[4-(6,7-Disubstituted quinazolin-4-ylamino)phenyl] carbamic acid esters: a novel series of dual EGFR/VEGFR-2 tyrosine kinase inhibitors†
Antonio
Garofalo
ab,
Laurence
Goossens
*ab,
Amélie
Lemoine
ab,
Séverine
Ravez
ab,
Perrine
Six
ab,
Michael
Howsam
ac,
Amaury
Farce
ab and
Patrick
Depreux
ab
aInstitut de Chimie Pharmaceutique Albert Lespagnol, Univ Lille Nord de France, 3 rue du Professeur Laguesse, B.P.83, F–59006 Lille, France. Tel: +33 (0)3 20 96 47 02; Fax: +33 (0)3 20 96 49 06; E-mail: laurence.goossens@univ–lille2.fr
bUDSL, ICPAL, EA 4481, F–59006 Lille, France
cUDSL, UFR de Pharmacie, F–59006 Lille, France
First published on 14th December 2010
Abstract
Investigating a series of anilinoquinazoline derivatives substituted by carbamic acid esters, we have established the importance of the carbamate functional group and the substitution on the arylamino ring by a donor/acceptor group such as halide or methyl. All the newly-synthesized compounds described were evaluated for both their in vitroEGFR and VEGFR-2 kinase inhibition and antiproliferative activities against various cancer cells. These novel compounds were effective tyrosine kinase inhibitors (TKIs) for these two enzymes with in vitro IC50 values in the submicromolar range, but showed a moderated inhibitory activity on cancer cells. Modification of the ether linkage at the 6- or 7- position of the quinazoline core with a basic or aliphatic side chain (70–80) was investigated and it was demonstrated that introduction of aminoalkyl substituents such as morpholinoethoxy is a key modification that increases antiproliferative activity.
1. Introduction
Protein kinases play important roles in regulating most of the cellular functions (i.e. proliferation, cell cycle, cell metabolism, survival, apoptosis, DNA damage/repair) and their overexpression is involved in many cancer cells.1,2 Most of the signal transduction pathways are mediated by protein kinases and aberrant kinase signalling may lead not only to the proliferation of cancer cells but also to angiogenesis and the growth of solid tumors, such as in prostatic, colon, breast and gastric cancers.3,4 The epidermal growth factor receptor (ErbB-1/HER-1/EGFR) and vascular endothelial growth factor receptor-2 (VEGFR-2/Flk-1/KDR) are examples of the receptors tyrosine kinases (RTKs) that are under investigation as potential targets for the development of cancer therapeutics.5Activation of ErbB family receptor tyrosine kinases is involved in numerous biological responses including proliferation, survival, cell migration, and angiogenesis.6,7EGFR dysregulation by overexpression, upactivation (auto- or paracrine loop ligand binding) or mutation is associated with tumorigenesis, through the activation of several signal transduction pathway (Ras/MAPK, PI3K/Akt, Jak/STAT) and cellular processes, and is correlated with poor prognosis in cancer patients.8–10 VEGFs and their receptors are key intermediates in tumor angiogenesis, and in the formation of new blood vessel networks supplying nutrients and oxygen for the tumor's growth.11–14 VEGFR-2, associated with VEGF ligand, is the main mediator of several physiological and pathological effects in endothelial cells such as proliferation, migration, survival, and vascular permeability.15 The intracellular signalling pathways (p38MAPK, PI3K/Akt) mediating these effects are crucial for tumour development and their inhibition has become a new therapeutic target against cancer.
It is well-established that tumor cells express EGFR and VEGFR-2, particularly in tumor endothelial cells. These two glycoproteins were identified in many cancers and their functional relationship is well established: targeting EGFR inhibits tumor growth by decreasing the production of VEGF, while inhibition of VEGFR-2 increases the antitumoral effect of EGFR inhibitors.16,17 Thus, the combined inhibition of both EGFR and VEGFR-2 signalling pathways represents a promising approach to cancer treatment with a synergistic effect.18,19
