DOI:
10.1039/C005311B
(Review Article)
Mol. BioSyst., 2011,
7, 29-37
Received
5th May 2010
, Accepted 10th September 2010
First published on 8th October 2010

Victor L. Davidson
| Victor Davidson received his BS in Biochemistry from the University of Illinois in 1973 and PhD in Chemistry from Texas Tech University in 1987. After postdoctoral training at Purdue University from 1982–1984 and a research position at the University of California at San Francisco from 1984–1988 he joined the faculty at the University of Mississippi Medical Center where he is currently Professor of Biochemistry. |
I. Introduction
Redox enzymes catalyze the oxidation and reduction of substrates. Given the limited chemical functionality of amino acid side-chains, these enzymes typically employ redox-active transition metals or exogenous organic molecules as cofactors at their active sites to assist with catalysis. In some cases posttranslational modification is required to covalently attach organic cofactors, such as flavin and heme, to amino acid residues. There are also examples of significant structural modification during posttranslational assembly of some metal-binding sites, as with iron–sulfur clusters. This review describes redox enzymes which possess catalytic and redox-active centers that are formed by posttranslational modification of one or more amino acid residues on the polypeptide chain. In some cases these posttranslational modifications are autocatalytic self-processing events, while in some cases they are catalyzed by accessory enzymes. Understanding the mechanisms of biosynthesis of these protein-derived cofactors1,2 expands our views of the diversity of types of posttranslational modifications that occur in nature, and the range of chemical and redox reactions that may be catalyzed by enzymes. The best characterized protein-derived redox cofactors are those that are derived from posttranslational modification of tyrosine3 and tryptophan residues.4 Many of these posttranslational modifications require the presence of a metal, usually copper or iron. However, despite this common feature, the mechanisms by which these posttranslational modifications are catalyzed are quite diverse. Since the residues which are modified are within the protein structure rather than exposed on the surface, this poses a particularly challenging task for enzymes that catalyze these posttranslational modifications.
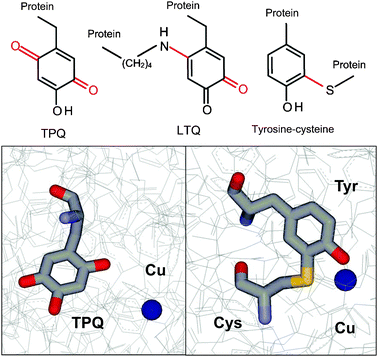
Three redox cofactors have been described which are derived from posttranslational modification of tyrosine residues. Each functions in a different class of oxidase enzymes (Fig. 1). Topaquinone (2,4,5-trihydroxyphenylalanine quinone or TPQ)5 is present in amine oxidases which also contain copper. These enzymes are ubiquitous in nature and participate in a wide range of biological activities from metabolism in bacteria to the inflammatory response in humans. Lysine tyrosylquinone (LTQ)6 is present in lysyl oxidase, an enzyme required for maturation of collagen and elastin in mammals.7 A tyrosine–cysteine cross-linked cofactor8 has been reported in oxidative enzymes in yeast, fungi and mammals. For each of these tyrosine-derived cofactors the posttranslational modifications by which they are generated are autocatalytic, self-processing events that do not require accessory enzymes. In each case, copper is required for in the posttranslational modification, and then remains in the active site of the enzyme after cofactor biosynthesis where it plays a role in the catalytic mechanism of the mature enzyme.
A Topaquinone (TPQ)
1 Occurrence and function.
TPQ-bearing enzymes are ubiquitous in nature and participate in a wide range of biological activities from metabolism in bacteria to the inflammatory response in humans.9 These enzymes have been given various names including benzylamine oxidase, diamine oxidase, histamine oxidase, plasma amine oxidase, retinal amine oxidase, and semicarbazide-sensitive amine oxidase. These names are typically based on the tissue from which the enzyme was isolated or one of the substrates for the enzyme, but do not necessarily describe a distinct subclass of the enzyme family. In microorganisms TPQ-dependent copper amine oxidases play a nutritional role, allowing primary amines to be utilized as the sole source of carbon and nitrogen for growth. In plants they play roles in the biosynthesis of hormones, cell walls, and alkaloids.10 In animals the physiological roles of these amine oxidases are quite varied and include regulation of glucose homeostasis, lymphocyte adhesion, and adipocyte maturation.11–13 In fact it has been shown that human endothelial vascular adhesion protein 1, whose cell surface expression is induced under inflammatory conditions, is a member of this class of TPQ-containing amine oxidases.11 The overall reaction catalyzed by the TPQ-dependent copper amine oxidases is the conversion of a primary amine substrate to its corresponding aldehyde plus ammonia with molecular oxygen and water as co-substrates and H2O2 as an additional product.14 The catalytic mechanism consists of two half reactions. In the reductive half-reaction (eqn (1)) the amine substrate forms a covalent adduct with TPQ which is reduced by two electrons and then releases the aldehyde product yielding an aminoquinol intermediate. In the oxidative half-reaction (eqn (2)) the aminoquinol is re-oxidized to TPQ by O2, releasing NH3 and H2O2.| | E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(quinone) + R–CH2NH2 → E–NH2(aminoquinol) + R–CHO O(quinone) + R–CH2NH2 → E–NH2(aminoquinol) + R–CHO | (1) |
