DOI:
10.1039/C0MB00216J
(Review Article)
Mol. BioSyst., 2011,
7, 38-47
Received
30th September 2010
, Accepted 21st October 2010
First published on 19th November 2010
Introduction
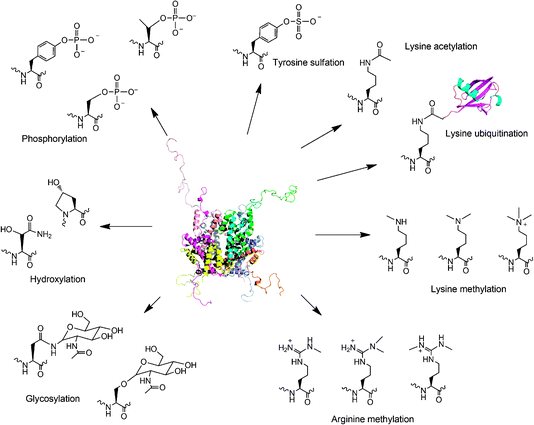
With few exceptions, the genetic code of all known organisms specifies the same 20 amino acid building blocks. However, it is clear that many proteins require additional chemical groups beyond what 20 amino acid building blocks can provide to carry out their native functions.1,2 Many of these chemical groups are installed on proteins through covalent posttranslational modifications of amino acid side chains. These modifications greatly expand the coding capacity especially of eukaryotic genomes and lead to proteomes two to three orders of magnitude more complex than the encoding genomes would predict. Prominent posttranslational modifications include serine, threonine, and tyrosine phosphorylation, tyrosine sulfation, lysine acetylation, lysine and arginine methylation, lysine ubiquitination, glycosylation, hydroxylation, etc. (Fig. 1). These modifications grant proteins with expanded opportunities for catalysis, regulating signal transduction, controlling gene expression, integration of information at many metabolic intersections, and alternation of cellular locations. Studies of posttranslational modifications will provide fundamental understanding of many cellular processes but have been greatly hampered by the difficulty to attain homogenously modified proteins. In cells, most posttranslational modifications are reversible and delicately regulated. It is usually impossible to drive a protein completely to a modified form.3 Separation of the modified form from the unmodified counterpart is generally difficult because most posttranslational modifications provide small size changes to proteins and do not dramatically change the proteins' biophysical properties. For a lot of eukaryotic proteins, multiple modification sites and multiple modification types coexist, adding a great complexity not only to functional investigation of these proteins but also to separation of a specific modified form from a largely heterogeneous protein pool in their natural sources.4–6 It is obvious that obtaining a homogeneously modified protein poses a big challenge for efforts to elucidate the precise regulatory role of a posttranslational modification. To resolve this major obstacle, several approaches have been developed to synthesize proteins with defined posttranslational modifications. A straightforward method is to directly incubate a target protein together with its corresponding enzyme or coexpress the two proteins together in living cells. Modified protein forms for both histones and p53 have been synthesized in this way.7,8 However, the separation of a modified form from its original protein is a big challenge for aforementioned reasons. For large-size modifications such as ubiquitination,9,10 the separation is readily achievable. But small-size modifications including acetylation and methylation often lead to modified protein forms inseparable from their unmodified precursors. Although antibodies have been used to isolate some modified protein forms, not every modified protein form has a specific antibody that is readily available.11,12 Substrate promiscuity of an enzyme (e.g. CBP/p300 catalyses acetylation of p53 at more than 8 sites) may lead to formation of heterogeneous protein forms that are hard to be isolated from each other even using antibodies.3,7,13–15 One additional problem is that not every protein modification has an identified corresponding enzyme. The advances of proteomic techniques have led to the discovery of thousands of protein modifications. But identifications of enzymes responsible for these modifications have largely fallen behind. Alternative approaches for synthesis of proteins with defined modifications include selective chemical modifications of proteins, native chemical ligation, and the genetic NAA incorporation technique. Shokat and coworkers developed a semi-synthetic method in which a protein cysteine selectively reacts with an alkyl halide to form a Nε-mono-, Nε,Nε-di- or Nε,Nε,Nε-trimethyllysine analog.16,17 Recently, Cole and coworkers extended this method to synthesize proteins with a lysine acetylation mimic.18 Modified histones synthesized in this way function similarly as their native counterparts and have been applied to study regulatory roles of histone methylation in chromatin.17 One limitation associated with the cysteine modification approach is that only a few posttranslational modification mimics can be obtained. In addition, non-targeted cysteine residues in a protein have to be mutated to other residues to avoid nonspecific modifications. Another semi-synthetic method, native chemical ligation together with its extension, expressed protein ligation has also been applied to synthesize proteins with posttranslational modifications and their mimics.19 This method has been extensively used to synthesize modified histones for investigating epigenetic regulation of chromatin. Using this method, an elegant study carried out by Muir and coworkers demonstrated that H2B ubiquitination directly stimulates intranucleosomal methylation mediated by hDot1L.20 Although powerful and straightforward, native chemical ligation requires synthesis of large quantities of peptide thioesters and is still challenging for most molecular biology and biochemistry labs. In addition, both the cysteine modification method and native chemical ligation are not applicable in living cells. Modified proteins have to be synthesized in vitro and introduced into cells by invasive approaches such as microinjection for their cellular function studies. In the past several years, Schultz, Yokoyama, Chin and Liu groups have focused on applying the genetic NAA incorporation approach to synthesize proteins with defined posttranslational modifications. One advantage of the NAA incorporation technique is that proteins with defined posttranslational modifications can be directly produced in living cells. This allows the direct characterization of phenotypic consequences of protein posttranslational modification events. Another advantage of the NAA incorporation technique is its simplicity. It combines the recombinant DNA technique and relatively simple organic synthesis. Most molecular biology and biochemistry groups can easily adopt it. In addition, genetic incorporation of a NAA is site-specific regardless of the incorporation site or protein size. It is an optimal choice for synthesis of large proteins with posttranslational modifications in internal sites, a process that is generally cumbersome for native chemical ligation. This review primarily focuses on applications of the genetic NAA incorporation approach to synthesize proteins with defined posttranslational modifications. We apologize for not citing most outstanding work related to other techniques.