Many therapies targeting EGFR/VEGFR-2 and their ligands have been discovered and subsequently developed and marketed as new drugs such as monoclonal antibodies (ErbituxTM, AvastinTM)20,21 or tyrosine kinase inhibitors (TarcevaTM, NexavarTM).22–24 The 4-anilinoquinazoline class of inhibitors has led to commercial compounds, among which the EGFR-selective Gefitinib (IressaTM), from AstraZeneca, received approval by the United States Food and Drug Administration in 2005 for the treatment of non-small cell lung cancer (NSCLC), is a example (Fig. 1).25Vandetanib (ZD6474), also from AstraZeneca, a once-daily oral anticancer drug in phase III, is considered to be a dual tyrosine kinase inhibitor targeting EGFR and VEGFR-2.26 This quinazoline substituted with a halide at the 2- and 4-positions on the phenyl group shows a strong inhibitory activity (IC50 = 500 nM for EGFR and 40 nM for VEGFR-2, in ELISAs with recombinant enzymes).27 Thus, concomitant inhibition of these two glycoproteins appears to be a new therapy to decrease progression of tumors.28
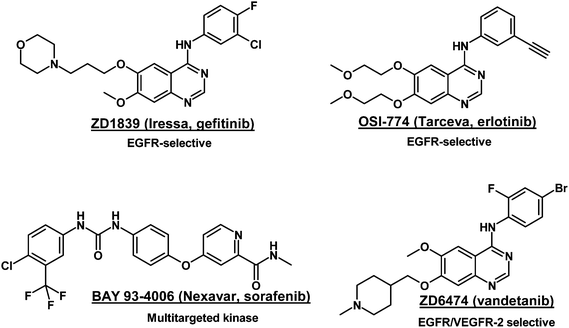 | ||
| Fig. 1 EGFR and/or VEGFR-2 tyrosine kinase inhibitors. | ||
In the work presented here, we report the identification of quinazoline-carbamic acid esters as a novel class of dual EGFR and VEGFR-2 inhibitors.
We recently described the Structure Activity Relationship (SAR) developed around an anilinoquinazoline skeleton leading to 1 and 2 endowed with a dual in vitroEGFR/VEGFR-2 inhibitory activity (Fig. 2).29 It was concluded that introduction of halides or a carbamic acid methyl ester group on the arylamino ring enhances the binding affinity when compared with urea or amide derivatives for the ATP pocket of these two proteins. Based on our previous findings, we explore herein the substitution by halide or alkyl groups on the phenyl group of the carbamic acid ester. Quinazolines containing an amide or urea group were also prepared to confirm the importance of the carbamic acid ester entity. These compounds were evaluated for their antiproliferative activity towards the hormone-independent PC3 prostate cancer cells, HT29 colon cancer cells, MCF-7 breast cancer cells and by measuring inhibition of various kinase activities, notably EGFR, VEGFR-2, and compared with the activity of the reference compounds gefitinib (Iressa) and Vandetanib.
 | ||
| Fig. 2 4-Anilinoquinazolines 1, 2 and pharmacological evaluation.29 | ||
2. Chemistry
The various key intermediates and final compounds were synthesized according to the general method described in Scheme 1. 4-Chloro-6,7-dimethoxyquinazoline 4 was prepared from commercial 4,5-dimethoxymethyl anthranilate, as starting material, in two steps.30 The synthesis of (aminophenyl)carbamic acid esters 5–21 was performed by a one-pot reduction procedure of the corresponding nitrophenyl isocyanate in the presence of various alcohols in a CH2Cl2–THF mixture. This approach has been recently reported to be efficient and provides good yields and short reaction times.31 Preparation of dichloroaniline analogues 22–24 was realized from the corresponding isocyanate and ethyl chloroformate. The synthesis of amide analogues 25–28 was prepared from 2-methyl-4-nitroaniline, according to a two step procedure: acetylation and reduction under a hydrogen atmosphere. Displacement of the chloro substituent of 4 with previously synthesized anilines 5–28 in refluxing 2-propanol provided the target quinazolines 29–50 that were isolated as hydrochloride salts. | ||
| Scheme 1 Synthesis of intermediates. Reagents and conditions: (a) NaOCH3, formamide, DMF–MeOH, reflux (85%); (b) POCl3, reflux (92%); (c) RANEY® nickel, H2, CH2Cl2, ROH (80–90%); (d) RANEY® nickel, H2, CH2Cl2–THF (1/1), ROH (80–90%); (e) isocyanate, CHCl3 (70–80%); (f) ClCOOC2H5, triethylamine, THF, 0 °C at 20 °C (80%); (g) RCOCl, toluene, reflux (70–85%); (h) RANEY® nickel, H2, MeOH (80–90%); (i) anilines 5–28, 2–propanol, reflux (60–80%). | ||
