Long-term subcutaneous microdialysis sampling and qRT-PCR of MCP-1, IL-6 and IL-10 in freely-moving rats
Erika C.
von Grote
a,
Venkat
Venkatakrishnan
ab,
Jia
Duo†
a and
Julie A.
Stenken
*a
aDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR 72701, USA. E-mail: jstenken@uark.edu; Fax: +1 479-575-4049; Tel: +1 479-575-7018
bCellular and Molecular Biology Program, University of Arkansas, Fayetteville, AR 72701, USA
First published on 23rd August 2010
Abstract
Cytokines are important mediators of the wound healing response. However, sampling of cytokines from the interstitial fluid at a healing wound site in experimental animals is a challenge. Microdialysis sampling is an in vivo collection option for this purpose as it permits continuous sampling, while remaining contiguous with the wound microenvironment. The polymeric membrane of the microdialysis probe is a foreign material thus allowing a unique approach to sample cytokines generated during a foreign body response (FBR). The focus of these studies was to use microdialysis sampling to collect cytokines from a microdialysis probe implant site in a rat model of a FBR up to 6 days post implantation. Fluorescent bead-based immunoassays (Luminex™) were used to quantify monocyte chemoattractant protein-1 (MCP-1/CCL2), interleukin-6 (IL-6) and interleukin-10 (IL-10) in the dialysates. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was used to cross validate the protein measurements obtained via micorodialysis sampling. A histological examination of tissue was also performed to assess the progression in leukocyte extravasation and collagen deposition surrounding implanted probes. Our findings demonstrate that in vivomicrodialysis sampling can be used to collect temporal concentrations of cytokines which are consistent with wound healing and the development of a FBR.
Introduction
The host response to surgical implantation of medical prostheses and devices is characterized by acute inflammation which does not completely resolve, but develops into a state of chronic inflammation evoked by the presence of a foreign material. The foreign body response (FBR) is distinguished by the localized persistence of macrophages and collagenous deposition at the tissue–material interface.1,2 The cytokines generated at a wound site direct the progress and outcome of the FBR via either the recruitment, activation and proliferation of leukocytes, lymphocytes or resident cells.3,4 A key aspect of biomaterials research is focused on the dynamic chemokine, cytokine, and growth factor profiles associated with the FBR in an effort to temper the host response which jeopardizes the structural integrity and function of medical prostheses and biosensors. However, a challenge with this type of research is the inability to perform real-time collections or measurements within the wound site itself for determination of signaling molecules associated with the wound healing event. The general stages of normal tissue repair include a short-term acute inflammatory response followed by matrix deposition, tissue remodeling and neovascularization.5,6 In the case of biomaterial implantation, the acute inflammatory response does not completely resolve, but transitions into a chronic response as the biomaterial-adherent macrophages fuse into multinucleated foreign body giant cells (FBGC).1 Ultimately, the normal repair process is redirected by the chemical signaling molecules secreted by these cells, and the remodeling of the damaged tissue replaced by an avascular collagenous deposition at the tissue–material interface.7Cytokines are a class of signaling molecules released by numerous cell types at a wound site including keratinocytes, endothelial cells, neutrophils, macrophages, lymphocytes and fibroblasts.4,8,9 These proteins generally function over short time spans, at picomolar concentrations, and propagate both redundant and pleiotropic effects. Accumulations of cytokine gradients result in the recruitment and activation of specific leukocytes, neutrophils and monocytes/macrophages, which are the initial mediators of the inflammation process. Although the typical function of both neutrophils and macrophages includes the amplified secretion of pro-inflammatory cytokines and clearance of cellular debris, it is the role of the activated macrophage which predominantly mediates the transition from normal wound healing to a FBR.10–12
Macrophages promote wound healing by secreting a tremendous variety of growth factors, pro- and anti-inflammatory cytokines which expedite cell proliferation, angiogenesis and tissue remodeling; however in a sustained state of activation evoked by a foreign material, the culmination of cytokine secretion results in increased fibrogenesis and encapsulation of the implanted material.1,13 The FBR not only has a destructive impact on biomaterials, but ultimately impedes biosensor function by forming a barrier and limiting the diffusion of analytes in the surrounding tissue.14
To date, the collection of cytokines in interstitial fluid has been successfully carried out using nylon wicks, hollow tubes and sponge implants.15–19 However, the practicality of these in vivo collection techniques causes analysis challenges. The integrity of the cytokine milieu in an actively-healing wound microenvironment may be influenced by the introduction of the collection device, and collected sample volumes may fall below that which is required for a typical immunoassay (∼100 μL). Small sample volume also limits the number of cross-validation assays which can be employed to analyze a single sample.