The genetic NAA incorporation approach
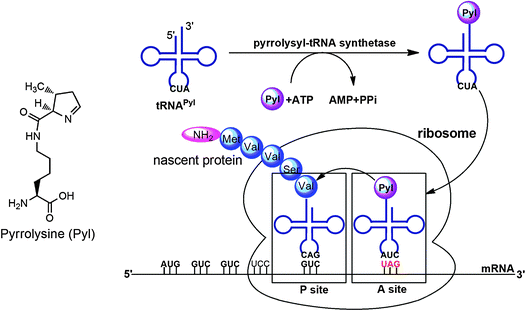
A general method for genetic incorporation of a NAA in living cells was developed by Furter, Schultz and coworkers.21–24 This method relies on the read-through of an in-frame amber (UAG) stop codon in mRNA by an amber suppressor tRNA (tRNACUA) that is specifically acylated with a NAA by an evolved aminoacyl-tRNA synthetase (aaRS). This system is equivalent to the naturally occurring pyrrolysine (Pyl) incorporation machinery discovered in some methanogenic archaea and a Gram positive bacterium Desulfitobacterium hafniense.25,26 In these organisms, Pyl is cotranslationally inserted into proteins by an in-frame amber codon (Fig. 2). Suppression of this amber codon is mediated by the Pyl amber suppressor tRNA (tRNAPylCUA), which has a CUA anticodon and is acylated with Pyl by pyrrolysyl-tRNA synthetase (PylRS). The PylRS-tRNAPylCUA pair in these organisms is orthogonal to and therefore does not cross-interact with other aaRS-tRNA pairs in cells, ensuring the fidelity of the Pyl incorporation at amber codon. Similarly to the Pyl incorporation machinery, an orthogonal aaRS-tRNACUA pair can be developed in which an aaRS is evolved to specifically charge its cognate tRNACUA with a NAA and used to site-specifically incorporate this NAA into a protein at amber codon with high fidelity and efficiency in living cells. Since both the orthogonal aaRS-tRNACUA pair and the mRNA with a defined amber codon are genetically encoded, a protein incorporated with a NAA can be expressed by just simply growing cells in a medium supplemented with the NAA. Using an evolved MjTyrRS-MjtRNATyrCUA pair that was derived from Methanococcus jannaschii tyrosyl-tRNA synthetase-tRNATyr pair, Schultz and coworkers successfully achieved genetic incorporation of sulfotyrosine into proteins in E. coli and synthesized hirudin, a naturally occurring sulfated peptide secreted by leeches.27,28 This was the first successful demonstration that proteins with defined posttranslational modifications can be directly synthesized cotranslationally. Following this outstanding work, proteins with other modifications or their mimics have been synthesized using evolved MjTyrRS-MjtRNATyrCUA pairs and PylRS-tRNAPylCUA pairs, as detailed in the following sections.
The posttranslational tyrosine sulfation is an enzymatic process catalyzed by two tyrosylprotein sulfotransferases, TPST1 and TPST2 in human.29 The sulfation occurs in the Golgi apparatus before newly synthesized proteins are sorted and shipped to their correct destinations. The natural occurrence of tyrosine sulfation was first discovered in 1954 in a peptide derived from bovine fibrinogen.30 In the following four decades, sulfated peptide hormones and proteins were only sporadically identified. Owing to the emergence of new proteomic tools, many proteins with sulfated tyrosine residues have been found in the past decade, revealing that tyrosine sulfation is prevalent in human cells. It was also recently discovered that tyrosine sulfation plays a crucial role in human physiology and pathology. Numerous secreted and integral membrane proteins in human contain sulfated tyrosine residues. Among these proteins, CCR5 and CXCR4 are probably the most prominent because of their roles in the entry of human immunodeficiency virus type 1 (HIV-1) into host cells.31,32 Entry of HIV-1 requires its gp120 envelop glycoprotein to bind to two cell-surface receptors, CD40 and a coreceptor, either CCR5 or CXCR4. The interaction of either CCR5 or CXCR4 with gp120 requires sulfation of several tyrosine residues at its N-terminus. Coincidently, several human monoclonal HIV-1 antibodies also contain sulfated tyrosine residues and target the same coreceptor binding site on gp120.33,34 Other human proteins containing tyrosine sulfation include P-selectin glycoprotein ligand-1, most chemokine receptors, and some other G-protein coupled receptors.35,36 It is evident that tyrosine sulfation in these proteins is critical for their interactions with binding partners. Although tyrosine sulfation is important in both physiology and pathology of human, biochemical studies of this modification is very difficult to pursue because of low expression levels of most sulfated proteins. Synthesis of sulfated peptides using organic chemistry is also challenging because of the instability of the sulfate group.37 A major technology breakthrough for biosynthesis of sulfated proteins was achieved in 2006. Using an evolved MjTyrRS-MjtRNATyrCUA pair, Schultz and coworkers successfully incorporated sulfotyrosine into proteins at amber mutation sites in E. coli and achieved overexpression of the naturally occurring hirudin. Hirudin is secreted by leeches and contains 65 residues.28 Its Tyr63 is naturally sulfated. Hirudin is the most potent naturally occurring direct thrombin inhibitor known and is used clinically as an anticoagulant. It