
| 1af | F | F | N+−O− | 18 |
| 1ag | Cl | H | N+−O− | 19 |
| 1ah | MeO | H | N+−O− | 80 |
| 1ai | OH | H | N+−O− | 29 |
| 1aj | F | H | CH | 10,000 |
| 1ak | F | H | CCF3 | 10,000 |
| 2a | 6 | |||
| 2b | 29 | |||
The TNFα inhibition assay was carried out in human peripheral blood cells (PBMC) stimulated with LPS.22PBMC were isolated from healthy volunteers by centrifugation and suspended in RPMI-1640 medium supplemented with 10% FCS, 100 U/mL penicillin, and 100 pg/mL streptomycin. The cells were plated in the wells of a 96-well plate at a concentration of 5 × 105cells/well and stimulated by adding LPS (1 ug/mL) in the presence or absence of test compounds. Cell-free supernatants were taken 18 h after incubation for the measurement of the amount of TNFα using an ELISA assay with anti-human TNFα antibodies. IC50 data for selected compounds are summarized in Table 3.
In the meta-pyridyl series (Table 1), the unsubstituted compound 1a is moderately active. A variety of substituents (R1), with electron-withdrawing, electron-donating, or steric hindrance properties, on the phenyl group do not cause significant change in activity, with 4-Cl anologue (1c) as the most potent and the 4-NH2 one (1g) as the least potent. The hydroxy (1k) and chloro (1l) groups at the 6-position of the indolizine core (the R2group) have only slight affect on activity. Yet the methoxy group results in more than 6-fold weaker activity (1mvs.1c), which suggests that the PDE4 binding pocket is sensitive to the size at this position of the inhibitors. More profound affect can be seen with the R3 substituents on the meta-pyridyl moiety. Thus, the 6-methoxy group (para to the amido group) result in 2-fold decrease of activity (1svs.1h); and the 2-chloro in 1n causes more than 6-fold reduction in activity when compared with 1h. The best results is obtained with 2,4-dichloro group (both ortho to the amido group). Thus, compound 1o, with an IC50 of 84 nM, is the most potent inhibitor in the meta-pyridyl series. In contrast, compounds with other 2,4-bissubstituted meta-pyridyl group (1q and 1r), do not possess notable activities.23
In the para-pyridyl series (Table 2), the 3,5-dichloro group (ortho to the amido group) was chosen for the SAR study as they are significantly more active than any other substituents and at any other position on the pyridine moiety.24 Comparing data in Tables 1 and 2, it is evident that the para-pyridyl derivatives are in general considerably more active than their meta-pyridyl counterparts. For instance, compound 1v is about 5-fold more active than its isomer 1o. As seen in Table 2, in agreement with the meta-pyridinyl series the cyano derivative (1t, R1 = CN) is among the less active inhibitors. In comparison with other R1groups (fluoro in 1u, chloro in 1v, and hydroxy in 1x) which all achieve low double-digit nM IC50, the methoxy derivative 1w is about twice less active with IC50 of 40 nM. The R2groups, however, have less predictable impact on the inhibitory activity. Bis-halogenated derivatives 1y, 1z, and 1aa seem to have comparable activity to their mono-halogenated analogues 1u and 1v, with IC50 ranging from 13 to 28 nM. Interestingly, significant enhancement of activity is achieved when the R2group in compound 1ab and 1ac is chloro group. Thus, Compound 1ab, in which R2 is chloro and R1 is hydroxy, is the most potent indolizine type PDE4 inhibitor with IC50 of 2 nM. Pyridines are commonly known to undergo P450 mediated oxidation at the nitrogen and form their nitrogen oxides as potential metabolites. The N-oxide derivatives 1ae to 1ai are all notably potent PDE4 inhibitors with IC50 ranging from 18 to 80 nM, indicating at most only about 2-fold decrease of activity compared to their parent compounds (1ahvs.1w, and 1aivs.1x). Compound 2a, the indolizine core 1,3-position isomer of 1u in which the positions of the 4-fluorobenzyl group and the oxalamide moiety are exchanged, is about 2-fold more active than 1u. The N-oxide 2b has an IC50 of 29 nM. This data suggests that the indolizine core, in addition to its contribution to the pi-pi interaction with the target protein residues, serves primarily as a template to optimally orient the pyridinyl and the benzyl groups thus facilitate their interactions with the target PDE4 enzyme. This hypothesis is supported by the docking analyses detailed below. In contrast, the N atom on the pyridyl moiety is imperative to the PDE4 inhibitory activity. Thus, both 1aj and 1ak, where the pyridyl group is replaced by similarly substituted phenyl groups, do not have measurable activities at 10 uM.