| | | E–NH2(aminoquinol) + O2 + H2O → EO(quinone) + NH3 + H2O2 | (2) |
2 Cofactor biogenesis.
The posttranslational modifications to create TPQ from tyrosine are the insertion of oxygens at positions 2 and 5 on the aromatic ring. The tyrosine which is modified is present in a conserved sequence, Thr-X-X-Asn-Tyr-Asp/Gln.15 The modifications are self-processing autocatalytic events that require molecular oxygen as well as copper, but no other enzymes.16,17
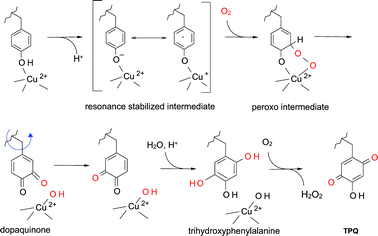
Several mechanisms have been proposed for TPQ biosynthesis18–21 and a consensus mechanism is shown in Fig. 2. While the exact chemical mechanism is not fully understood there is general agreement on the nature of the intermediate species which are indicated. It requires the participation of nearby amino acid residues, as well as a bound copper which is coordinated by three strictly conserved histidine residues.22 The tyrosine which is modified is deprotonated by a nearby basic amino acid residue and then reacts with O2 at the C5 position to yield a dopaquinone intermediate.23 This reaction is believed to proceed via a resonance stabilized intermediate pair; a charge transfer complex between the tyrosinate and Cu2+, and a Cu+–tyrosyl radical species. The intermediate reacts with O2 to yield superoxide, which then reacts with the tyrosine. Then fragmentation of the oxygen–oxygen peroxide bond yields dopaquinone. A highly unusual aspect of the biogenesis of TPQ is that at this point in the biosynthetic process the side-chain of the dopaquinone intermediate rotates 180° around its Cβ–Cγ bond. Evidence for the rotation of the modified tyrosine during cofactor biosynthesis is supported by crystal structures of the unmodified apoprotein, and a zinc-substituted protein in which the posttranslational modification has not occurred.20,24 After this conformational change, hydroxide from water adds at the C2 position to yield 2,4,5-trihydroxyphenylalanine, which then is oxidized by a second molecule of O2 to form the mature TPQ plus H2O2. The copper remains bound at this site and then plays a role in catalysis. In addition to copper, other residues assist in catalysis of TPQ. Site-directed mutagenesis of Tyr305 of the amine oxidase from Hansenula polymorpha revealed that this residue confers critical control of oxidation chemistry involved and provided direct evidence of peroxy intermediates in TPQ.25
2 Cofactor biogenesis.
The posttranslational modifications to create LTQ from tyrosine are the insertion of oxygen at position 5 on the aromatic ring and formation of a covalent crosslink between the carbon at position 2 and the side-chain nitrogen of a lysine residue (Fig. 1).6 Mechanistic studies of LTQ biosynthesis have been hampered by difficulties in expressing the recombinant mammalian lysyl oxidase preprotein. The mechanism of biosynthesis is not known with certainty but it is a self-processing event.29 The covalent bond with the lysine is formed at the same position on the modified tyrosine at which the second oxygen atom is inserted into the mature TPQ cofactor in the amine oxidases, which was discussed above. Given the presence of copper at the active site of both TPQ and LTQ enzymes, it has been assumed that the initial steps in the mechanism of biosynthesis of LTQ are similar to those described earlier for TPQ, but with the lysine nitrogen adding to the dopaquinone intermediate (see Fig. 2).23 By analogy with the proposed mechanism for TPQ biosynthesis (see Fig. 2), following nucleophilic attack at the C2 position by the amino nitrogen, aromatization yields a reduced lysylquinol intermediate which is then oxidized by a second molecule of O2 to form LTQ plus H2O2.