exhibits a Ki towards thrombin of approximately 25 fM. Although the therapeutic form of hirudin have been produced recombinantly in E. coli and yeast, the recombinant hirudin is lacking sulfation at Tyr63 and exhibits a Ki of approximately 300 fM. The activity difference between the natural form and the recombinant form clearly arises from the additional sulfate group on the natural form. It was proposed that the enhanced binding of the natural hirudin to thrombin is due to the salt bridge interaction between the sulfate group and K81H of thrombin, but there was no direct structural evidence to support the claim. The natural hirudin is extracted from leech heads in very low yields, causing a huge obstacle to directly crystallize its complex with thrombin. Although structures of synthetic sulfated C-terminal peptide fragments of hirudin bound to thrombin were solved, they either showed distorted structures around the sulfate group or did not reveal direct interactions between the sulfate group and thrombin. Since the natural hirudin could be simply expressed in E. coli, a large quantity of this protein was later obtained and used directly to undergo X-ray crystallographic studies of its interactions with thrombin. The structure determined clearly shows a sulfate group covalently linked to Tyr63 and revealed a network of hydrogen bonding/salt bridge interactions between the sulfate group and thrombin (Fig. 3).38 This work unprecedentedly demonstrated potential applications of the genetic NAA incorporation approach in basic biochemistry studies of protein posttranslational modifications. Given the prevalence of tyrosine sulfation in higher organisms, it is expected that the genetic sulfotyrosine incorporation will be used broadly to understand many basic biological questions related to tyrosine sulfation. In addition to its applications in basic biochemistry studies, the genetic sulfotyrosine incorporation may also be applied to evolve synthetic antibodies with enhanced binding potentials toward antigens. By coupling phage display and the genetic sulfotyrosine incorporation, Schultz and coworkers recently identified a single-chain variable fragment that shows a 10 fold higher binding potential toward gp120 than the monoclonal antibody 412d.27
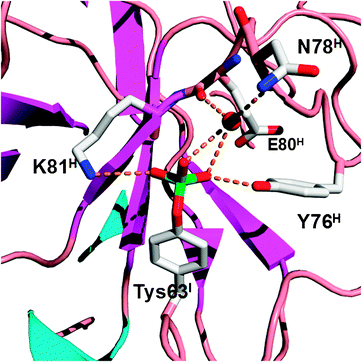 |
| | Fig. 3 Structure of sulfotyrosine (Tys) and amino acid residues that directly or indirectly interact with the sulfate group of Tys in the hirudin–thrombin complex. The structure is derived from the PDB entry: 2PW8. The superscripts in labels indicate which chains the residues are located at (I: hirudin; H: the heavy chain of thrombin). | |
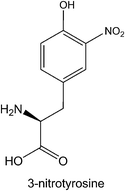
Protein tyrosine nitration is a nonenzymatic covalent modification, introducing a nitro group at one of the ortho carbons of the tyrosine phenolic ring. This modification primarily arises from reactive nitrosative species such as peroxynitrite that are generated by nitric oxide. The addition of a nitro group confers particular physiological properties to tyrosine such as a much lower pKa at the corresponding proteins. The impact of tyrosine nitration may affect protein function and structure, and change the rate of proteolytic degradation of nitrated proteins.45,46 Many recent studies have shown that tyrosine nitration is a pivotal physiological event implicated in numerous biological processes and a number of diseases such as cancer, neurodegenerative diseases, and cardiovascular injury.47 The availability of a method for synthesis of proteins with defined tyrosine nitration is critical to elucidate detrimental effects of most tyrosine nitration events. Although protein tyrosine nitration can be achieved by chemically modifying an expressed protein with tetranitromethane in ambient conditions, this modification is not selective and preferably nitrates most solvent accessible tyrosine residues.48 Alternatively, a 3-nitrotyrosine can be selectively incorporated into a protein at then amber codon using the genetic NAA incorporation approach. Chin and coworkers recently showed that proteins with defined tyrosine nitration can be expressed to large quantities in E. coli using evolved MjTyrRS-MjtRNATyrCUA pairs.49 Human manganese superoxide dismutase incorporated with 3-nitrotyrosine at Tyr34 synthesized using this method displays a diminished activity which is only 3% of that for the wild-type enzyme. Manganese superoxide dismutase is known to undergo near-quantitative nitration on Tyr34 in cardiovascular disease and aging and is also nitrated in acute and chronic inflammatory processes in both animal models and human diseases. The work by Chin and coworkers clearly showed a deleterious effect of a single nitration to an enzyme's activity. Since many other proteins such as p53 and tau undergo tyrosine nitration in neurodegenerative disease and atherosclerosis, applications of the genetic 3-nitrotyrosine incorporation to understand basic functions of tyrosine nitration in these proteins are expected.50