To further understand the ligand-receptor interactions and to guide the lead optimization process, we conducted docking analyses of selected inhibitors into published PDE4 crystal structure. The top figure in Fig. 1 shows the favorable orientation of compound 1ab in the substrate binding site of PDE4B in complex with piclamilast (PDB code: 1XM4).25,26 The superimposition between docked compound 1ab and piclamilast in its cocrystal structure is shown at the bottom in Fig. 1.
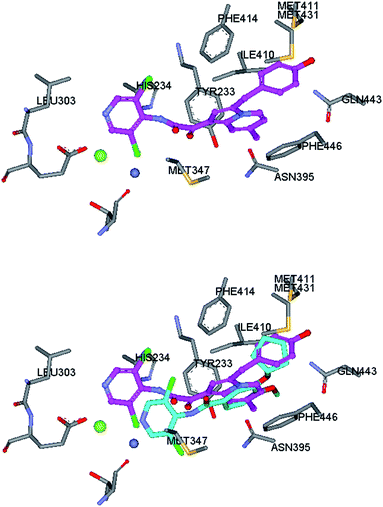 | ||
| Fig. 1 Top: the docking pose of compound 1ab in the PDE4B catalytic site. Its indolizine ring is sandwiched by the hydrophobic clamp between Phe446 and Ile410. The hydroxybenzyl moiety is buried in the so called Q2 pocket that accommodates the cyclopentyl ring of piclamilast. The dicloropyridyl moiety is tilted slightly up towards top region of metal pocket on the left of the figure. Bottom: the superimposition of compound 1ab and piclamilast. For clarity, carbon atoms of compound 1ab and piclamilast were colored with purple and cyan, respectively. Two ions in the metal site are represented by green (Mg2+) and blue (Zn2+). | ||
As illustrated, the indolizine ring sits in the hydrophobic pocket formed by residues Tyr233, Lue393, Ile410, Phe414. And in particular, the indolizine ring is sandwiched between the side-chain of Phe446 and Ile410 and satisfied with one of the common recognition elements between the PDE4 ligand and receptor. Several H-bond acceptors or donors, including Asn395 and Tyr233, are in close proximity to the 5-position of indolizine ring (R2group) and may only allow small substituents such as Cl or OH groups and may also form a potential H-bond with those substituents. The benzyl moiety occupies another hydrophobic pocket surrounded by Phe446, Met411, Met431, Ser442 and Gln443, where it also accommodates the cyclopentyl or cyclopropyl groups in catechol type PDE4 inhibitors, such as rolipram and piclamilast. In the proposed binding mode for compound 1ab, the hydroxyl group on benzyl ring may form H-bond with the sulfur atom in the side chain of Met411. On the other hand, we didn't observe the other common recognition element between other types of PDE4 inhibitors and the conserved Gln443 side chain. Finally, our modeling results suggest that the dichloropyridyl group extend and tile slightly up toward the top region of the metal pocket. There are several nearby hydrophobic residues, Met347, His234, and Leu303 that may form hydrophobic interactions with the pyridinyl group. The pyridinyl N atom and the 2-oxoacetamides carbonyl groups may form H-bond interactions with water molecules coordinated to the ion Mg2+ and His234. The N-oxide may displace one of the coordinated water while maintaining the H-bonding interactions, thus retain most of the activity. However, displacement of structural water is not favoured due to entropy loss hence results in decrease of activity.
A number of potent PDE4 inhibitors were selected and tested for their inhibitory activity against TNF-α production in human PBMC stimulated by LPS (Table 3). All of the tested PDE4 inhibitors demonstrate significant activity in the inhibition of TNFα production, which in general correlates to their PDE4 inhibitory activities. Among them, the relatively low activity of 1ab (the most potent PDE4 inhibitor in this chemical series) in TNFα assay is likely due to lower cellular membrane permeability caused by the polar hydroxy group.