Modulation of ether linkage at the 6- or 7- position of the quinazoline core with a basic or aliphatic side chain led to 53–55 according to described procedures29,32 respectively with methyl vanillate, isovanillate 51 or methyl 3,4-dihydroxybenzoate 52 as starting material (Scheme 2).
 | ||
| Scheme 2 Synthesis of intermediates 53–55 | ||
A different synthetic route for the synthesis of 67–69 is outlined in Scheme 3. The allylic protection of methyl vanillate, followed by the selective nitration procedure previously described, allowed access to compound 57, which was deprotected in TFA in the presence of LiClO4 (58). Etherification of the phenol by piperidine (59) and morpholine (60) derivatives was realized in refluxing acetone in basic conditions (Scheme 3). Chemical reduction of the nitro group with SnCl2 led to anthranilate derivatives 61 and 62, while compound 63 was directly obtained from catalytic hydrogenation of 57. Chloroquinazolines 67–69 were then synthesized using the conditions previously described.29a
 | ||
Scheme 3 Reagents and conditions: (a) BrCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, K2CO3, acetone, reflux (79%); (b) SnCl4, HNO3, CH2Cl2, −70 °C (79%); (c) LiClO4, TFA, 60 °C (75%); (d) Cl–R, K2CO3, acetone, reflux (75–85%); (e) SnCl2·2H2O, HCl, 0 °C at 60 °C (70–80%); (f) RANEY® nickel, H2, CH2Cl2–MeOH (67%); (g) formamide, ammonium formate, reflux (60–70%); (h) POCl3, reflux (80–85%). CH2, K2CO3, acetone, reflux (79%); (b) SnCl4, HNO3, CH2Cl2, −70 °C (79%); (c) LiClO4, TFA, 60 °C (75%); (d) Cl–R, K2CO3, acetone, reflux (75–85%); (e) SnCl2·2H2O, HCl, 0 °C at 60 °C (70–80%); (f) RANEY® nickel, H2, CH2Cl2–MeOH (67%); (g) formamide, ammonium formate, reflux (60–70%); (h) POCl3, reflux (80–85%). | ||
The target compounds (70–80) were obtained by nucleophilic substitution with previously synthesized ethyl(anilino)carbamates 10 and 14 in DMF in the presence of sodium hydride (40–60%) (Scheme 4).
 | ||
| Scheme 4 Nucleophilic substitution leading to compounds 70–80. Reagents and conditions: (a) anilines 10 or 14, DMF, NaH (40–60%). | ||
3. Results and discussion
Table 1 shows the enzymatic and cellular activities of compounds whose 6,7-dimethoxyquinazoline skeleton was substituted by various anilines with amide, carbamic acid ester or urea groups. Introduction of methyl or chlorine in the ortho-position of the carbamate function led to dual active inhibitors of both EGFR and VEGR-2 with in vitro IC50 values in the submicromolar range (compounds 34, 36 and 40) whereas substitution with urea (44, 46) or amide (47–50) derivatives showed reduced activity against the two enzymes. As in previous work,29 the oxygen of the carbamic acid ester plays an important role in the interaction of the compound with the binding sites of EGFR and VEGFR-2. Varying the substitution on the central phenyl ring was also studied. The para-position of carbamic acid ester is better tolerated than the meta–position (33, 35, 39 and 41) for the activity against EGFR and VEGFR. The size of bulky substituents such as ethyl or propyl carbamate (compared to methyl or butyl carbamates) is a key modification to increase kinase inhibition in both EGFR and VEGFR-2. The bi-substitution by two chlorides (44) on the aniline ring resulted in a significant decrease in activity against EGFR and VEGFR-2 caused by the steric hindrance.|
|
||||||
|---|---|---|---|---|---|---|
| Cpd | Y | Enzymatic inhibitory (IC50/μM)a,b | Proliferative inhibitory % inhibitiona or (IC50/μM)b | |||
| EGFR | VEGFR–2 | PC3 | HT29 | MCF–7 | ||
| a Cell proliferation was realized by MTS assay at 10 μM from at least three independent determinations. Higher concentrations were not used to avoid precipitation of the compounds in the culture medium. b The values are the mean ± SD of at least three independent experiments (SD < 10%). c N.D.: Not determined. | ||||||
| Iressa® | 0.07 | 14.80 | 7.40 | N.D. | 30% | |
| Vandetanib | 0.80 | 0.10 | 33% | 4.20 | 26% | |
| 1 |