Protein lysine acetylation is a reversible enzymatic process catalyzed by histone acetyltransferases and histone deacetylases (HDACs). Originally identified in histones 40 years ago, lysine acetylation is now known to occur in more than 80 transcription factors such as p53 and p300/CBP, many other nuclear regulators, and various cytoplasmic proteins.6 As a result, lysine acetylation is both crucial for the transcription regulation in the nucleus and important for regulating different cytoplasmic processes, including cytoskeleton dynamics, energy metabolism, endocytosis, autophagy, and even signaling from the plasma membrane. Recently the histone code that involves histone acetylation has garnered considerable attention for its role in regulating the chromatin structure and gene expression. To date, biochemical studies of histone acetylation have mainly been attributed to native chemical ligation. Recently genetic incorporation of Nε-acetyllysine in E. coli for synthesis of proteins with lysine acetylation was also developed. The incorporation of Nε-acetyllysine takes advantage of an evolved PylRS-tRNAPylCUA pair originally from Methanosarcina barkeri.51 Using this evolved pair, Chin and coworkers expressed homogenous histone proteins with acetylation at different sites and reconstituted nucleosomes bearing defined acetylated lysine residues. Studies of H3 Lys56 acetylation revealed that this modification does not directly affect the compaction of chromatin, has modest effects on remodeling by SWI/SNF and RSC, and increases DNA breathing.52 A most recent application of this technique was to elucidate the regulatory role of Lys125 acetylation in cyclophilin A (CypA), a ubiquitous cis–trans prolyl isomerase. CypA has a key role in immunity and is required for effective HIV-1 replication in host cells. Chin & James showed that Lys125 acetylation markedly inhibits CypA catalysis of cis to trans isomerization and stabilizes cis rather than trans forms of the HIV-1 capsid.53 This modification also antagonizes the immunosuppressive effects of cyclosporine by inhibiting cyclosporine binding and calcineurin inhibition. Given that a lot of proteins including all four histone proteins, p53, p300/CBP, PGC-1α, and RIP140 contain multiple lysine acetylation sites, one intriguing interest is to study regulatory roles of concomitant lysine acetylation at multiple sites of a single protein.6 To this end, our group has extended the genetic Nε-acetyllysine incorporation technique to incorporate three Nε-acetyllysine residues into a single green fluorescent protein in E. coli.54 This was achieved by overexpressing a truncated large ribosomal protein L11 to enhance amber suppression in E. coli. In the future, we will apply this technique to study concomitant lysine acetylation at multiple sites in four histone proteins. One advantage of the PylRS-tRNAPylCUA pair is its orthogonality in yeast and mammalian cells. This makes it possible to apply the evolved PylRS-tRNAPylCUA pair specific for Nε-acetyllysine directly in eukaryotic cells for cellular functional analysis of certain lysine acetylation events. However, lysine acetylation is a reversible process. After genetic incorporation of Nε-acetyllysine into proteins in eukaryotic cells, its deacetylation by HDACs is anticipated. This will certainly complicate the in vivo functional analysis of lysine acetylation events. To resolve this obstacle, we designed an unhydrolysable Nε-acetyllysine analog, 2-amino-8-oxononanoic acid and took advantage of the evolved PylRS-tRNAPylCUA pair specific for Nε-acetyllysine to incorporate it into proteins.55 Further tests need to be carried out to assess whether this analog can closely mimic Nε-acetyllysine in mammalian cells.
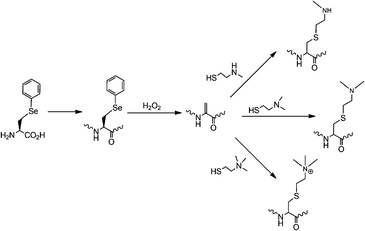
Protein lysine methylation, a reversible enzymatic process catalyzed by histone methyltransferases and histone demethylases, was also originally discovered in histones.56 It represents one of the most important posttranslational modifications and is crucial in modulating chromatin-based transcriptional control and shaping inheritable epigenetic programs in the eukaryotic cells.57 Similar to lysine acetylation, lysine methylation was also found in non-histone transcription factors such as p53 and pCAF in the nucleus.58–61 A recent discovery of histone methyltransferase Ezh2 in the cytosol of various mouse and human T cells has led to expectation that lysine methylation is also prevalent in the cytosol.62 There are three lysine methylation patterns, monomethylation, dimethylation and trimethylation, which may serve different regulatory roles.63 As evidenced in the literature, distinctive histone methyltransferases are responsible for these three methylation patterns. So far, studies of histone lysine methylation have been attributed to native chemical