In summary, we have designed and synthesized a series of novel indolizine type PDE4 inhibitors. SAR studies revealed key structural attributes to activity; and chemical modification led to the discovery of PDE4 inhibitors with IC50 as low as 2 nM. Our data is in agreement with the literature reports demonstrating the 3,5-dichloro-4-aminopyridyl moiety as an key structural feature for activity.27Lead compounds demonstrated strong TNFα inhibitory activity in human PBMC. Docking analyses also revealed key ligand-receptor interactions that are critical for maintaining high activities. This focused SAR study led to the discovery of potent and selective PDE4 inhibitors with excellent oral bioavailability and efficacy in multiple animal models. In vitro and in vivo pharmacological and DMPK profiling of lead compounds will be reported separately.
Notes and references
- (a) D. T. Manallack, R. A. Hughes and P. E. Thompson, J. Med. Chem., 2005, 48, 3449–3462 CrossRef CAS; (b) A. Kodimuthali, S. S. Lal Jabaris and M. Pal, J. Med. Chem., 2008, 51, 5471–5489 CrossRef CAS.
- S. Malhotra, S. F. Man and D. D. Sin, Expert Opin. Emerging Drugs, 2006, 11, 275–291 Search PubMed.
- M. D. Houslay, P. Schafer and K. Y. J. Zhang, Drug Discovery Today, 2005, 10, 1503–1519 CrossRef CAS.
- Z. Huang and J. A. Mancini, Curr. Med. Chem., 2006, 13, 3253–3262 CrossRef CAS.
- K. Y. J. Zhang, P. N. Ibrahim, S. Gillette and G. Bollag, Expert Opin. Ther. Targets, 2005, 9, 1283–1305 CrossRef CAS.
- W. Baeumer, J. Hoppmann, C. Rundfeldt and M. Kietzmann, Inflammation Allergy: Drug Targets, 2007, 6, 17–26 Search PubMed.
- A. L. O Hebb and H. A. Robertson, Curr. Opin. Pharmacol., 2007, 7, 86–92 CrossRef.
- A. Blokland, R. Schreiber and J. Prickaerts, Curr. Pharm. Des., 2006, 12, 2511–2523 CrossRef CAS.
- M. S. Barnette, S. B. Christensen and D. Essayan, J. Pharmacol. Exp. Ther, 1988, 284, 420–426.
- R. Draheim, U. Egerland and C. Rundfeldt, J. Pharmacol. Exp. Ther., 2003, 308, 555 CrossRef.
- R. Draheim, U. Egerland and C. Rundfeldt, J. Pharmacol. Exp. Ther., 2003, 308, 555–563 CrossRef.
- A. J. Duplantier, E. L. Bachert, J. B. Cheng, V. L. Cohan, T. H. Jenkinson, K. G. Kraus, M. W. McKechney, J. D. Pillar and J. W. Watson, J. Med. Chem., 2007, 50, 344–349 CrossRef CAS.
- R. W. Friesen, Y. Ducharme, R. G. Ball, M. Blouin, L. Boulet, B. Cote, R. Frenette, M. Girard, D. Guay, Z. Huang, T. R. Jones, J. J. LaliberteLynch, J. Mancini, E. Martins, P. Masson, E. Muise, D. J. Pon, P. D. S Siegl, A. Styhler, N. N. Tsou, M. J. Turner, R. N. Young and Y. J. Girard, J. Med. Chem., 2003, 46, 2413–2426 CrossRef CAS.
- (a) D. A. James, K. Koya, H. Li, S. Chen, Z. Xia, W. Ying, J. Wu and L. Sun, Bioorg. Med. Chem. Lett., 2006, 16, 5164 CrossRef CAS; (b) D. A. James, K. Koya, H. Li, S. Chen, Z. Xia, W. Ying, J. Wu and L. Sun, Abstr. of Papers, 232nd ACS National Meeting, San Francisco, CA, United States, Sept. 10–14, 2006, MEDI-039 Search PubMed; (c) K. Koya, L. Sun, H. Li, D. A. James, N. Tatsuta, T. Korbut, D. Zhou, G. Liang, Y. Wu, Z. Du, S. Chen, J. Barsoum, D. A. Dahl, M. Ono and L.-B. Chen, Proc. Am. Assoc Cancer Res., 2004, 45: A, 5416 Search PubMed; (d) H. Li, Z. Xia, S. Chen, K. Koya, M. Ono and L. Sun, Org. Process Res. Dev., 2007, 11, 246–250 Search PubMed.