For biomaterials studies, a popular method known as the “cage system”, typically used in the rat model of the FBR, involves the use of a cylindrical stainless steel mesh enclosure surrounding an implanted material. The cage system facilitates repeated collection of exudate from the microenvironment surrounding the tissue–material interface.20 Several features of this technique may confer possible complications such as the repeated fluid extraction by hypodermic syringe insertion and the necessity to discriminate between the immune response elicited by the stainless steel cage and that of the target biomaterial. Furthermore, the spatial integrity of collection location in relation to implanted material may not precisely represent the conditions at the tissue–material interface.
An ideal approach to the sampling of interstitial fluid during a FBR would be to use a minimally-invasive implanted collection device which also elicits an immunological response. Microdialysis sampling is based on the passive diffusion of soluble analytes from the interstitial spaces of tissues or organs using an implanted probe and has been recently reviewed.21,22Microdialysis sampling has been successfully used to collect cytokines from the cutaneous interstitial fluids in a variety of applications.22–24 The system includes concentrically-aligned inlet and outlet tubing, at the junction of which is a hollow polymeric probe. The terminus of the probe houses a semi-permeable membrane which permits the passive diffusion of analytes from the interstitial fluid across the probe membrane and into the out-circulating dialysate. The flow of the perfusion fluid is maintained at a continuous rate by a syringe pump. The perfusion fluid is designed to isotonically match the extracellular fluid. This characteristic minimizes loss of perfusion fluid across the membrane into the tissue, and promotes passive diffusion as the primary mode of solute transport across the membrane into the probe. Passive diffusion is supported by a fluid flow-rate of 0.5–2 μL min−1 and the selection of cytokine molecules in the appropriate molecular weight range is attained using a microdialysis probe membrane with a molecular weight cut off value (MWCO) of at least 100 kDa.23 Relative recovery (RR) across the membrane is defined as the ratio between the analyte concentration in the dialysate divided by the sample concentration, i.e., RR = Cdialysate/Csample.
Comparatively this collection technique is advantageous for multiple reasons; it facilitates continuous sampling over time, its small size (∼500 μm) is less disruptive to the wound site, and the dialysate sample typically requires no further processing prior to chemical analysis of its contents. Additionally, the advantage of the awake and freely moving animal system allows any potential effect of anesthetic or suppression of the immune response due to lower body temperatures incurred during anesthesia to be minimized.25,26
With respect to a FBR, cytokines are categorized as either pro-inflammatory/anti-wound healing or anti-inflammatory/pro-wound healing. In our studies we chose to measure monocyte chemoattractant protein-1 (MCP-1/CCL2) and interleukin-6 (IL-6) based on their abilities to promote macrophage infiltration and inflammation, respectively. We also chose to measure interleukin-10 (IL-10) based on its ability to suppress the transcription of pro-inflammatory cytokines.4,7,27
The goal of the present study was to use microdialysis sampling in a rat model to characterize a subcutaneous FBR to an implanted probe. Cytokine concentrations in dialysates were quantified using a bead-based fluorescence sandwich immunoassay. Additionally, the cytokine gene expression in tissue at the probe interface was also determined by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) to cross validate data. Histological analysis of tissue at the probe interface was also performed to demonstrate the localized infiltration of leukocytes and increase of collagenous deposition which corresponds with a FBR.
Results
In vitro relative recovery values for PES membrane (10 mm)