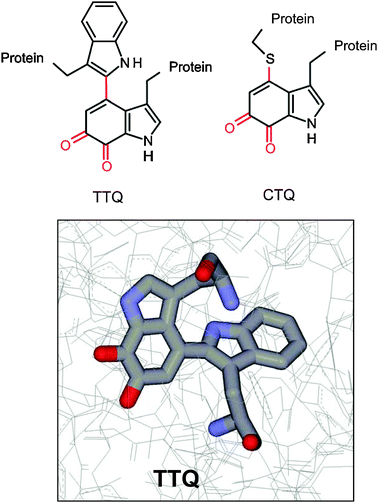
Tryptophan-derived quinone cofactors4 (Fig. 4) are present in two independently evolved bacterial dehydrogenases possessing either tryptophan tryptophylquinone (2′,4-bitryptophan-6,7-dione or TTQ)38 or cysteine tryptophylquinone (CTQ).39,40
A Tryptophan tryptophyquinone (TTQ)
2 Cofactor biogenesis.
In contrast to the tyrosine derived cofactors described above, the posttranslational modifications which form TTQ are not self-processing events but require the action of accessory enzymes. The posttranslational modifications to create TTQ are the insertion of oxygen at positions 6 and 7 on the indole ring of a specific tryptophan and formation of a covalent crosslink to the indole ring of another tryptophan residue. The mechanism of biosynthesis of TTQ has been studied most extensively in methylamine dehydrogenase (MADH) from Paracoccus denitrificans.47 The mechanism by which TTQ biosynthesis occurs has not been completely elucidated, but it is known to require the action of other enzymes that are subject to the same genetic regulation as the structural genes for the enzyme.48,49 The genes that encode the two MADH subunits, mauA and mauB, are clustered in the methylamine utilization (mau) locus.49 A role specifically in TTQ biosynthesis has been demonstrated for one of these genes, mauG. Its gene product, MauG, is a diheme enzyme.50 Inactivation of mauG allowed isolation of a biosynthetic precursor of MADH (preMADH) that contains an incompletely synthesized TTQ with a single hydroxyl group on βTrp57 and no covalent cross-link to βTrp108.51 Incubation of this intermediate in vitro with purified MauG and oxidation equivalents provided by a reductant plus O2, or by H2O2, resulted in completion of TTQ biosynthesis and formation of active MADH.51–53 Thus, while the mechanism by which the first oxygen is inserted into βTrp57 is unknown, MauG is able to complete the biosynthetic process.
Completion of TTQ biosynthesis is a six-electron oxidation process to achieve oxygen insertion, crosslinking of the βTrp57 and βTrp108 side-chains, and oxidation to the quinone (Fig. 5A). These biosynthetic oxidation reactions proceed via a relatively stable, high valent bis-Fe(IV) MauG intermediate, with one heme as Fe(IV)O and the other as Fe(IV) with two axial ligands from the protein.54 The two hemes of MauG have been shown to exhibit cooperative redox behavior and behave as a single diheme unit.54,55 The crystal structure of MauG56 reveals that the sole axial ligand for the ferryl heme is histidine, and the axial ligands for the other heme are histidine and tyrosine. This is the first example of a c-type heme with tyrosine providing an axial ligand. Replacement of this tyrosine with histidine demonstrated that the tyrosine ligation was critical for stabilization of the Fe(IV) state on the six-coordinate heme which was critical for TTQ biosynthesis activity.57
![(A) The six-electron oxidation reaction that is catalyzed by MauG TTQ biosynthesis from the preMADH precursor of methylamine dehydrogenase. [O] represents oxidizing equivalents that may be provided by H2O2 or a reductant plus O2. (B) The structure of the preMADH–MauG complex. MauGs are colored pink, the preMADH α subunits are colored blue and the β subunits are colored green. The hemes and residues β57 and β108, which are converted to TTQ, are colored black and indicated by arrows. The structure is PDB entry 3L4M.](/image/article/2011/MB/c005311b/c005311b-f5.gif) |
| | Fig. 5 (A) The six-electron oxidation reaction that is catalyzed by MauG TTQ biosynthesis from the preMADH precursor of methylamine dehydrogenase. [O] represents oxidizing equivalents that may be provided by H2O2 or a reductant plus O2. (B) The structure of the preMADH–MauG complex. MauGs are colored pink, the preMADH α subunits are colored blue and the β subunits are colored green. The hemes and residues β57 and β108, which are converted to TTQ, are colored black and indicated by arrows. The structure is PDB entry 3L4M. | |
Despite the cooperative redox behavior, the heme irons are separated by 21 Å, although an intervening tryptophan residue is within 4 Å of each heme. Further surprising insight into the catalytic mechanism was obtained from the crystal structure of the MauG–preMADH protein complex.56 This structure revealed that βTrp57 and βTrp108 of preTTQ do not make direct contact with either heme of MauG but are actually separated by very long distance requiring long range ET for catalysis (Fig. 5B). The oxygen-binding heme iron is more than 40 Å from the site of oxygen insertion into residue βTrp57. The relevance of this structure was confirmed by the demonstration that TTQ biosynthesis could be achieved in crystallo by addition of H2O2 to the crystal.56 Thus, it appears that the role of MauG is to provide an extremely high potential oxidant to extract electrons from the preMADH substrate, generating reactive radical intermediates that then insert oxygen atom from solvent and form the covalent crosslink between the two side-chains.