ligation.64 But several technological breakthroughs in the past one year could drive applications of the genetic NAA incorporation approach in this important research. Given the structural similarity between Nε-methyllysine and Nε-acetyllysine, one would think it is possible to evolve the PylRS-tRNAPylCUA pair for genetic incorporation of Nε-methyllysine. However, creating a synthetase that recognizes Nε-methyllysine but discriminates against lysine is difficult since the two amino acids differ only by a single methyl group. For this reason, we and other two groups developed indirect biosynthesis methods for production of proteins with lysine monomethylation as shown in Fig. 5. Directly using the wild-type PylRS-tRNAPylCUA pair from Mathanosarsina barkeri, Chin and coworkers managed to incorporate Nε-Boc-Nε-methyllysine site-specifically at Lys9 of histone H3.65 The deprotection of Boc from the incorporated Nε-Boc-Nε-methyllysine to recover Nε-methyllysine was achieved using 2% TFA. Although the deprotection condition denatures proteins, small proteins such as histones can be easily refolded afterward. H3K9me1 (monomethylation at Lys 9 of H3) synthesized in this way can be specifically recognized by heterochromatin protein 1, a chromodomain protein that binds H3K9me1 but not unmethylated H3. We and the Schultz group developed alternative methods that undergo deprotection to recover Nε-methyllysine at mild conditions.66,67 Both groups independently evolved the PylRS-tRNAPylCUA pair for genetic incorporation of Nε-(o-nitrobenzylcarbamoyl)-Nε-methyllysine into proteins in E. coli. The o-nitrobenzylcarbamoyl group is photolytic. Deprotection of this group from the incorporated NAA to recover Nε-methyllysine can be simply achieved using UV light. This photo deprotection process (360 nm UV light, 30 min, and room temperature) is very mild to proteins and has been performed on proteins incorporated with Nε-(o-nitrobenzylcarbamoyl)-Nε-methyllysine in living mammalian cells. We have also achieved the incorporation of Nε-Cbz-Nε-methyllysine into proteins using an evolved PylRS-tRNAPylCUA pair. Deprotection of Cbz to recover Nε-methyllysine in mild conditions is in theory feasible by catalytic hydrogenation but has not been achieved yet in practice. Another protected Nε-methyllysine that has been genetically incorporated into proteins is Nε-allylcarbamoyl-Nε-methyllysine. Its incorporation was achieved using a modified PylRS-tRNAPylCUA pair. Deprotection of the allylcarbamoyl group can be carried out using chloro-pentamethylcyclopentadienyl-cyclooctadiene-ruthenium(II) ([Cp*Ru(cod)Cl]) under conditions compatible with living cells.68 Besides the direct installation of Nε-methyllysine in proteins, Schultz and coworkers also developed a method to site-specifically install a Nε-methyllysine analog into proteins. This method takes advantage of the unique chemical reactivity of phenylselenocysteine that can be genetically incorporated into proteins in E. coli using evolved MjTyrRS-MjtRNATyrCUA pairs.69 After its incorporation into proteins, phenylselenocysteine can be efficiently converted to dehydroalanine under relatively mild conditions, followed by Michael addition reaction with a corresponding thiol-containing nucleophilic compound to form a Nε-methyllysine analog (Fig. 6).70 Using this method, histone H3 with a Nε-methyllysine analog at Lys9 was homogenously synthesized. The same method has also been extended to synthesize proteins incorporated with Nε,Nε-dimethyllysine and Nε,Nε,Nε-trimethyllysine analogs. Although the oxidation of phenylselenocysteine to dehydroalanine requires a high concentration of H2O2 that also modifies cysteine and methionine residues, this method is a very useful tool to study proteins in which mutating cysteine and methionine to other residues does not affect protein functions.
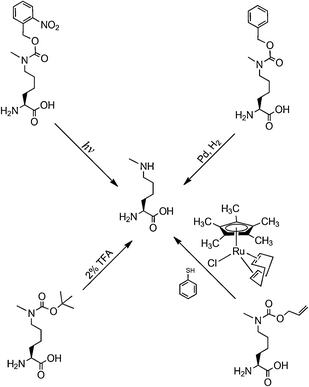 |
| | Fig. 5
N
ε
-methyllysine precursors and their deprotection. | |
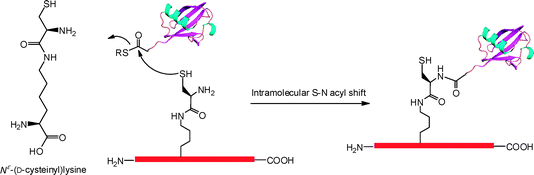 |
| | Fig. 7 Genetic incorporation of Nε-(D-cysteinyl)lysine that undergoes expressed protein ligation to crosslink with a ubiquitin mol. | |
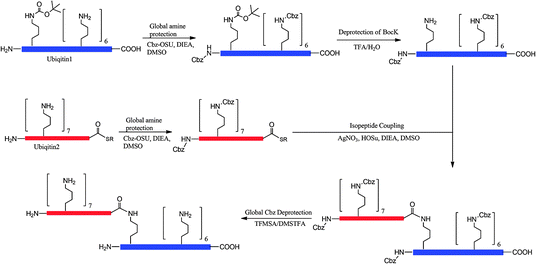 |
| | Fig. 8 Genetic incorporation of Nε-Boc-lysine and its application to synthesize a diubiquitin protein by GOPAL. | |
Challenges and future directions
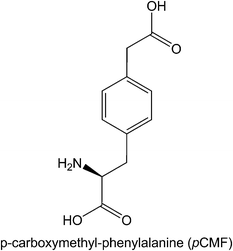
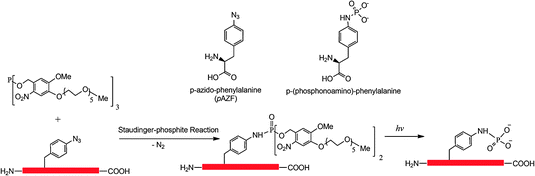
Although considerable efforts have made several methods available for synthesis of proteins with defined posttranslational modifications and their mimics using the genetic NAA incorporation approach, challenges still exist in this research area. Both sulfotyrosine and phosphotyrosine mimics can only be incorporated into proteins in E. coli. Studies of cellular functions of these modifications require the use of invasive methods to introduce them to their eukaryotic cell hosts. A method that allows genetic incorporation of these NAAs directly into eukaryotic cells is necessary for functional studies of these modifications in cells for which microinjection is not readily available. We have recently demonstrated that the PylRS-tRNAPylCUA pair can be evolved for genetic incorporation of phenylalanine derivatives (Wang and Liu, unpublished data), p-iodophenylalanine and p-bromophenylalanine, and will evolve this pair for genetic incorporation of sulfotyrosine and pCMF. Since the PylRS-tRNAPylCUA pair is orthogonal in both yeast and mammalian cells, direct applications of the evolved pairs in these cells will allow synthesizing proteins with defined tyrosine sulfation and phosphorylation mimics for their cellular function analysis. So far we have achieved genetic incorporation of Nε-methyllysine using indirect methods. Despite the efforts of other groups and us, direct incorporation of Nε-methyllysine is not successful. We may argue that it is difficult to evolve an aminoacyl-tRNA synthetase that thermodynamically favors the binding of Nε-methyllysine rather than lysine. But another explanation is also possible. Nε-methyllysine may have a difficulty to be transported into E. coli cells by ABC transporters. Since no NAA with a positively charged side chain has been evolved so far, we cannot rule out this possibility. We have tried to evolve PylRS mutants specific for Nε,Nε-dimethyllysine and Nε,Nε,Nε-trimethyllysine but no positive clones have been identified. We think a thorough investigation of membrane transportation of these NAAs is necessary before further efforts are carried out. Since arginine also undergoes methylation with several distinctive patterns,79,80 an interesting question is whether we could evolve an aaRS-tRNA pair for genetic incorporation of methylated arginines. Given the structure similarity between lysine and arginine, it is highly possible to evolve PylRS mutants that are specific for protected or unprotected methylated arginines. Another challenge in this research area is the installation of multiple different modifications to proteins. Concomitant modifications in one protein are prevalent in cells. It is an interesting question whether these modifications crosstalk to each others, reinforce each others' effects, or counteract each others' effects. To synthesize a protein with two different modifications, two orthogonal aaRS-tRNA pairs and two different blank codons have to be used. Recently, we and Chin et al. independently developed two systems for genetic incorporation of two different NAAs.81,82 The method developed by Chin et al. uses one amber codon and one quadruple AGGA codon to code two different NAAs. Our method uses one amber codon and one ochre codon. Two orthogonal pairs in both systems were derived from the MjTyrRS-MjtRNATyrCUA pair and the PylRS-tRNAPylCUA pair. These two methods make it possible to synthesize a protein containing one sulfotyrosine/phosphotyrosine mimic and one Nε-acetyllysine/Nε-methyllysine. However, it will require developing an additional aaRS-tRNA pair orthogonal to the PylRS-tRNAPylCUA pair for synthesis of a protein containing both Nε-acetyllysine and Nε-methyllysine. Searching additional orthogonal aaRS-tRNA pairs may be necessary.
In summary, multiple methods based on the genetic NAA incorporation approach have been developed for synthesis of proteins with defined posttranslational modifications and their mimics. We expect the intrinsic simplicity of these methods will promote their adoption by many scientists and accelerate our understanding of protein posttranslational modifications.
Acknowledgements
We thank Dr Zhiyong Wang for critically reading the manuscript. Financial support was provided by Texas A&M Chemistry Department and Welch grant A-1715.
References
-
C. T. Walsh, Posttranslational Modification of Proteins: Expanding Nature's Inventory, Roberts & Company Publishers, Englewood, CO, 2005 Search PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Jr., Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS.
- J. Luo, M. Li, Y. Tang, M. Laszkowska, R. G. Roeder and W. Gu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2259–2264 CrossRef CAS.
- M. Jansson, S. T. Durant, E. C. Cho, S. Sheahan, M. Edelmann, B. Kessler and N. B. La Thangue, Nat. Cell Biol., 2008, 10, 1431–1439 CrossRef CAS.
- J. A. Latham and S. Y. Dent, Nat. Struct. Mol. Biol., 2007, 14, 1017–1024 CrossRef CAS.
- X. J. Yang and E. Seto, Mol. Cell, 2008, 31, 449–461 CrossRef CAS.
- W. Gu and R. G. Roeder, Cell (Cambridge, Mass.), 1997, 90, 595–606 CrossRef CAS.
- B. Dastugue, L. Tichonicky and J. Kruh, Biochimie, 1972, 54, 1435–1441 CrossRef CAS.
- L. Hicke, Nat. Rev. Mol. Cell Biol., 2001, 2, 195–201 CrossRef CAS.
- J. S. Thrower, L. Hoffman, M. Rechsteiner and C. M. Pickart, EMBO J., 2000, 19, 94–102 CrossRef CAS.
- A. J. Hoffhines, E. Damoc, K. G. Bridges, J. A. Leary and K. L. Moore, J. Biol. Chem., 2006, 281, 37877–37887 CrossRef CAS.