The laboratory frequently performs in vitro recovery studies of cytokines for other studies. Our results for cytokine recovery for the current lot of PES membrane probes are within the range of those reported by our group in ref. 23. For the cytokines studied in this work, the mean relative recovery values at 1.0 μL min−1 are approximately 3%, 1%, and 13%, for IL-6, IL-10, and MCP-1, respectively.Cytokine concentrations quantified by Luminex
The concentrations of cytokines from dialysates at 1, 3 and 6 days post implantation are presented in Fig. 1–3. During collection from the 1 day implant group the “active collection period” was designated as 0 to 160 min. During collection from the 3 and 6 day implant groups, initial dialysate was designated as “void volume” (3 μL min−1, 0–10 min), the subsequent “active collection period” at a flow rate of 1 μL min−1 was designated from 10 to 160 min. The void volume contains cytokines that have accumulated into the static perfusion fluid that remains in the probe tip during the long-term implantation.28 Since the Luminex immunoassays require 25 μL, the 10 min sweep at 3 μL min−1 is sufficient to remove this plug of fluid. Determination of void volume was based on the diameter and length estimates of implanted probe/tubing, and is presented in separate figures due to the markedly higher cytokine concentrations relative to that which was detected during the active collection period. | ||
| Fig. 1 MCP-1 concentrations in collected dialysates from 1 (a), 3 (b) and 6 day (c) microdialysis probe implants. Dialysis probes implanted in the subcutaneous space were perfused at 3 μL min−1 in the first 10 min (void volume) followed by a 1 μL min−1 collection period to 160 min. The “n” values indicate the number of probes that provided detectable protein concentrations from 11 working probes. N.D. = not detectable. Symbols are as follows: the box represents the 25–75 percentile; the line through the box represents the median, the whiskers represent the fence, and ■ represents the mean, and ▼ represents the outliers. | ||
 | ||
| Fig. 2 IL-6 concentrations in collected dialysates from 1 (a), 3 (b) and 6 day (c) microdialysis probe implants. Dialysis probes implanted in the subcutaneous space were perfused at 3 μL min−1 in the first 10 min (void volume) followed by a 1 μL min−1 collection period to 160 min. The “n” values indicate the number of probes that provided detectable protein concentrations from 11 working probes. N.D. = not detectable. Symbols are as follows: the box represents the 25–75 percentile; the line through the box represents the median, the whiskers represent the fence, and ■ represents the mean, and ▼ represents the outliers. | ||
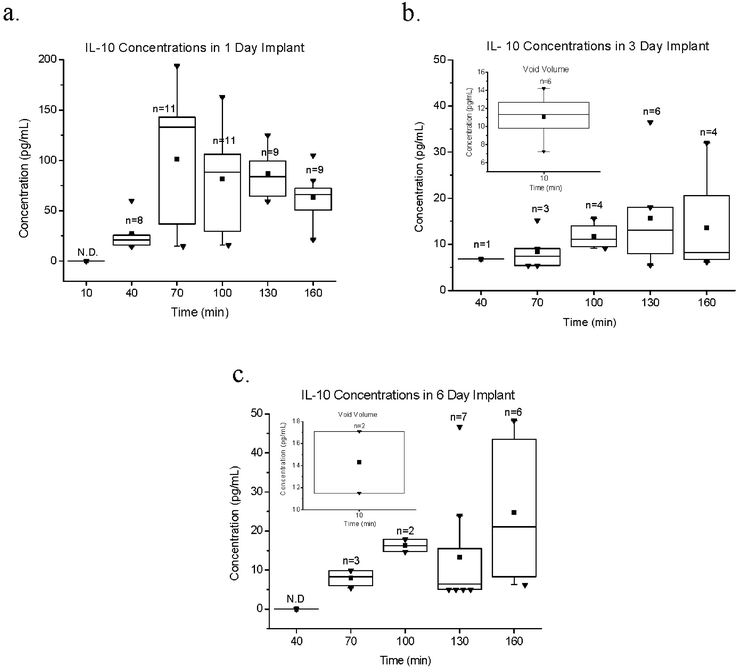 | ||
| Fig. 3 IL-10 concentrations in collected dialysates from 1 (a), 3 (b) and 6 day (c) microdialysis probe implants. Dialysis probes implanted in the subcutaneous space were perfused at 3 μL min−1 in the first 10 min (void volume) followed by a 1 μL min−1 collection period to 160 min. The “n” values indicate the number of probes that provided detectable protein concentrations from 11 working probes. N.D. = not detectable. Symbols are as follows: the box represents the 25–75 percentile; the line through the box represents the median, the whiskers represent the fence, and ■ represents the mean, and ▼ represents the outliers. | ||
Sample values which fell below the established limit of detection were designated as non-detectable (N.D.). Additionally, at least one of the 12 probes in each group (1, 3 and 6 days) failed to deliver dialysate after implantation. The non-parametric box and whisker plot diagram was chosen as the most suitable presentation for these data. Boxes indicate the distribution of 75th and 25th data percentile (the interquartile range), upper and lower quartiles respectively. Whiskers indicate the values for the upper (75th percentile + 1.5 × interquartile range) and lower (25th percentile − 1.5 × interquartile range) fence values. Sample concentrations that fall out of the fence area are deemed outliers. The total number of quantifiable dialysates (total denoted by “n” number) at each time increment within each group is denoted on the graphs. The median and mean values are represented by the crosshatch and square within each box, respectively.