The kinetic mechanism of initial two-electron oxidation of preMADH by bis-Fe(IV) MauG has been characterized58 and it was demonstrated that the product of the reaction is a radical species which was formed on preMADH,54 and has yet to be fully characterized. In contrast to other heme-dependent oxygenases such as cytochrome P450s which require oxygen binding to prime the enzyme for substrate binding,59,60 the kinetic mechanism of MauG was shown to be random as the order of binding of the oxygen or H2O2 did not matter.58 However, cycling between the bis-Fe(IV) and diferric state in the absence of the preMADH substrate did cause inactivation of MauG.61 The order of oxygen insertion and crosslink formation during TTQ biosynthesis has not yet been determined. However, it is believed that the final reaction is the two-electron oxidation of quinol MADH to quinone (TTQ) MADH, and this step has been enzymatically characterized.62 Thus, while the overall process of MauG-dependent TTQ biosynthesis has not yet been fully elucidated, it is clear that these unusual posttranslational modifications comprise a novel mechanism for cofactor biogenesis which requires a diheme enzyme with unprecedented physical and catalytic properties.
1 Occurrence and function.
CTQ (Fig. 4) has been identified as the protein-derived cofactor of bacterial quinohemoprotein amine dehydrogenase (QHNDH).39,40 QHNDH contains two covalently bound c-type hemes in addition to CTQ. The overall reaction catalyzed by QHNDH is similar to that catalyzed by the TTQ-dependent dehydrogenases. The primary amine substrate is converted to its corresponding aldehyde plus ammonia. However, in QHNDH the immediate electron acceptor for the two substrate-derived electrons are the two hemes which are present in the α subunit. These electrons are then donated to either azurin or a cytochrome c, depending upon the host bacterium, to reoxidize QHNDH.63,64
2 Cofactor biogenesis.
CTQ is formed by posttranslational modifications during which two atoms of oxygen are incorporated into the indole ring of a tryptophan residue and a covalent bond is formed between the modified indole ring and the sulfur of a cysteine residue. While CTQ- and TTQ-dependent enzymes possess the same tryptophylquinone moiety at their active sites, the overall structures of the CTQ- and TTQ-dependent enzymes are quite different. The TTQ-bearing amine dehydrogenases are α2β2 heterodimeric proteins.41 The CTQ-bearing quinohemoprotein amine dehydrogenase is an αβγ heterotrimeric protein.39,40 Another highly unusual structural feature of QHNDH is that in addition to CTQ, the γ subunit contains three novel thioether crosslinks. Each is formed between the sulfur of a cysteine residue and either the β-methylene carbon of an aspartic acid residue or the γ-methylene carbon of a glutamic acid residue. These crosslinked amino acid residues appear not to participate in catalysis. Instead they play a structural role in determining the tertiary structure of the γ subunit. It is interesting to note that the TTQ-dependent methylamine dehydrogenase does not have these thioether crosslinked residues, but does have six intrasubunit disulfide bonds between cysteine residues which similarly play a role in determining the tertiary structure of the TTQ-bearing β subunit of that enzyme.65
The mechanism of biogenesis of CTQ is much less understood than that of TTQ. The gene cluster which encodes QHNDH39 is completely different from that which encodes MADH. The QHNDH gene cluster does not contain a mauG equivalent. It does contains an open reading frame (ORF2) which encodes a putative radical S-adenosylmethione protein,66,67 which is not present in the MADH gene cluster. The ORF2 protein is believed to be required for the posttranslational formation of the intra-peptidyl thioether crosslinks.68 While an accessory gene like mauG is not present, the α subunit of QHNDH contains two c-type hemes. This may be coincidental but it raises the possibility that this diheme protein subunit may perform a role in CTQ biosynthesis similar to that of MauG during TTQ biosynthesis; but at present this is purely speculation.
IV. Other posttranslationally modified redox sites
In addition to the posttranslationally modified enzymes discussed thus far, there are also enzymes and proteins which use either copper or heme iron that possess posttranslationally modified amino acid residues in the active sites which the metals inhabit. The extent to which this phenomenon occurs is still an open question, and the mechanisms of posttranslational modifications have not yet been established for most of the modified proteins thus far described. Described below are of few of these proteins with posttranslationally modified redox sites which have been structurally characterized.