- L. G. Puente and L. A. Megeney, Methods Mol. Biol., 2008, 424, 365–372 CAS.
- R. H. Goodman and S. Smolik, Genes Dev., 2000, 14, 1553–1577 CAS.
- N. A. Barlev, L. Liu, N. H. Chehab, K. Mansfield, K. G. Harris, T. D. Halazonetis and S. L. Berger, Mol. Cell, 2001, 8, 1243–1254 CrossRef CAS.
- Y. Tang, W. Zhao, Y. Chen, Y. Zhao and W. Gu, Cell (Cambridge, Mass.), 2008, 133, 612–626 CrossRef CAS.
- M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, Cell (Cambridge, Mass.), 2007, 128, 1003–1012 CrossRef CAS.
- X. Lu, M. D. Simon, J. V. Chodaparambil, J. C. Hansen, K. M. Shokat and K. Luger, Nat. Struct. Mol. Biol., 2008, 15, 1122–1124 CrossRef CAS.
- R. Huang, M. A. Holbert, M. K. Tarrant, S. Curtet, D. R. Colquhoun, B. M. Dancy, B. C. Dancy, Y. Hwang, Y. Tang, K. Meeth, R. Marmorstein, R. N. Cole, S. Khochbin and P. A. Cole, J. Am. Chem. Soc., 2010, 132, 9986–9987 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453, 812–816 CrossRef CAS.
- R. Furter, Protein Sci., 1998, 7, 419–426 CAS.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2004, 44, 34–66.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS.
- G. Srinivasan, C. M. James and J. A. Krzycki, Science, 2002, 296, 1459–1462 CrossRef CAS.
- S. Herring, A. Ambrogelly, C. R. Polycarpo and D. Soll, Nucleic Acids Res., 2007, 35, 1270–1278 CrossRef CAS.
- C. C. Liu, H. Choe, M. Farzan, V. V. Smider and P. G. Schultz, Biochemistry, 2009, 48, 8891–8898 CrossRef CAS.
- C. C. Liu and P. G. Schultz, Nat. Biotechnol., 2006, 24, 1436–1440 CrossRef CAS.
- M. J. Stone, S. Chuang, X. Hou, M. Shoham and J. Z. Zhu, Nat. Biotechnol., 2009, 25, 299–317 CAS.
- F. R. Bettelheim, J. Am. Chem. Soc., 1954, 76, 2838–2839 CrossRef CAS.
- K. A. Martin, R. Wyatt, M. Farzan, H. Choe, L. Marcon, E. Desjardins, J. Robinson, J. Sodroski, C. Gerard and N. P. Gerard, Science, 1997, 278, 1470–1473 CrossRef CAS.
- C. C. Huang, S. N. Lam, P. Acharya, M. Tang, S. H. Xiang, S. S. Hussan, R. L. Stanfield, J. Robinson, J. Sodroski, I. A. Wilson, R. Wyatt, C. A. Bewley and P. D. Kwong, Science, 2007, 317, 1930–1934 CrossRef CAS.
- C. C. Huang, M. Venturi, S. Majeed, M. J. Moore, S. Phogat, M. Y. Zhang, D. S. Dimitrov, W. A. Hendrickson, J. Robinson, J. Sodroski, R. Wyatt, H. Choe, M. Farzan and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2706–2711 CrossRef CAS.
- H. Choe, W. Li, P. L. Wright, N. Vasilieva, M. Venturi, C. C. Huang, C. Grundner, T. Dorfman, M. B. Zwick, L. Wang, E. S. Rosenberg, P. D. Kwong, D. R. Burton, J. E. Robinson, J. G. Sodroski and M. Farzan, Cell (Cambridge, Mass.), 2003, 114, 161–170 CrossRef CAS.
- T. Pouyani and B. Seed, Cell (Cambridge, Mass.), 1995, 83, 333–343 CrossRef CAS.
- P. P. Wilkins, K. L. Moore, R. P. McEver and R. D. Cummings, J. Biol. Chem., 1995, 270, 22677–22680 CrossRef CAS.
- T. Young and L. L. Kiessling, Angew. Chem., Int. Ed., 2002, 41, 3449–3451 CrossRef CAS.
- C. C. Liu, E. Brustad, W. Liu and P. G. Schultz, J. Am. Chem. Soc., 2007, 129, 10648–10649 CrossRef CAS.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- J. Xie, L. Supekova and P. G. Schultz, ACS Chem. Biol., 2007, 2, 474–478 CrossRef CAS.
- L. Tong, T. C. Warren, S. Lukas, J. Schembri-King, R. Betageri, J. R. Proudfoot and S. Jakes, J. Biol. Chem., 1998, 273, 20238–20242 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782–11783 CrossRef.
- R. Serwa, I. Wilkening, G. Del Signore, M. Muhlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234–8239 CrossRef CAS.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS.
- K. Yokoyama, U. Uhlin and J. Stubbe, J. Am. Chem. Soc., 2010, 132, 8385–8397 CrossRef CAS.
- F. J. Schopfer, P. R. Baker and B. A. Freeman, Trends Biochem. Sci., 2003, 28, 646–654 CrossRef CAS.
- M. Sokolovsky, J. F. Riordan and B. L. Vallee, Biochemistry, 1966, 5, 3582–3589 CrossRef CAS.