MCP-1, IL-6 and IL-10 gene expression quantified by real-time RT-PCR
The expression of the MCP-1, IL-6 and IL-10 cytokine genes detected in tissue at 1, 3 and 6 days post implantation is presented in Fig. 4. A ratio representing each gene's expression in control vs. treated tissue shows a significant increase in gene expression at 1, 3 and 6 days post implantation. The relative increase of each gene's expression at 1, 3 and 6 days post implantation shared similarity with the corresponding protein concentrations detected in dialysates during the same time points. | ||
| Fig. 4 Gene expression of MCP-1, IL-6 and IL-10 expression in tissue surrounding 1, 3 and 6 day microdialysis probe implants. Total RNA was extracted from subcutis and dermis tissue surrounding probes in 1 day (a), 3 day (b) and 6 day (c) implant groups. Raw data were normalized with expression values for the endogenous control 18S rRNA. The log base 2 transformed expression ratios (fold-change) represent the mean values of four animals per treatment group ± SEM (represented by error bars). Significance is denoted as *p < 0.05, **p < 0.01. | ||
The PCR amplification efficiencies of each primer set were evaluated in both control and treatment tissue samples and the efficiency correction technique was used to calibrate the data.29 A template of 200 ng cDNA was determined as the most suitable concentration for reactions with each of the primer sets. All gene expression data were normalized against expression values for the 18S rRNA gene in the same tissues. By evaluating the PCR cycle threshold (Ct) value equivalence in both control and treatment tissues it was determined that the 18S rRNA gene was a suitable endogenous control for dermis and subcutis tissues. It was also determined that the commonly used actin-beta (ACTB) and hypoxanthine phosphoribosyltransferase-1 (HPRT-1) endogenous control genes were not appropriate controls for our tissue type.
Expression ratio values were tested for significance by a Pair Wise Fixed Reallocation Randomization Test© using the REST© (Relative Expression Software Tool). The log base 2 transformed expression ratios represent the mean values of four animals per treatment group ± SEM (represented by error bars).
Histology
The histopathology of the healing tissue surrounding the probe implant was evaluated at 1 and 6 days post implantation for characteristic phases of a wound healing response. Between day 1 and day 6 post implantation a gradual increase in extracellular matrix (ECM) and inflammatory cell infiltration was observed by hematoxylin and eosin (H & E) staining (Fig. 5 and 6). Nuclei of leukocytes or lymphocytes were observed as round intracellular purple-stained structures, while disrupted collagen in the ECM was observed as ribbon-like pink-stained structures. Using Masson's Trichrome stain, the overall density and organization of the fibrin and collagen deposition (light blue) surrounding the probe on day 6 appeared much greater than on day 1 (Fig. 7 and 8). The histological observations were consistent with the typical time-dependent stages of normal tissue repair which include an immune cell response followed by formation of a provisional fibrin and collagen matrix. | ||
| Fig. 5 Localization of inflammatory cell infiltrate and disrupted collagen surrounding a microdialysis probe explanted after 1 day. Light micrographs of hematoxylin and eosin (H & E) stained axially-sectioned probe ((a) 100×, (b) 400×) showed red blood cells (A, dark pink), damaged connective tissue fibers (B, light pink), and inflammatory cells (C, dark blue) which appeared embedded in a loose ECM. | ||
 | ||
| Fig. 6 Dense localization of inflammatory cell infiltrate, and organization of provisional fibrin and collagen matrix surrounding a microdialysis probe explanted after 6 days. Light micrographs of hematoxylin and eosin (H & E) stained axially-sectioned probe ((a) 100×, (b) 400×) showed an increased aggregation of inflammatory cells (A, dark/med blue nuclei), fibroblasts (B, dark blue elongated nuclei), and fibrin/collagen (pink) in a more tightly organized ECM surrounding the probe. | ||
 | ||
| Fig. 7 Loose organization of ECM and disrupted collagen surrounding a microdialysis probe explanted after 1 day. Light micrographs of a Masson's Trichrome stained axially-sectioned probes ((a) 100×, (b) 400×) showed damaged collagen and connective tissue fibers (A, light blue), and a minimal number of inflammatory cells (B, dark blue) organized in the ECM surrounding the probe. | ||
 | ||
| Fig. 8 Formation of an organized ECM with fibrin/collagen surrounding a microdialysis probe explanted after 6 days. Light micrographs of a Masson's Trichrome stained axially-sectioned probes ((a) 100×, (b) 400×) showed denser and more organized fibrin/collagen deposition (A, bright blue) and numerous inflammatory cells (B, dark/med blue) aggregated within close proximity to the probe–tissue interface. | ||
Discussion
The natural progression of cutaneous wound healing is altered by the presence of a foreign material; therefore, the measurement of its associated cytokine profile is of significant benefit to the fields of immunology, biomaterials and biosensor research.1,4,13 In the present study, we have used microdialysis sampling of interstitial fluids in a freely moving rat model to collect MCP-1, IL-6 and IL-10 at 1, 3 and 6 days during a FBR. To cross validate our microdialysis results we used quantitative RT-PCR on tissue excised from the implant site to measure the corresponding gene expression activity. A substantial number of research studies have evaluated the in vivo sampling of FBR-related cytokines by single time point syringe collection of static accumulated wound exudates.18,20,30–32The cytokine activity generated in the very early stages (∼first week) of healing is an important predictor of the ultimate rate and outcome of the reparative and fibrotic activity in response to a specific foreign material.20,33–35 Serum proteins, neutrophils and recruited monocytes, which initially coat the implanted material, give rise to a population of biomaterial-adherent activated macrophages.3,36 The specific combination of cytokines secreted by macrophages localized to the surrounding ECM and those which are biomaterial-adherent is the primary source of pro/anti-inflammatory cytokine signaling determining the course of the FBR. Furthermore, the cytokine profile of adherent macrophages is uniquely influenced by the surface chemistry of the material.31,36–40 Commonly used hydrophobic biomaterials, similar to the hydrophobic/neutral surface chemistry of the PES membrane of the microdialysis sampling probe, are known to evoke fibrinogen deposition, platelet aggregation, and promote increased cytokine concentrations and fibrosis.2,3,41–43