Posttranslationally modified amino acid residues have been identified in the proximity of the catalytic heme of certain redox enzymes (Fig. 6). Catalase-peroxidases (KatGs) are bifunctional hemoproteins that exhibit both catalatic and peroxidatic activities. Of particular interest is the KatG of Mycobacterium tuberculosis which contributes to pathogenesis through the ability to catabolize the peroxides generated by the phagocyte NADPH oxidase.69 Crystal structures of KatG from three bacterial sources revealed that it possesses a crosslinked triad of amino acid residues at the heme active site.70–72 In this case, there are two covalent crosslinks to a tyrosine residue, both to the aromatic ring. One is with a tryptophan residue and one is with a methionine residue. It was possible to gain insight into the mechanism of formation of these crosslinks from studies on the expression of the recombinant enzyme from M. tuberculosis. A protein lacking the crosslinks was prepared from cells deficient in heme by growth in iron-depleted media. After reconstitution with heme in vitro, formation of the crosslinks was observed after addition of peroxyacetic acid.73 On the basis of these results, it was proposed that the formation of these crosslinks was catalyzed by the endogenous heme, which initially functions as a peroxidase using these amino acid residues as co-substrates in the reaction. Formation of amino acid-based radicals followed by radical coupling generates each covalent crosslink. Once the formation of the mature active site with the methionine–tyrosine–tryptophan crosslinked species is complete in the mature enzyme, the heme then reacts with its natural substrates. Crosslinked amino acid residue pairs have been observed in the heme sites of two other catalases. A crosslink between a histidine and a tyrosine residue has been identified in catalase HPII,74 and a crosslink between a cysteine and a tyrosine residue has been identified in catalase-1.75 In each case, the crosslink is formed at the β-carbon of the tyrosine residue rather than to the aromatic ring. The crosslinked residues are not directly coordinated to the heme iron but in close proximity. While mechanisms by which these posttranslational modifications occur are not known, a process similar to that which was elucidated for KatG is a distinct possibility.
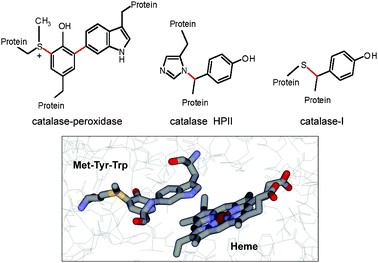 |
| | Fig. 6 Posttranslationally modified amino acid residues at heme sites of enzymes. (top panel) Structures are shown of three sets of amino acid residues at the heme sites of their respective enzymes which possess posttranslational modifications (colored in red) that are required for the enzymatic activity. (bottom panel) The structure of the heme site of KatG from M. tuberculosis (PDB entry 1sj2) with residues Met255, Tyr229 and Trp107 and the nearby heme displayed as sticks colored gray for carbon, red for oxygen, blue for nitrogen, yellow for sulfur and dark red for iron. | |
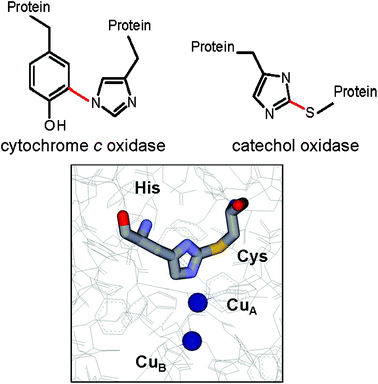
B Posttranslationally modified copper ligands
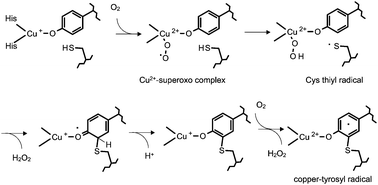
Posttranslationally modified amino acid residues have been identified which provide ligands for copper at the catalytic site of certain redox enzymes (Fig. 7). A tyrosine–histidine crosslink is present in cytochrome c oxidase, which is a component of the membrane bound respiratory electron transport chains of mammals, plants and microorganisms. The crystal structures of bacterial and mammalian cytochrome c oxidase76,77 revealed that a histidine residue which provides one of the copper ligands of the binuclear CuB component of the CuB–hemea3 center is covalently crosslinked to a tyrosine residue. Analysis by mass spectrometry revealed that this same crosslinked amino acid species is present and a common feature of each of the three distinct families of heme–copper oxygen reductases in nature.78 A cysteine–histidine crosslink is present in catechol oxidase and hemocyanin, two enzymes that possess type 3 dinuclear copper centers. Catechol oxidase catalyzes the oxidation of phenols by molecular oxidation. The product of the oxidation of catechol is benzoquinone, which is responsible for the browning of fruit. The crystal structure of a plant catechol oxidase revealed that the CuA of the dinuclear CuA–CuB metal site is coordinated by three histidine ligands, one of which is covalently cross-linked to a cysteine residue by a thioether bond.79 Hemocyanins carry oxygen in the blood of some mollusks and crustaceans. Their biological function is similar to that of hemoglobin in animals but rather than using heme iron to bind O2, hemocyanins possess two copper atoms that reversibly bind O2. A cysteine–histidine bond to one of the copper ligands in the dinuclear metal site of hemocyanin has been identified in its crystal structure.80
V. Concluding remarks
A distinctive aspect of the posttranslational modifications discussed in this review is that the modified residues in each case are buried within the protein rather than exposed at the surface. Cofactor biosynthesis must precede catalytic function. Therefore, in the cases in which the posttranslational modification is self-processing, structural features of the protein must have initially evolved for some different function, which somehow created the environment for the posttranslational modifications that generated the protein-derived cofactor and a new catalytic function. It is also clearly evident that certain amino acid residues and bound metals at the active sites of these enzymes possess multiple functions: one in the posttranslational modification to create the protein-derived cofactor, and another in catalysis in the mature enzyme. In the cases where accessory enzymes are required to catalyze the posttranslational modifications, the nature of the substrate requires that catalysis occur somehow without direct contact between the active site of the modifying enzyme and the buried residues which are modified. The posttranslational modifications described in this review were discovered within the last 20 years, and it seems likely that more will be revealed in the coming years. The characterization of the mechanisms by which redox-active centers are generated by posttranslational modification raises interesting questions regarding the evolution of protein structures and functions, and is revealing unanticipated catalytic mechanisms for the formation of the protein-derived cofactors.
Abbreviations
Acknowledgements
Work from the author’s laboratory was supported by NIH grant GM41574.
References
- V. L. Davidson, Biochemistry, 2007, 46, 5283–5292 CrossRef CAS.
- N. M. Okeley and W. A. van der Donk, Chem. Biol., 2000, 7, R159–R171 CrossRef CAS.
- M. Mure, Acc. Chem. Res., 2004, 37, 131–139 CrossRef CAS.
- V. L. Davidson, Bioorg. Chem., 2005, 33, 159–170 CrossRef CAS.
- S. M. Janes, D. Mu, D. Wemmer, A. J. Smith, S. Kaur, D. Maltby, A. L. Burlingame and J. P. Klinman, Science, 1990, 248, 981–987 CAS.
- S. X. Wang, M. Mure, K. F. Medzihradszky, A. L. Burlingame, D. E. Brown, D. M. Dooley, A. J. Smith, H. M. Kagan and J. P. Klinman, Science, 1996, 273, 1078–1084 CAS.
- H. M. Kagan and P. C. Trackman, Am. J. Respir. Cell Mol. Biol., 1991, 5, 206–210 Search PubMed.
- N. Ito, S. E. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. Yadav and P. F. Knowles, Nature, 1991, 350, 87–90 CrossRef CAS.
-
Copper Amine Oxidases: Structures, catalytic mechanisms, and roles in pathophysiology, ed. G. Floris and B. Mondovi, CRC Press, Boca Raton, Florida, 2009 Search PubMed.
- A. Cona, G. Rea, R. Angelini, R. Federico and P. Tavladoraki, Trends Plant Sci., 2006, 11, 80–88 CrossRef CAS.
- T. T. Airenne, Y. Nymalm, H. Kidron, D. J. Smith, M. Pihlavisto, M. Salmi, S. Jalkanen, M. S. Johnson and T. A. Salminen, Protein Sci., 2005, 14, 1964–1974 CrossRef CAS.
- C. M. Stolen, R. Madanat, L. Marti, S. Kari, G. G. Yegutkin, H. Sariola, A. Zorzano and S. Jalkanen, FASEB J., 2004, 18, 702–704 CAS.
- P. H. Yu, S. Wright, E. H. Fan, Z.-R. Lun and D. Gubisne-Harberle, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1647, 193–199 CrossRef CAS.
- M. Mure, S. A. Mills and J. P. Klinman, Biochemistry, 2002, 41, 9269–9278 CrossRef CAS.
- D. Mu, S. M. Janes, A. J. Smith, D. E. Brown, D. M. Dooley and J. P. Klinman, J. Biol. Chem., 1992, 267, 7979–7982 CAS.
- R. Matsuzaki, T. Fukui, H. Sato, Y. Ozaki and K. Tanizawa, FEBS Lett., 1994, 351, 360–364 CrossRef CAS.
- D. Cai and J. P. Klinman, J. Biol. Chem., 1994, 269, 32039–32042 CAS.
- B. J. Brazeau, B. J. Johnson and C. M. Wilmot, Arch. Biochem. Biophys., 2004, 428, 22–31 CrossRef CAS.
- M. C. Wilce, D. M. Dooley, H. C. Freeman, J. M. Guss, H. Matsunami, W. S. McIntire, C. E. Ruggiero, K. Tanizawa and H. Yamaguchi, Biochemistry, 1997, 36, 16116–16133 CrossRef CAS.
- M. Kim, T. Okajima, S. Kishishita, M. Yoshimura, A. Kawamori, K. Tanizawa and H. Yamaguchi, Nat. Struct. Biol., 2002, 9, 591–596 CAS.
- J. E. Dove and J. P. Klinman, Adv. Protein Chem., 2001, 58, 141–174 CAS.
- R. Li, J. P. Klinman and F. S. Mathews, Structure (London), 1998, 6, 293–307 Search PubMed.
- R. H. Moore, M. A. Spies, M. B. Culpepper, T. Murakawa, S. Hirota, T. Okajima, K. Tanizawa and M. Mure, J. Am. Chem. Soc., 2007, 129, 11524–11534 CrossRef CAS.