|
5.70 | 1.65 | 1% | 0% | 31% |
| 2 |

|
6.90 | 5.80 | 2% | 10% | 27% |
| 29 |

|
>10 | 5.60 | 12% | 16% | 6% |
| 30 |

|
7.05 | 5.00 | 1% | 18% | 29% |
| 31 |
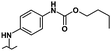
|
0.80 | 6.80 | 20% | 32% | 19% |
| 32 |

|
4.00 | 0.85 | 6% | 10% | 0% |
| 33 |

|
7.20 | 7.80 | 0% | 0% | 12% |
| 34 |
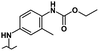
|
0.90 | 0.65 | 0% | 0% | 0% |
| 35 |
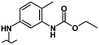
|
5.00 | 6.50 | 0% | 0% | 1% |
| 36 |
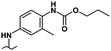
|
0.90 | 0.85 | 6% | 18% | 0% |
| 37 |

|
7.60 | 5.20 | 4% | 13% | 33% |
| 38 |
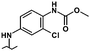
|
6.15 | 5.05 | 19% | 22% | 0% |
| 39 |

|
>10 | >10 | 3% | 0% | 0% |
| 40 |
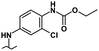
|
1.00 | 0.50 | 9.80 | 30% | 0% |
| 41 |
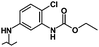
|
0.90 | 5.55 | 13% | 4% | 39% |
| 42 |

|
1.00 | 3.30 | 0% | 29% | 20% |
| 43 |
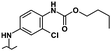
|
6.40 | 4.90 | 0% | 41% | 5% |
| 44 |

|
>10 | 9.90 | 0% | 5% | 6% |
| 45 |

|
>10 | 5.40 | 11% | 16% | 21% |
| 46 |

|
>10 | 9.20 | 0% | 3% | 0% |
| 47 |

|
>10 | >10 | 0% | 0% | 0% |
| 48 |

|
>10 | >10 | 0% | 0% | 28% |
| 49 |

|
>10 | 4.30 | 0% | 0% | 5% |
| 50 |

|
>10 | 6.80 | 0% | 0% | 0% |

Carbamic acid ester derivatives (34, 36, 40 and 42) showed less activity in the cellular assay (Table 1) than the reference compounds Iressa and Vandetanib, while other quinazolines were found to be poorly cytotoxic with IC50 >10 μM.
Replacement of one of the methoxy groups at the C-6 or C-7 position of the quinazoline core with an aminoalkyl functionality to provide a basic side chain may improve the physicochemical and pharmacological properties. We focused on the O-linked quinazoline series. We have evaluated these compounds on EAHY926 umbilical immortalized cancer cell lines, which overexpressed VEGFR-2 glycoprotein.
Table 2 shows the effect of varying the O-substituent at the 6- or 7-position on the quinazoline core. Replacement of the methoxy group by a diethylamino side chain at the 6- or 7-position of quinazoline (71, 72, 75, 76 and 80) led to a drastically reduced enzymatic activity. Replacement of the methoxy group by aminoalkyl at the 7-position of the quinazoline skeleton is reasonably well tolerated. Introduction of piperidine (72, 77) or morpholine (73, 78) side chains showed a significant decrease in activity against EGFR (IC50 > 1 μM) but increased inhibitory activity against VEGFR-2. For piperidine substituted derivatives, an antiproliferative activity was observed on HT29 (colon cancer cells; IC50 < 6 μM). Compound 80, which differs by the substitution of a butoxy side chain in the 6 position, exhibited an inhibitory effect on the growth of PC3 and EAHY926 cells (IC50 < 3 μM) but not against enzymes EGFR and VEGFR-2. Its cellular results are better than reference compounds Iressa and Vandetanib.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | X | Enzymatic inhibition (IC50/μM) a,b | Antiproliferative activity (IC50/μM)b | ||||
| EGFR | VEGFR–2 | PC3 | HT29 | MCF–7 | EAHY926 | ||||
| a The values are the mean ± SD of at least three independent experiments. b IC50 not determined because less than 50% inhibition was observed at the highest tested concentration (10 μM). Higher concentrations were not used to avoid precipitation of the compounds in the culture medium. c N.D.: Not determined. | |||||||||
| Iressa® | 0.07 | 14.80 | 7.40 | N.D. | 30% | N.D. | |||
| Vandetanib | 0.80 | 0.10 | 33% | 4.20 | 26% | 5.10 | |||
| 34 | CH3 | CH3 | CH3 | 0.90 | 0.65 | >10b | >10b | >10b | >10b |
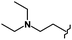
| 70 | CH3 |
|
CH3 | >10b | 3.70 | >10b | >10b | >10b | >10b |
| 71 |

|
–CH3 | CH3 | >10b | 7.20 | >10b | >10b | >10b | >10b |
| 72 | CH3 |

|
CH3 | >10b | 0.90 | >10b | 5.20 | >10b | 9.30 |
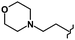
| 73 | CH3 |
|
CH3 | 8.00 | 0.40 | >10b | >10b | >10b | >10b |
| 74 | CH3 |

|
CH3 | 3.90 | 0.50 | N.D. | 5.35 | N.D. | >10b |
| 75 |

|

|
CH3 | >10b | 5.70 | >10b | 5,80 | >10b | 8.10 |
| 40 | –CH3 | –CH3 | Cl | 0.9 | 0.50 | 9.8 | >10b | >10b | >10b |
| 76 | –CH3 |

|
Cl | >10b | 3.30 | >10b | 5.40 | >10b | 6.20 |
| 77 | –CH3 |

|
Cl | >10b | 0.80 | >10b | 5.20 | 3.00 | 9.30 |
| 78 | –CH3 |

|
Cl | 8.50 | 0.30 | >10b | >10b | >10b | >10b |
| 79 | –CH3 |

|
Cl | 5.50 | 0.50 | N.D. | 9.00 | N.D. | >10b |
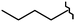
| 80 |
|