The concentration of chemoattractant MCP-1 at an acute-phase wound site strongly promotes the rate and the distribution of infiltrating monocytes.44–46MCP-1 concentrations also contribute to extracellular matrix remodeling and also correlate with an increased rate of wound site regeneration, which has been demonstrated by the use of anti-MCP-1 antibody.33,34,47,48 In normal wound healing, the increased MCP-1 concentrations secreted by monocytes/macrophages are confined to the first few days post injury.30,33,49
The modulating influence of IL-6 over the acute-phase of the inflammatory response is underscored by evidence demonstrating how IL-6 “knockout” mice have not only defects in their ability to promote necessary MCP-1 production by cells at a wound site, but also in the effective recruitment of leukocytes to a subcutaneous lesion.50,51 In normal wound healing, IL-6 concentrations (validated with gene expression data) peak within the first day post-injury, after which they begin to decline by the 3rd day, and are almost equivalent to controls by the 10th day.52
The anti-inflammatory cytokine IL-10 is secreted by a large number of activated cells in wound tissue including keratinocytes, mast cells, basophils, macrophages and TH2-activated T lymphocytes and acts primarily to suppress the production of pro-inflammatory cytokines produced by activated macrophages, induce anti-inflammatory/pro-wound healing growth factors, and inhibit further wound site infiltration by neutrophils and macrophages.27,53–56
In our studies we observed an increase in all cytokine levels on the 1st day of implantation. Sampling of the 3 and 6 day implant groups included void volume fluid—the static perfusion fluid inside the probe and tubing in which passively diffusing cytokines can accumulate. Measurement of the void volumes in the 3 day implant group revealed high accumulations of MCP-1 and IL-6 (330 pg mL−1 and 430 pg mL−1, respectively), but not IL-10 (10 pg mL−1). During the subsequent active collection period (10–160 min), MCP-1 and IL-10 concentrations declined to low but consistent levels, while IL-6 demonstrated an increase. IL-10 concentrations measured in void volumes were not high and did not change substantially during the active collection period. Sampling of the 6 day implant group also revealed high accumulation of MCP-1 and IL-6 concentrations (2305 pg mL−1 and 215 pg mL−1, respectively) but not IL-10 (15 pg mL−1) in the initial void volumes. However, unlike the 3 day group, the active collection period of the 6 day group showed an increase in all cytokine concentrations.
In previously reported data by this lab, IL-6 in void volumes sampled in 3 and 7 day probe implant groups were measured at slightly higher concentrations of 470 and 290 pg mL−1, respectively.28 The higher concentrations observed in this previous study are likely the result of the slower flow rate (1 μL min−1) that was used during the void volume collection period (0–30 min), which increases relative recovery of cytokines passively diffusing into the probe membrane.
A comparison of the tissue-associated gene expression activity with the trend in protein concentration (during 70–160 min of the active collection period) demonstrated a good correlation between the increase in both IL-6 and MCP-1 protein and gene expression (Table 1). There has been much written in the literature about the difficulties of correlating mRNA to protein expression.57 For cytokines there is some literature that has compared cytokine protein and mRNA levels. In one case a correlation was found,58 yet in another there was no correlation.59 Observed differences between mRNA and protein levels are frequently due to kinetic variations with respect to expression vs.translation and ultimately degradation of either the mRNA or the protein.
| Cytokine | Day 1 implant | Day 3 implant | Day 6 implant | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg. [protein]/pg mL−1 | Expression ratio (log 2) | Avg. [protein]/pg mL−1 | Expression ratio (log 2) | Avg. [protein]/pg mL−1 | Expression ratio (log 2) | |||||||
| Minutes | Fold increase | Minutes | Fold increase | Minutes | Fold increase | |||||||
| 70–100 | 100–130 | 130–160 | 70–100 | 100–130 | 130–160 | 70–100 | 100–130 | 130–160 | ||||
| MCP-1 | 60 | 110 | 135 | 4.0 | 25 | 20 | 20 | 2.1 | 30 | 50 | 80 | 4.9 |
| IL-6 | 660 | 925 | 950 | 5.2 | 45 | 70 | 85 | 6.5 | 15 | 60 | 110 | 6.0 |
| IL-10 | 80 | 85 | 65 | 1.8 | 10 | 15 | 15 | 1.5 | 15 | 15 | 25 | 2.7 |
In our studies, both the forms of measurement (expression vs. actual protein concentrations) demonstrate an initial increase in protein concentration and gene expression activity in the 1 day implant group, followed by a decrease in the 3 day implant group, followed by a rebound in the 6 day implant group. MCP-1 expression was observed to initially increase by 4.0-fold in the 1 day implant group, followed by a decrease to 2.1-fold in the 3 day implant group, matching a decrease in protein concentration. In tissue surrounding the 6 day implant MCP-1 expression appeared to rebound and peak at an increase of 4.9-fold. This rebound in expression activity is also matched with a gradual increase in MCP-1 protein concentration detected in dialysates from the 6 day implant.
IL-6 expression was observed to initially increase by 5.2-fold in the 1 day implant group, followed by a decrease to 3.5-fold in the 3 day implant group, matching a decrease in protein concentration. In tissue surrounding the 6 day implant, IL-6 expression appeared to rebound and peak at a 6-fold increase, which matched with an increase in IL-6 protein concentration detected in dialysates from the 6 day implant. The comparison of IL-10 gene expression activity with protein concentration trends was not as well correlated. When gene expression was observed to initially increase by 1.8-fold in the 1 day implant group, protein concentrations gradually declined over time. In the 3 day implant group, expression activity had decreased from 1.8 to 1.5-fold and overall protein concentration remained consistently low for the duration of the collection period. An explanation for this may be that under any given circumstance the level of mRNA transcription is not necessarily a direct corollary of protein concentration. Post-translational modifications and receptor-bound protein may alter detectable protein levels. In tissue surrounding the 6 day implant IL-10 expression increased to 2.7-fold, which also matched a slight increase in IL-10 protein concentration detected in dialysates.
Our results agree with a recent study which highlighted the temporal and spatial location of cytokines by immunostaining of explanted tissues at 2, 4 and 10 weeks post implantation.60 This study revealed several important pieces of information regarding the cellular events of the FBR. It showed that although IL-6, MCP-1 and IL-10 are all present at the site of implantation at all times, they are disparately localized as the FBR response progresses. IL-6 was extensively localized with fibroblasts and macrophages at the periphery of the lesions, and not with FBGCs close to the implant. Staining of IL-10 increased over time, but was only localized with macrophages and FBGCs within close proximity to the implant FBGCs. The staining of MCP-1 was to a lesser extent throughout the lesion, but was extensively stained with FBGCs. This evidence in combination with our results suggests that sustained secretion of pro-inflammatory cytokines occurs in a FBR, and their spatial location within the tissues is dependent on their proximity to the implanted material.
The possibility of this dynamic is supported by numerous in vitro and in vivo studies. The phenotype of activated macrophages has been characterized as classically-activated (host defense) or alternatively-activated (wound healing), where the former reflects a cytokine profile which is generally pro-inflammatory, and the latter describes one which reflects a more anti-inflammatory profile.10 The biomaterial adherent macrophages have a unique phenotype which has been characterized by a cytokine signature associated with both classically and alternatively-activated cytokines.31,39,40,60 It seems likely that the rebound in IL-6 and MCP-1 levels we observed in the 6 day implant group may reflect the cytokine profile of a gradual transition from wound healing to a FBR.
The histological evaluation of the explanted probes which compared immune cell recruitment and collagen accumulation surrounding the microdialysis probes at 1 and 6 days illustrated the spatial and cellular events that promote a FBR. Initially, immune cells infiltrate into the damaged tissue matrix surrounding the probe on the 1st day was marginal compared with the dense aggregation near the tissue/probe interface observed at 6 days post implantation. Although other research studies presenting histological analyses have mainly focused on the later stages of the FBR, our observations at 1 and 3 days seem consistent with the temporal progression of cellular activity which would precipitate into the later stages of a FBR.