- Z. Chen, B. Schwartz, N. K. Williams, R. Li, J. P. Klinman and F. S. Mathews, Biochemistry, 2000, 39, 9709–9717 CrossRef CAS.
- Z. W. Chen, S. Datta, J. L. Dubois, J. P. Klinman and F. S. Mathews, Biochemistry, 2010, 49, 7393–7402 CrossRef CAS.
- H. M. Kagan and W. Li, J. Cell. Biochem., 2003, 88, 660–672 CrossRef CAS.
- J. T. Erler, K. L. Bennewith, M. Nicolau, N. Dornhofer, C. Kong, Q. T. Le, J. T. Chi, S. S. Jeffrey and A. J. Giaccia, Nature, 2006, 440, 1222–1226 CrossRef CAS.
- S. R. Vora, Y. Guo, D. N. Stephens, E. Salih, E. D. Vu, K. H. Kirsch, G. E. Sonenshein and P. C. Trackman, Biochemistry, 2010, 49, 2962–2972 CrossRef CAS.
- J. A. Bollinger, D. E. Brown and D. M. Dooley, Biochemistry, 2005, 44, 11708–11714 CrossRef CAS.
- M. M. Whittaker, P. J. Kersten, D. Cullen and J. W. Whittaker, J. Biol. Chem., 1999, 274, 36226–36232 CrossRef CAS.
- P. J. Kersten and T. K. Kirk, J. Bacteriol., 1987, 169, 2195–2201 CAS.
- C. R. Simmons, Q. Liu, Q. Huang, Q. Hao, T. P. Begley, P. A. Karplus and M. H. Stipanuk, J. Biol. Chem., 2006, 281, 18723–18733 CrossRef CAS.
- J. E. Dominy, Jr., J. Hwang, S. Guo, L. L. Hirschberger, S. Zhang and M. H. Stipanuk, J. Biol. Chem., 2008, 283, 12188–12201 CrossRef.
- M. M. Whittaker and J. W. Whittaker, J. Biol. Chem., 2003, 278, 22090–22101 CrossRef CAS.
- M. S. Rogers, A. J. Baron, M. J. McPherson, P. F. Knowles and D. M. Dooley, J. Am. Chem. Soc., 2000, 122, 990–991 CrossRef CAS.
- S. J. Firbank, M. S. Rogers, C. M. Wilmot, D. M. Dooley, M. A. Halcrow, P. F. Knowles, M. J. McPherson and S. E. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12932–12937 CrossRef CAS.
- J. W. Whittaker, Arch. Biochem. Biophys., 2005, 433, 227–239 CrossRef CAS.
- W. S. McIntire, D. E. Wemmer, A. Chistoserdov and M. E. Lidstrom, Science, 1991, 252, 817–824 CrossRef CAS.
- S. Datta, Y. Mori, K. Takagi, K. Kawaguchi, Z. W. Chen, T. Okajima, S. Kuroda, T. Ikeda, K. Kano, K. Tanizawa and F. S. Mathews, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14268–14273 CrossRef CAS.
- A. Satoh, J. K. Kim, I. Miyahara, B. Devreese, I. Vandenberghe, A. Hacisalihoglu, T. Okajima, S. Kuroda, O. Adachi, J. A. Duine, J. Van Beeumen, K. Tanizawa and K. Hirotsu, J. Biol. Chem., 2002, 277, 2830–2834 CrossRef CAS.
- V. L. Davidson, Adv. Protein Chem., 2001, 58, 95–140 CAS.
- G. R. Bishop, E. J. Valente, T. L. Whitehead, K. L. Brown, R. T. Hicks and V. L. Davidson, J. Am. Chem. Soc., 1996, 118, 12868–12869 CrossRef CAS.
- M. Husain and V. L. Davidson, J. Biol. Chem., 1985, 260, 14626–14629 CAS.
- Y. L. Hyun and V. L. Davidson, Biochemistry, 1995, 34, 12249–12254 CrossRef CAS.
- G. R. Bishop, H. B. Brooks and V. L. Davidson, Biochemistry, 1996, 35, 8948–8954 CrossRef CAS.
- Z. Zhu and V. L. Davidson, Biochemistry, 1999, 38, 4862–4867 CrossRef CAS.
- C. M. Wilmot and V. L. Davidson, Curr. Opin. Chem. Biol., 2009, 13, 469–474 CrossRef CAS.
- C. J. van der Palen, W. N. Reijnders, S. de Vries, J. A. Duine and R. J. van Spanning, Antonie van Leeuwenhoek, 1997, 72, 219–228 CrossRef CAS.
- C. J. van der Palen, D. J. Slotboom, L. Jongejan, W. N. Reijnders, N. Harms, J. A. Duine and R. J. van Spanning, Eur. J. Biochem., 1995, 230, 860–871 CrossRef CAS.
- Y. Wang, M. E. Graichen, A. Liu, A. R. Pearson, C. W. Wilmot and V. L. Davidson, Biochemistry, 2003, 42, 7318–7325 CrossRef CAS.
- A. R. Pearson, T. de la Mora-Rey, M. E. Graichen, Y. Wang, L. H. Jones, S. Marimanikkupam, S. A. Aggar, P. A. Grimsrud, V. L. Davidson and C. W. Wilmot, Biochemistry, 2004, 43, 5494–5502 CrossRef CAS.
- Y. Wang, X. Li, L. H. Jones, A. R. Pearson, C. M. Wilmot and V. L. Davidson, J. Am. Chem. Soc., 2005, 127, 8258–8259 CrossRef CAS.
- X. Li, L. H. Jones, A. R. Pearson, C. M. Wilmot and V. L. Davidson, Biochemistry, 2006, 45, 13276–13283 CrossRef CAS.
- X. Li, R. Fu, S. Lee, C. Krebs, V. L. Davidson and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8597–8600 CrossRef CAS.
- X. Li, M. Feng, Y. Wang, H. Tachikawa and V. L. Davidson, Biochemistry, 2006, 45, 821–828 CrossRef CAS.
- L. M. Jensen, R. Sanishvili, V. L. Davidson and C. M. Wilmot, Science, 2010, 327, 1392–1394 CrossRef CAS.
-
N. Abu Tarboush, M. L. R. Jensen, M. Feng, H. Tachikawa, C. M. Wilmot and V. L. Davidson, 2010, submitted.
- S. Lee, S. Shin, X. Li and V. L. Davidson, Biochemistry, 2009, 48, 2442–2447 CrossRef CAS.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS.
- S. Shin, S. Lee and V. L. Davidson, Biochemistry, 2009, 48, 10106–10112 CrossRef CAS.
- S. Shin, N. Abu Tarboush and V. L. Davidson, Biochemistry, 2010, 49, 5810–5816 CrossRef CAS.
- O. Adachi, T. Kubota, A. Hacisalihoglu, H. Toyama, E. Shinagawa, J. A. Duine and K. Matsushita, Biosci., Biotechnol., Biochem., 1998, 62, 469–478 CrossRef CAS.
- K. Takagi, K. Yamamoto, K. Kano and T. Ikeda, Eur. J. Biochem., 2001, 268, 470–476 CrossRef CAS.
- L. Chen, M. Doi, R. C. Durley, A. Y. Chistoserdov, M. E. Lidstrom, V. L. Davidson and F. S. Mathews, J. Mol. Biol., 1998, 276, 131–149 CrossRef CAS.
- P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS.
- K. Ono, T. Okajima, M. Tani, S. Kuroda, D. Sun, V. L. Davidson and K. Tanizawa, J. Biol. Chem., 2006, 281, 13672–13684 CrossRef CAS.
- V. H. Ng, J. S. Cox, A. O. Sousa, J. D. MacMicking and J. D. McKinney, Mol. Microbiol., 2004, 52, 1291–1302 CrossRef CAS.
- T. Bertrand, N. A. Eady, J. N. Jones, Jesmin, J. M. Nagy, B. Jamart-Gregoire, E. L. Raven and K. A. Brown, J. Biol. Chem., 2004, 279, 38991–38999 CrossRef CAS.
- Y. Yamada, T. Fujiwara, T. Sato, N. Igarashi and N. Tanaka, Nat. Struct. Biol., 2002, 9, 691–695 CrossRef CAS.
- X. Carpena, S. Loprasert, S. Mongkolsuk, J. Switala, P. C. Loewen and I. Fita, J. Mol. Biol., 2003, 327, 475–489 CrossRef CAS.
- R. A. Ghiladi, G. M. Knudsen, K. F. Medzihradszky and P. R. de Montellano, J. Biol. Chem., 2005, 280, 22651–22663 CrossRef CAS.
- J. Bravo, I. Fita, J. C. Ferrer, W. Ens, A. Hillar, J. Switala and P. C. Loewen, Protein Sci., 1997, 6, 1016–1023 CrossRef CAS.
- A. Diaz, E. Horjales, E. Rudino-Pinera, R. Arreola and W. Hansberg, J. Mol. Biol., 2004, 342, 971–985 CrossRef CAS.
- C. Ostermeier, A. Harrenga, U. Ermler and H. Michel, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10547–10553 CrossRef CAS.
- S. Yoshikawa, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, E. Yamashita, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki and T. Tsukihara, Science, 1998, 280, 1723–1729 CrossRef CAS.
- J. Hemp, D. E. Robinson, K. B. Ganesan, T. J. Martinez, N. L. Kelleher and R. B. Gennis, Biochemistry, 2006, 45, 15405–15410 CrossRef CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef CAS.
- M. E. Cuff, K. I. Miller, K. E. van Holde and W. A. Hendrickson, J. Mol. Biol., 1998, 278, 855–870 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.