- (a) J. P. Michael, Nat. Prod. Rep., 2001, 18, 520 RSC; (b) W. Gao, W. Lam, S. Zhong, C. Kaczmarek, D. C. Baker and Y. C. Cheng, Cancer Res., 2004, 64, 678–688 CAS; (c) Y. F. Wang, C. H. Lu, G. F. Lai, J. X. Cao and S. D Luo, Planta Med., 2003, 69, 1066–1068 CrossRef CAS.
- L.-L. Gundersen, C. Charnock, A. H. Negussie, F. Rise and S. Teklu, Eur. J. Pharm. Sci., 2007, 30, 26 CrossRef CAS.
- T. Weide, L. Arve, H. Prinz, H. Waldmann and H. Kessler, Bioorg. Med. Chem. Lett., 2006, 16, 59 CrossRef CAS.
- S. Teklu, L.-L. Gundersen, T. Larsen, K. E. Malterud and F. Rise, Bioorg. Med. Chem., 2005, 13, 3127 CrossRef CAS.
- (a) T. Przewloka, S. Chen, Z. Xia, H. Li, S. Zhang, D. Chimmanamada, E. Kostik, D. James, K. Koya and L. Sun, Tetrahedron Lett., 2007, 48, 5739–5742 CrossRef CAS; (b) Z. Xia, T. Przewloka, K. Koya, M. Ono, S. Chen and L. Sun, Tetrahedron Lett., 2006, 47, 8817–8820 CrossRef CAS.
- Y. Tominaga, Y. Ichihara, T. Mori, C. Kamio and A. Hosomi, J. Heterocycl. Chem., 1990, 27, 263–268 CAS.
- (a) H. Tenor, A. Hatzelmann, R. Kupferschmidt, L. Stanciu, R. Djukanović, C. Schudt, A. Wendel, M. K. Church and J. K. Shute, Clin. Exp. Allergy, 1995, 25, 625–633 CrossRef CAS; (b) C. Burnouf, E. Auclair, N. Avenel, B. Bertin and C. Bigot, et al. , J. Med. Chem., 2000, 43, 4850–4867 CrossRef CAS.
- L. G. Corral, P. A. J. Haslett, G. W. Muller, R. Chen, L. Wong, C. J. Ocampo, R. T. Patterson, D. I. Stirling and G. Kaplan, J. Immunol., 1999, 163, 380–386 CAS.
- The isomeric ortho-pyrdinyl derivatives are significantly less active, hence not discussed in this article.
- The IC50 of the unsubstituted para-pydridyl analogue of 1v is 760 nM.
- G. L. Card, B. P. England, Y. Suzuki, D. Fong, B. Powell, B. Lee, C. Luu, M. Tabrizizad, S. Gillette, P. N. Ibrahim, D. R. Artis, G. Bollag, M. V. Milburn, S.-H. Kim, J. Schlessinger and K. Y. J. Zhang, Structure, 2004, 12, 2233–2247 CrossRef CAS.
- The docking program GLIDE (version 4.0) from Schrodinger's was employed in the docking studies. The ligand structure of 1ab was built and prepared by Schrodinger's Maestro interface and LigPrep with OPLS_2005 force field and other default parameters. Protein structure was prepared with protein preparation module in GLIDE and OPLS-2005 force field. All crystallographic waters were deleted while the metals (Zn++ and Mg++) are maintained. The geometry center of piclamilast present in 1XM4 was used to define docking grid with the length of 16 Å. Compounds were docked with Glide in extra-precision mode, with up to 10 poses per molecule. The best docking pose was presented in Fig. 1.
- M. J. Ashton, D. C. Cook, G. Fenton, J.-A. Karlsson, M. N. Palfreyman, D. Raeburn, A. J. Ratcliff, J. E. Souness, S. Thurairatnam and N. Vicker, J. Med. Chem., 1994, 37, 1696–1703 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |