Asymmetric synthesis of potent chroman-based Rho kinase (ROCK-II) inhibitors†
Yen Ting
Chen
,
Tomas
Vojkovsky
,
Xingang
Fang
,
Jennifer R.
Pocas
,
Wayne
Grant
,
Amiee M. W.
Handy
,
Thomas
Schröter
,
Philip
LoGrasso
,
Thomas D.
Bannister
* and
Yangbo
Feng
*
Translational Research Institute, Scripps Florida, 130 Scripps Way #2A1, Jupiter, FL 33458, USA. Tel: +1 561 228 2201; Tel: +1 561 228 2206; Fax: +1-561-228-3089; E-mail: tbannist@scripps.edu; yfeng@scripps.edu
First published on 6th December 2010
Abstract
Rho kinase (ROCK) is currently investigated as a target for various diseases such as glaucoma and spinal cord injury. Herein, we report the asymmetric synthesis of chroman 1, a highly potent ROCK inhibitor, and its analogs. The inhibitory properties of these compounds for ROCK-II and a selected set of highly homologous kinases are also discussed.
There is considerable interest in the inhibition of Rho kinase (ROCK) to treat a variety of diseases. ROCK is a member of the AGC kinase family of serine/threonine kinases and is comprised of two highly homologous isoforms, ROCK-I and ROCK-II.1 As a downstream effector of RhoA, ROCK regulates actin cytoskeletal organization and stress fiber formation by phosphorylation of myosin light chain (MLC). Hence, ROCK plays an important role in many aspects of cell motility, such as vascular smooth muscle contraction and neurite growth.2 Currently, one ROCK inhibitor, fasudil, has been clinically approved in Japan for the treatment of cerebral vasospasm following subarachnoid hemorrhage with reasonable safety.3 Several groups have demonstrated the blood pressure-reducing effects of ROCK inhibitors.4 In addition to implications in cardiovascular diseases, several ROCK inhibitors have entered clinical trials for the treatment of glaucoma5 and spinal cord injury.6
In a recent report, we presented a series of chroman-3-amides as highly effective inhibitors of ROCK-II.7 In particular, chroman 1 (Fig. 1) was a sub-nanomolar ROCK-II inhibitor with excellent to moderate selectivity over related kinases studied as a preliminary assessment, such as protein kinase A (PKA), AKT1, and the highly homologous Cdc42-binding kinase (MRCKα). Furthermore, this compound exhibited good activity in the functional cell-based myosin light chain bis-phosphorylation (ppMLC) assay,8 and a reasonable pharmacokinetic profile. These properties encouraged further investigation of this series of inhibitors for development. Chroman 1 was previously prepared and evaluated as a racemate. Therefore, the scalable synthesis of the enantiomers of this lead compound became a high priority within our optimization strategy.
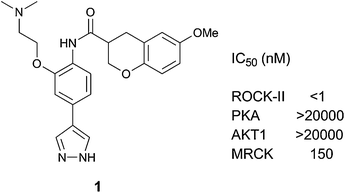 | ||
| Fig. 1 Structure of chroman 1. | ||
The enantiomerically pure chroman acids 7a and 7b, needed for single enantiomer synthesis of inhibitor 1, were prepared as illustrated in Scheme 1. Cyclization of salicaldehyde 2 with acrylonitrile under Baylis–Hillman conditions afforded nitrile 3, which was subsequently hydrolyzed to chromene acid 4.9Acylation of acid 4 with Oppolzer's (−)-camphorsultamvia acid chloride formation, followed by asymmetric reduction of intermediate 5a with L-selectride10 formed the (R)-chromane derivative (6a). Similarly, the use of the (+)-camphorsultam as