|
Cl | 9.80 | 4.80 | >10b | 2.50 | 7.10 | 2.80 |

These observations demonstrated that replacement of one of the methoxy groups (34 and 40) at the C-6 or C-7 position of the quinazoline core did not lead to a dual EGFR/VEGFR active compound while these modifications allow an increase of antiproliferative activity. Introduction of hydrophilic aminoalkyl groups at the 7 position of the quinazoline is the key modification to increase VEGFR-2 kinase inhibition while these modifications resulted in a complete loss of EGFR activity. We also investigated the inhibitory activity of several compounds substituted with a propoxy side chain (74 and 79). Compound 79 did positively impact significantly on the enzyme inhibition (IC50 < 1 μM against VEGFR–2).
4. Molecular modelling
Docking simulations were carried out in order to predict the binding mode of the best ethyl-(6-7-dimethoxy-anilinoquinazoline)-carbamate (40) into the ATP active site of EGFR and VEGFR (Fig. 3). In the ATP active site of EGFR, compound 40 interacts via H-bond interactions with Met769 (between a methionine NH and the N1 nitrogen of the quinazoline from 40), Thr766 (via a bridge between a water molecule, the OH threonine side chain and the N3 nitrogen of quinazoline from 40), Glu738 and Thr830 (with the NH nitrogen and the sp2 oxygen from the carbamate group of 40). The results are in good agreement with the biological data. | ||
| Fig. 3 Docking mode of compound 40 into the ATP-site of EGFR (left) and VEGFR-2 (right). | ||
In the ATP active site of VEGFR-2, the quinazoline core adopts essentially volumes and conformations in the same directions. We observed interactions between the CO carbamate group of 40 and the backbone of Asp1046 and interactions between the NH carbamate group and the Lys866–Glu883 salt bridge. H-bond interaction with Cys919 (between a cystein NH and the N1 nitrogen of quinazoline from 40) was established by these molecular modelling studies. Introduction of amide or urea instead of carbamate with different substituents, involved the loss of interactions with these active sites and this explains the low affinity of this class of compounds (data not shown).
We have shown that the carbamate group substituted on its aryl core is a good entity to interact in these two active sites, EGFR and VEGFR-2.
5. Conclusion
In summary, investigation of SAR around the lead carbamic acid methyl ester 2 revealed a range of potent dual EGFR/VEGFR-2 inhibitors. The substitution by a halide or methyl substituent on the anilino group of the carbamic acid ester increased the in vitro inhibition against both EGFR and VEGFR-2. Varying the methoxy groups on the quinazoline scaffold by addition of a basic side chain led to a loss of activity against EGFR and a better affinity against VEGFR-2 (72 and 77) with an increase of inhibitory activity on HT29 cancer cells. This study provides a new promising scaffold for further development of new anticancer drugs targeting EGFR and VEGFR tyrosine kinases. Some compounds, particularly compound 80, exhibited a significant antiproliferative activity on cancer cells not in relation with EGF or VEGF inhibition. Consequently, further pharmacological studies are required to determine their exact mechanism of action.Acknowledgements
The authors are grateful to the “Ligue Nationale Contre le Cancer” (Comity of Pas–de–Calais–France) for its financial support. We are grateful to Dr Nicolas Lebegue for many discussions on the drug design of inhibitors targeting it.Bibliographic references and notes
- A. Levitzki, Acc. Chem. Res., 2003, 36, 462–469 CrossRef CAS.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS.
- P. Cohen, Eur. J. Biochem., 2001, 268, 5001–5010 CrossRef CAS.
- E. Zwick, J. Bange and A. Ullrich, Trends Mol. Med., 2002, 8, 17–22 CrossRef CAS.
- P. Traxler, G. Bold, E. Buchdunger, G. Caravatti, P. Furet, P. Manley, T. O'Reilly, J. Wood and J. Zimmermann, Med. Res. Rev., 2001, 21, 499–512 CrossRef CAS.