Experimental
Microdialysis equipment and perfusion fluid
The microdialysis sampling of interstitial fluid from 1, 3 and 6 day probe implants was carried out using the CMA-120 Microdialysis system for freely moving animals (PN 8309049 CMA Microdialysis, North Chelmsford, MA). Two types of syringe pumps were used, the CMA 402 dual channel (PN 8003100 CMA Microdialysis, North Chelmsford, MA) and the Baby Bee dual channel (BASi, W. Lafayette, IN). Both syringe pumps were equally calibrated. Additionally, two types of microsyringes were used with the appropriate pump: the 1000 μL CMA glass syringe (PN 8309020 CMA Microdialysis, North Chelmsford, MA) and the 1000 μL BAS glass syringe (PN MD-0100, W. Lafayette, IN). The CMA-20 (10 mm × 0.5 mm (o.d.)) 100 kDa MWCO polyethersulfone (PES) microdialysis probe was used with all subjects (PN 8309671 CMA Microdialysis, North Chelmsford, MA). Fluorinated ethylene polypropylene (FEP) tubing ∼0.4 mm (i.d.) diameter (PN 840 9501 CMA Microdialysis, North Chelmsford, MA) was used to connect the microsyringes to the inlet/outlet tubings of the implanted microdialysis probe and 0.25 mL polypropylene micro vials were used to collect sample (PN 02-681-230, Fisher Scientific, Pittsburgh, PA).The perfusion fluid consisted of phosphate buffered saline (PBS), pH 7.2, supplemented with 6% (w/v) Dextran 70 (31390, Sigma-Aldrich, St. Louis, MO) as an osmotic agent and 0.1% (w/v) bovine serum albumin (A7906, Sigma-Aldrich, St. Louis, MO). Perfusion fluid was prepared daily as needed and filter-sterilized with a 0.2 μm PES membrane filter (Whatman, Florham Park, NJ).
Animals and surgical procedures
Sprague-Dawley rats were purchased from Harlan Laboratories, Inc (Madison, WI). Three weight ranges were selected to accommodate a staggered experimental design, such that the mean group weight during cytokine sampling would be relatively equivalent. Six rats were assigned to each of three groups designated as 1, 3 and 6 day implants. At the time of microdialysis sampling rats had a weight range of 260 to 280 g. Rats were housed in an environmentally-controlled facility with a 12-hour on/off light cycle and ad libitum access to food and water. Under the approval of the University of Arkansas, IACUC, each rat was surgically implanted with two microdialysis probes. While under ∼2.5% (in oxygen) inhalational isoflurane anesthesia, the dorsum was shaved and disinfected, and probes were subcutaneously inserted parallel to the spine ∼2 cm on either side of the midline. The inlet/outlet tubing of each probe was tunneled subcutaneously to within ∼2 cm of the neck where it exited via two additional bilateral incisions. Incisions near the neck were closed with reflex clips, and incisions at the implant site were sealed with Vetbond tissue adhesive (3M Animal Care Products, St. Paul, MN).Microdialysis sampling procedures
Prior to microdialysis sampling, all FEP tubing was flushed for 20 min with perfusion fluid at a flow rate of 5 μL min−1. Following surgical implantation of rats in the 1 day implant group, microdialysis was initiated at a flow rate of 3 μL min−1 for 10 min to rapidly flush the tubing lines. A subsequent flow rate of 1 μL min−1 for 150 min was used for the remaining collection period. Following surgical probe implantation of rats in the 3 day implant group, rats were returned to housing for 3 days. Three days post implantation microdialysis was carried out as described for the first group. Following implantation of rats in the 6 day implant group, rats were returned to housing for 6 days. Six days post implantation microdialysis was carried out as described for the first group.In all groups, temporal collection of dialysate from 0–10 min (3 μL min−1) followed by 10–40 min, 40–70 min, 70–100 min, 100–130 min and 130–160 min at 1 μL min−1 was performed. Six samples, 30 μL each, were obtained from each probe between 0–160 min. Samples were frozen at −80 °C, no longer than 4 days prior to analysis. Each group consisted of 6 rats and each rat was implanted with 2 probes.
Cytokine quantification
Dialysate samples (25 μL aliquots) were loaded onto a multiscreen filter reaction plate and processed according to the manufacturer's instructions supplied with the Milliplex™ MAP Kit for the bead-based sandwich immunoassay detection of rat MCP-1, IL-6 and IL-10 (Cat# RCYTO-80K-05 Millipore, Billerica, MA). Using the Luminex 100™ IS analyzer, sample concentrations (pg mL−1) were quantified using the median fluorescence intensity (MFI) compared against a standard curve of known protein concentrations (Luminex Corp, Austin, TX). Standard, control and limit of detection values were included with each reaction plate. The concentration range of each assay was 4.88–5000 pg mL−1.Tissue harvest and probe explantation
Immediately following microdialysis sampling of cytokines, each rat was euthanized by CO2 asphyxiation. From four rats in each group, 25–50 mg of tissue (including dermis and subcutis layer) surrounding the probe was quickly excised, immediately preserved in RNAlater stabilization reagent, and stored at 2–8 °C according to manufacturer's instructions (Qiagen, Hilden, Germany). Control tissue samples were also excised in a similar manner from a location at least 3–4 cm from the implant site. From the remaining two rats in each group, probes were carefully explanted (including interfacing subcutaneous tissue) and preserved in 10% neutral buffered formalin.Total RNA extraction and reverse-transcription
Tissue samples were homogenized using the Bullet Blender™ Blue (Next Advance, Inc, Averill Park, NY). RNA was extracted using TRI Reagent® Soln (AM9738 Applied Biosystems/Ambion, Inc, Austin, TX). Extracts were then column-cleaned using RNeasy Mini Kit (Qiagen, Valencia, CA). RNA concentration and purity were determined by measuring spectrophotometric absorbance at 260 nm (A260) and the 260 nm/280 nm ratio using a NanoVue™ spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). To assess RNA quality, a 2 μg concentration of each extract was denatured in RNA Sample Loading Buffer (R4268 Sigma, St. Louis, MO) and evaluated by formaldehyde gel electrophoresis.61A total of 2 μg RNA was used to generate cDNA in 20 μL reaction volumes using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). Reverse transcriptase reactions were performed in a Techne TC-3000 thermocycler for 60 min at 37 °C, followed by 5 min at 95 °C.