the chiral auxiliary led to (S)-chromane 6b, and the diastereomeric purity of both 6a and 6b were determined to be >95% by 1H NMR. Finally, the chiral auxiliary was removed with a lithium hydroxide–hydrogen peroxide mixture11 to furnish the enantiomerically pure chroman-3-acids 7a and 7b as the (R)- and (S)-enantiomers, respectively.12 The enantiomeric excess for both were determined as >98% by chiral reverse phase HPLC (Chiralcel AD-RH). While this method provided sub-gram quantities of each chroman enantiomer, enabled assignment of the absolute stereochemistry, and allowed initial exploration of the effects of the chroman stereocenter on ROCK inhibition, an alternative method which would be amenable to multi-gram synthesis was needed. We therefore turned to asymmetric hydrogenation of chromene 4. After exploring several reaction conditions,13 we found that a commercially available variant of H8-BINAP·Ru(II)Cl2 complex, [(RuCl(H8-BINAP))2(μ-Cl)3][NH2Me2],14 was the most effective catalyst, offering a combination of low catalyst loading, acceptable yield, and high enantiomeric excess. Optimal conditions involved the treatment of chromene 4 with 0.001 equiv. of (R)-H8-BINAP·RuCl2 and caesium formate (4 equiv.) in methanol under hydrogen atmosphere at 100 psi and at 40 °C to provide the (S)-acid (7b) in quantitative yield and 89% ee. Further enrichment to 99% ee was achieved by recrystallization with (S,S)-chloramphenicol base in acetonitrile.15 The (R)-enantiomer of 7 was also obtained with similar yield and ee by the same method, except with the use of the (S)-H8-BINAP-Ru(II) as the catalyst and (R,R)-chloramphenicol base as the chiral resolving agent.
![Reagents and conditions: (a) Acrylonitrile, DABCO, 110 °C, 5 h, 73%; (b) NaOH, H2O, reflux, 5 h, 92%; (c) (i) oxalyl chloride, DMF, CH2Cl2, rt, overnight; (ii) 5a: (−)-camphorsultam, NaH, toluene, rt, 3 h, 95% (two steps); 5b: (+)-camphorsultam, NaH, toluene, rt, 6 h, 92% (two steps); (d) l-selectride, THF, −50 °C, 45 min, 6a (60% (>98% de) from 5a), 6b (70% (>98% de) from 5b); (e) LiOH, H2O2, THF, H2O, 0 °C, 20 min, (R)-7a (93%), (S)-7b (94%); (f) 7a: (i) (S)-[(RuCl(H8-BINAP))2(μ-Cl)3][NH2Me2], caesium formate, 100 psi H2, MeOH, 40 °C, 20 h; (ii) (R,R)-chloramphenicol base, acetonitrile (86%, 99% ee); 7b: (i) (R)-[(RuCl(H8-BINAP))2(μ-Cl)3][NH2Me2], caesium formate, 100 psi H2, MeOH, 40 °C, 20 h; (ii) (S,S)-chloramphenicol base, acetonitrile (87%, 99% ee).](/image/article/2011/MD/c0md00194e/c0md00194e-s1.gif) | ||
| Scheme 1 Reagents and conditions: (a) Acrylonitrile, DABCO, 110 °C, 5 h, 73%; (b) NaOH, H2O, reflux, 5 h, 92%; (c) (i) oxalyl chloride, DMF, CH2Cl2, rt, overnight; (ii) 5a: (−)-camphorsultam, NaH, toluene, rt, 3 h, 95% (two steps); 5b: (+)-camphorsultam, NaH, toluene, rt, 6 h, 92% (two steps); (d) L-selectride, THF, −50 °C, 45 min, 6a (60% (>98% de) from 5a), 6b (70% (>98% de) from 5b); (e) LiOH, H2O2, THF, H2O, 0 °C, 20 min, (R)-7a (93%), (S)-7b (94%); (f) 7a: (i) (S)-[(RuCl(H8-BINAP))2(μ-Cl)3][NH2Me2], caesium formate, 100 psi H2, MeOH, 40 °C, 20 h; (ii) (R,R)-chloramphenicol base, acetonitrile (86%, 99% ee); 7b: (i) (R)-[(RuCl(H8-BINAP))2(μ-Cl)3][NH2Me2], caesium formate, 100 psi H2, MeOH, 40 °C, 20 h; (ii) (S,S)-chloramphenicol base, acetonitrile (87%, 99% ee). | ||
Our previous synthesis of racemic inhibitor 17 involved several steps with bases present that could potentially racemize compounds arising from chroman acids 7a and 7b. To avoid this possibility, a new method to prepare the chroman-containing inhibitors was devised. In this revised route (Scheme 2), 4-bromo-2-fluoronitrobenzene (8) was converted to aryl ethers with general structure 9via nucleophilic substitution with the appropriate alcohol, followed by microwave assisted Suzuki heteroarylation and Boc protection. The nitro group of pyrazole 9 was then reduced to obtain aniline 10 by hydrogenation. Finally, a non-basic carbodiimide amide coupling with chromane acid 7 and Boc deprotection provided the desired kinase inhibitors with general structure 11.
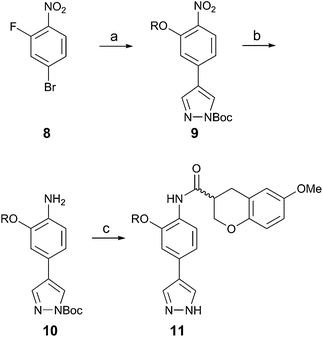 | ||
| Scheme 2 Reagents and conditions: (a) (i) ROH, NaH or KOt-Bu, THF, rt, overnight; (ii) 1H-pyrazole-4-boronic acid pinacol ester, Pd(PPh3)4, Na2CO3, toluene, ethanol, H2O, 1 h, 135 °C, μW; (iii) Boc2O, DMAP, dioxane, rt, overnight, 61–72% (three steps); (b) H2, 10% Pd/C, EtOAc, MeOH, rt, 20 h, 72–87%; (c)(i) 7a or 7b, HOAt, EDC, CH2Cl2, rt, overnight; (ii) TFA, CH2Cl2, rt, 2 h, 45–66% (two steps). | ||
The versatility of this synthetic route permitted not only the synthesis of single enantiomers of chroman 1, but also a variety of analogs with various aryl ether substituents (11a–d). These compounds were evaluated against ROCK-II, PKA, and MRCKα by methods described previously.16 As shown in Table 1, the (S)-chroman was the eutomer for this series of inhibitors, with regard to ROCK-II and also the related kinases. The (S)-enantiomer of chroman 1 displayed more than a 10-fold improvement in the inhibition of ROCK-II and potency in the cell-based ppMLC assay over (R)-1. However, (S)-1 also showed greater affinity for PKA and MRCKα. The (S)-chromans of all the analogs of compound 1 (11a–11d) also displayed subnanomolar IC50 values against ROCK-II and at the lowest limits of detection (4 nM) in the ppMLC assay. From a selectivity perspective, the (R)-chroman series were interesting due to their ability to effectively inhibit ROCK-II with low or no affinity for PKA and MRCKα. However, potency in the cell-based assay was compromised. Modest gains in selectivity in the (S)-chroman series were obtained when pyrrolidines were present in the aryl ether side chain. In particular, the pyrrolidine analog (11a) showed the highest selectivity against PKA and MRCKα in this study (>9500- and >1200-fold against PKA and MRCKα, respectively). Curiously, potency against ROCK-II was maintained when replacing a tertiary amine with an alcohol and an additional methylene group. However, selectivity of this compound over the other kinases was slightly reduced. Compound (S)-11d was also one of the most potent MRCKα inhibitor we have identified in our series of kinase inhibitors.
| Cmpd | R | IC50/nMa | |||
|---|---|---|---|---|---|
| ROCK-II | PKA | MRCKα | ppMLCb | ||
| a Average of two or more measurements. The error in these values is within ±30% of the average. b Cell-based assay. c Not determinable. | |||||
| (S)-1 |
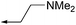
|
<1 | 1740 | 127 | <4 |
| (R)-1 |
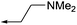
|
7 | >20000 | 8640 | 43 |
| (S)-11a |

|
<1 | 9600 | 1275 | <4 |
| (R)-11a |
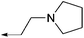
|
30 | >20000 | >20000 | 411 |
| (S)-11b |

|
<1 | 3464 | 261 | <4 |
| (R)-11b |

|
14 | >20000 | >20000 | 32 |
| (S)-11c |

|
<1 | 7380 | 294 | <4 |
| (R)-11c |

|
34 | >20000 | ndc | 900 |
| (S)-11d |

|
<1 | 652 | 74 | <4 |
| (R)-11d |

|
35 | >20000 | ndc | 519 |
Conclusions
We have developed a method for the asymmetric synthesis of chroman-3-carboxylic acids and used these building blocks to prepare a series of highly potent and cell-permeable ROCK-II inhibitors. The compounds in the (S)-chroman series had higher affinity for ROCK-II with low nanomolar potency in the cell-based assay. While this series also moderately inhibited selected kinases related to ROCK-II, introduction of pyrrolidines in the side chain slightly enhanced selectivity. Further pharmacological studies of (S)-11a and related compounds, as well as the synthesis and evaluation of other series of ROCK-II inhibitors that incorporate single chroman enantiomers will be presented in future communications.Notes and references
- O. Nakagawa, K. Fujisawa, T. Ishizaki, Y. Saito, K. Nakao and S. Narumiya,, FEBS Lett., 1996, 392, 189 CrossRef CAS.
- K. Riento and A. J. Ridley, Nat. Rev. Mol. Cell Biol., 2003, 4, 446 CrossRef CAS.
- M. Shibuya, Y. Suzuki, K. Sugita, I. Saito, T. Sasaki, K. Takakura, I. Nagata, H. Kikuchi, T. Takemae, H. Hidaka and M. Nakashima, J. Neurosurg., 1992, 76, 371.
- (a) M. Löhn, O. Plettenburg, Y. Ivashchenko, A. Kannt, A. Hofmeister, D. Kadereit, M. Schaefer, W. Linz, M. Kohlmann, J.-M. Herbert, P. Janiak, S. E. O'Connor and H. Ruetten, Hypertension, 2009, 54, 676 CrossRef; (b) R. Kast, H. Schirok, S. Figueroa-Pérez, J. Mittendorf, M. J. Gnoth, H. Apeler, J. Lenz, J. K. Franz, A. Knorr, J. Hütter, M. Lobell, K. Zimmermann, K. Münter, K. H. Augstein, H. Ehmke and J. P. Stasch, Br. J. Pharmacol., 2007, 152, 1070 CAS; (c) C. Doe, R. Bentley, D. J. Behm, R. Lafferty, R. Stavenger, D. Jung, M. Bamford, T. Panchal, E. Grygielko, L. L. Wright, G. K. Smith, Z. Chen, C. Webb, S. Khandekar, T. Yi, R. Kirkpatrick, E. Dul, L. Jolivette, J. P. Marino, Jr., R. Willette, D. Lee and E. Hu, J. Pharmacol. Exp. Ther., 2007, 320, 89 CAS.
- H. Tanihara, M. Inatani, M. Honjo, H. Tokushige, J. Azuma and M. Araie, Arch. Ophthalmol., 2008, 126, 309 CrossRef CAS.
- C. Hahmann and T. Schroeter, Cell. Mol. Life Sci., 2010, 67, 171 CrossRef CAS.
- Y. T. Chen, T. D. Bannister, A. Weiser, E. Griffin, L. Lin, C. Ruiz, M. D. Cameron, S. Schurer, D. Duckett, T. Schroter, P. LoGrasso and Y. Feng, Bioorg. Med. Chem. Lett., 2008, 18, 6406 CrossRef CAS.
- T. Schroter, E. Griffin, A. Weiser, Y. Feng and P. LoGrasso, Biochem. Biophys. Res. Commun., 2008, 374, 356 CrossRef.
- L. D. Wise, H. A. DeWald, E. S. Hawkins, D. M. Reynolds, T. G. Heffner, L. T. Meltzer and T. A. Pugsley, J. Med. Chem., 1988, 31, 688 CrossRef CAS.
- W. Oppolzer and G. Poli, Tetrahedron Lett., 1986, 27, 4717 CrossRef CAS.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141 CrossRef CAS.
- Assignment of absolute stereochemistry was based on the predictive model for asymmetric induction described in ref. 10. For another example of the use of the Oppolzer auxiliary to obtain similar stereoselectivity, please refer to: K. Takao, N. Hayakawa, R. Yamada, T. Yamaguchi, H. Saegusa, M. Uchida, S. Samejima and K. Tadano, J. Org. Chem., 2009, 74, 6452 Search PubMed.
- A table of other conditions examined during efforts to optimize the asymmetric hydrogenation reaction is provided in the ESI.
- T. Uemura, X. Zhang, K. Matsumura, N. Sayo, H. Kumobayashi, T. Ohta, K. Nozaki and H. Takaya, J. Org. Chem., 1996, 61, 5510 CrossRef CAS.
- Second re-crystallization of the chloramphenicol base salt of chroman 7b further improved the enantiomeric purity to 99.6%. However, the re-crystallization procedure requires a large volume of acetonitrile, and 99% ee was acceptable for our purposes.
- Y. Feng, Y. Yin, A. Weiser, E. Griffin, M. D. Cameron, L. Lin, C. Ruiz, S. C. Schürer, T. Inoue, P. V. Rao, T. Schröter and P. LoGrasso, J. Med. Chem., 2008, 51, 6642 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of novel compounds and biological assays. See DOI: 10.1039/c0md00194e |
| This journal is © The Royal Society of Chemistry 2011 |