- R. Jr. Roskoski, Biochem. Biophys. Res. Commun., 2004, 319, 1–11 CrossRef.
- R. S. Herbst, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, 21–26 CrossRef CAS.
- T. B. Deb, L. Su, L. Wong, E. Bonvini, A. Wells, M. David and G. R. Johnson, J. Biol. Chem., 2001, 276, 15554–15560 CrossRef CAS.
- Y. Yarden and M. X. Sliwkowski, Nat. Rev. Mol. Cell. Biol., 2002, 2, 127–137 CrossRef.
- R. Bianco, T. Gelardi, V. Damiano, F. Ciardiello and G. Tortora, Int. J. Biochem. Cell Biol., 2007, 39, 1416–1431 CrossRef CAS.
- N. Rahimi, Exp. Eye Res., 2006, 83, 1005–1016 CrossRef CAS.
- R. Jr. Roskoski, Biochem. Biophys. Res. Commun., 2008, 375, 287–291 CrossRef.
- M. Lohela, M. Bry, T. Tammela and K. Alitalo, Curr. Opin. Cell Biol., 2009, 21, 154–165 CrossRef CAS.
- N. M. Pandaya, N. S. Dhalla and D. D. Santani, Vasc. Pharmacol., 2006, 44, 265–274 CrossRef.
- K. Holmes, O. L. Roberts, A. M. Thomas and M. J. Cross, Cell. Signalling, 2007, 19, 2003–2012 CrossRef CAS.
- A. Maity, N. Pore, J. Lee, D. Solomon and D. M. O'Rourke, Cancer Res., 2000, 60, 5879–5888 CAS.
- D. N. Amin, D. R. Bielenberg, E. Lifshits, J. V. Heymach and M. Klagsbrun, Microvasc. Res., 2008, 76, 15–22 CrossRef CAS.
- C. Bianco, E. Giovannetti, F. Ciardiello, V. Mey, S. Nannizzi, G. Tortora, T. Troiani, F. Pasqualetti, G. Eckhardt, M. de Liguoro, S. Ricciardi, M. Del Tacca, D. Raben, L. Cionini and R. Danesi, Clin. Cancer Res., 2006, 12, 7099–7107 CrossRef CAS.
- C. Le Tourneau, S. Faivre and E. Raymond, Cancer Treat. Rev., 2008, 34, 37–48 CrossRef CAS.
- J. Capdevilla, E. Elez, T. Macarulla, F. J. Ramos, M. Ruiz–Echarri and J. Tabernero, Cancer Treat. Rev., 2009, 35, 354–363 CrossRef.
- N. Ferrara, K. J. Hillan and W. Novotny, Biochem. Biophys. Res. Commun., 2005, 333, 328–335 CrossRef CAS.
- M. Ranson and S. Wardell, J. Clin. Pharm. Ther., 2004, 29, 95–103 CrossRef CAS.
- F. A. Shepherd, R. J. Pereira, T. Ciuleanu, E. H. Tan, V. Hirsh, S. Thongprasert, D. Campos, S. Maoleekoonpiroj, M. Smylie, R. Martins, M. van Kooten, M. Dediu, B. Findlay, D. Tu, D. Johnston, A. Bezjak, G. Clark, P. Santabárbara and L. Seymour, N. Engl. J. Med., 2005, 353, 123–132 CrossRef CAS.
- B. Escudier, T. Eisen, W. M. Stadler, C. Szczylik, S. Oudard, M. Siebels, S. Negrier, C. Chevreau, E. Solska, A. A. Desai, F. Rolland, T. Demkow, T. E. Hutson, M. Gore, S. Freeman, B. Schwartz, M. Shan, R. Simantov and R. M. Bukowski, N. Engl. J. Med., 2009, 49, 332–334.
- M. H. Cohen, G. A. Williams, R. Sridhara, G. Chen and R. Pazdur, Oncologist, 2003, 8, 303–306 CrossRef CAS.
- F. Ciardiello, R. Bianco, R. Caputo, R. Caputo, V. Damiano, T. Troiani, D. Melisi, F. De Vita, S. De Placido, A. R. Bianco and G. Tortora, Clin. Cancer Res., 2004, 10, 784–793 CrossRef CAS.
- S. R. Wedge, D. J. Ogilvie, M. Dukes, J. Kendrew, R. Chester, R. Boffey, P. Valentine, J. O. Curwen, H. L. Musgrove, G. A. Graham, P. Thomas, B. Curry, P. F. Wadsworth and L. F. Hennequin, Cancer Res., 2002, 62, 4645–4655 CAS.
- P. Dent, D. T. Curiel, P. B. Fisher and S. Grant, Drug Resist. Updates, 2009, 12, 65–73 CrossRef CAS.
- (a) A. Garofalo, L. Goossens, A. Lemoine, A. Farce, Y. Arlot and P. Depreux, J. Enzyme Inhib. Med. Chem., 2010, 25, 158–171 Search PubMed; (b) M. Desroses, G. Laconde, P. Depreux and J.-P. Hénichart, Synthesis of unsymmetrical dialkoxy quinazolines, Org. Prep. Proced. Int., 2004, 36, 445–452 CrossRef CAS.
- T. Furuta, T. Sakai, T. Senga, T. Osawa, K. Kubo, T. Shimizu, R. Suzuki, T. Yoshino, M. Endo and A. Miwa, J. Med. Chem., 2006, 49, 2186–2192 CrossRef CAS.
- A. Garofalo, L. Goossens, N. Lebegue and P. Depreux, Synth. Commun., 2010 Search PubMed , in press.
- A. Telliez, M. Desroses, N. Pommery, O. Briand, A. Farce, G. Laconde, A. Lemoine, P. Depreux and J. P. Hénichart, ChemMedChem, 2007, 2, 318–332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0md00183j |
| This journal is © The Royal Society of Chemistry 2011 |