- H. Neumann, J. L. Hazen, J. Weinstein, R. A. Mehl and J. W. Chin, J. Am. Chem. Soc., 2008, 130, 4028–4033 CrossRef CAS.
- V. A. Yakovlev, A. S. Bayden, P. R. Graves, G. E. Kellogg and R. B. Mikkelsen, Biochemistry, 2010, 49, 5331–5339 CrossRef CAS.
- H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Chem. Biol., 2008, 4, 232–234 CrossRef CAS.
- H. Neumann, S. M. Hancock, R. Buning, A. Routh, L. Chapman, J. Somers, T. Owen-Hughes, J. van Noort, D. Rhodes and J. W. Chin, Mol. Cell, 2009, 36, 153–163 CrossRef CAS.
- M. Lammers, H. Neumann, J. W. Chin and L. C. James, Nat. Chem. Biol., 2010, 6, 331–337 CrossRef CAS.
- Y. Huang, W. K. Russell, W. Wan, P. J. Pai, D. H. Russell and W. Liu, Mol. Biosyst., 2010, 6, 683–686 RSC.
- Y. Huang, W. Wan, W. K. Russell, P. J. Pai, Z. Wang, D. H. Russell and W. Liu, Bioorg. Med. Chem. Lett., 2010, 20, 878–880 CrossRef CAS.
- V. G. Allfrey, R. Faulkner and A. E. Mirsky, Proc. Natl. Acad. Sci. U. S. A., 1964, 51, 786–794 CrossRef CAS.
- P. Cheung and P. Lau, Mol. Endocrinol., 2005, 19, 563–573 CAS.
- S. Chuikov, J. K. Kurash, J. R. Wilson, B. Xiao, N. Justin, G. S. Ivanov, K. McKinney, P. Tempst, C. Prives, S. J. Gamblin, N. A. Barlev and D. Reinberg, Nature, 2004, 432, 353–360 CrossRef CAS.
- J. Huang, L. Perez-Burgos, B. J. Placek, R. Sengupta, M. Richter, J. A. Dorsey, S. Kubicek, S. Opravil, T. Jenuwein and S. L. Berger, Nature, 2006, 444, 629–632 CrossRef CAS.
- J. Huang, R. Sengupta, A. B. Espejo, M. G. Lee, J. A. Dorsey, M. Richter, S. Opravil, R. Shiekhattar, M. T. Bedford, T. Jenuwein and S. L. Berger, Nature, 2007, 449, 105–108 CrossRef CAS.
- T. Masatsugu and K. Yamamoto, Biochem. Biophys. Res. Commun., 2009, 381, 22–26 CrossRef CAS.
- J. C. Nolz, T. S. Gomez and D. D. Billadeau, Trends Cell Biol., 2005, 15, 514–517 CrossRef CAS.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CrossRef CAS.
- S. He, D. Bauman, J. S. Davis, A. Loyola, K. Nishioka, J. L. Gronlund, D. Reinberg, F. Y. Meng, N. Kelleher and D. G. McCafferty, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12033–12038 CrossRef CAS.
- D. P. Nguyen, M. M. Garcia Alai, P. B. Kapadnis, H. Neumann and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 14194–14195 CrossRef CAS.
- D. Groff, P. R. Chen, F. B. Peters and P. G. Schultz, Chembiochem, 2010, 11, 1066–1068 CrossRef CAS.
- Y. S. Wang, B. Wu, Z. Wang, Y. Huang, W. Wan, W. K. Russell, P. J. Pai, Y. N. Moe, D. H. Russell and W. R. Liu, Mol. Biosyst., 2010, 6, 1557–1560 RSC.
- H. W. Ai, J. W. Lee and P. G. Schultz, Chem. Commun., 2010, 46, 5506–5508 RSC.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS.
- V. Chau, J. W. Tobias, A. Bachmair, D. Marriott, D. J. Ecker, D. K. Gonda and A. Varshavsky, Science, 1989, 243, 1576–1583 CrossRef CAS.
- V. Nagy and I. Dikic, Biol. Chem., 2010, 391, 163–169 CrossRef CAS.
- W. Li and Y. Ye, Cell. Mol. Life Sci., 2008, 65, 2397–2406 CrossRef CAS.
- C. Chatterjee, R. K. McGinty, J. P. Pellois and T. W. Muir, Angew. Chem., Int. Ed., 2007, 46, 2814–2818 CrossRef CAS.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS.
- J. Chen, Y. Ai, J. Wang, L. Haracska and Z. Zhuang, Nat. Chem. Biol., 2010, 6, 270–272 CrossRef CAS.
- X. Li, T. Fekner, J. J. Ottesen and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 9184–9187 CrossRef CAS.
- S. Virdee, Y. Ye, D. P. Nguyen, D. Komander and J. W. Chin, Nat. Chem. Biol., 2010, 6, 750–757 CrossRef CAS.
- M. T. Bedford and S. G. Clarke, Mol. Cell, 2009, 33, 1–13 CrossRef CAS.
- M. Litt, Y. Qiu and S. Huang, Biosci. Rep., 2009, 29, 131–141 CrossRef CAS.
- W. Wan, Y. Huang, Z. Wang, W. K. Russell, P. J. Pai, D. H. Russell and W. R. Liu, Angew. Chem., Int. Ed., 2010, 49, 3211–3214 CrossRef CAS.
- H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441–444 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.