Quantitative real-time RT-PCR
An aliquot of 200 ng cDNA (in quadruplicate for each gene) was amplified in 50 μL reactions containing the manufacturer's recommended combination of TaqMan® Gene Expression Master Mix and pre-developed TaqMan® probe/primer assay reagents (Rn00580555_m1/MCP-1, Rn99999011_m1/IL-6, Rn99999012_m1/IL-10 and Hs99999901_s1/18S rRNA) (Applied Biosystems, Foster City, CA). A relative quantification assay was designed to evaluate the ratio of each cytokine in tissue surrounding the probeversus control tissue site. Raw data values in each treatment and control group were normalized with the expression values detected for the internal reference gene 18S rRNA. Standard curves for each gene primer set were generated with treatment and control cDNA to determine primer efficiency values, which were then used to calibrate the data by the Pfaffl efficiency correction method.29Cytokine PCR sample wells were prepared in quadruplicate, and PCR plates were prepared in duplicate for each treatment group. All PCR reactions were performed using an ABI Prism 7300 Sequence Detection System (PE Applied Biosystems) with a reaction program consisting of 2 min at 50 °C, 10 min at 95 °C, followed by 40 cycles of the following: 15 s at 95 °C and 1 min at 60 °C.Histology, microscopy and imaging
Explanted probes were preserved in 10% neutral buffered formalin at room temperature. Processing of samples was performed by the Histology Laboratory at the University of Arkansas, Department of Poultry Science (Fayetteville, AR). Briefly, samples were embedded in paraffin, axially thin-sectioned and stained with hematoxylin and eosin (H & E) stains to distinguish the degree of cell infiltration and Masson's Trichrome stain to examine collagen and fibrous deposition at the probe/tissue interface.Photographs of histological preparations were taken with a Zeiss Axioplan 2 microscope with 10×/30 mm ocular lenses and both 10× and 40× objectives. The images were recorded with a high-resolution charge-coupled device camera and Auto-Montage digital imaging software (Zeiss, Thornwood, NY, USA) and Microsoft Power Point (2007).
Statistical analysis
The normality of the concentrations found in microdialysis samples was determined using the Shapiro–Wilk test. The Kruskal–Wallis ANOVA was used for significance testing among the time points for each cytokine. Cytokine concentration data were analyzed and graphed as box and whisker plot diagrams using Origin 8 software program (OriginLab Corp., Northampton, MA, USA). A box and whisker plot is a non-parametric representation of the range of the data. Relative expression ratio data were analyzed for significance by a Pair Wise Fixed Reallocation Randomization Test© using the Relative Expression Software Tool (version REST-MCS©). The log base 2 transformed expression ratios were used to represent the mean values ± SEM of 4 replicates (performed in duplicate) of each biological sample, 4 rats per treatment group.Conclusions
By using the microdialysis technique, the microdialysis probe served as both a minimally invasive collection device, and a hydrophobic/neutral polymeric material which evoked the foreign body response.2,31,36,40 Using a combined protein and gene expression measurement approach, our findings suggest that in vivomicrodialysis sampling in a freely-moving animal model can be effectively used to evaluate cytokine concentrations during the early stage wound healing/foreign body response. The high concentrations of proteins in the 3 and 6 day implant void volumes illustrates an important limitation in cytokine sampling. The cytokine levels detected in static interstitial fluids, possibly similar to single time point syringe collection of wound exudates, may not necessarily be reflective of proteins which are being actively generated.Our results substantiate the current model of in vivocytokine response proposed for a wound healing/FBR. In this model the typical transition from acute pro-inflammatory to an anti-inflammatory phase, which corresponds with the down-regulation of IL-6 and MCP-1 and the up-regulation of IL-10, is instead characterized by a rebound in IL-6 and MCP-1 levels accompanied by a fibrotic response localized around the implanted material. Because a variety of cell types localized within the ECM throughout a wound site are a contributing source of cytokines in the FBR, in vitro FBR assay results need to be corroborated with in vivo assays. Our results show that microdialysis sampling has specific potential for in vivo applications, and our data contribute to the current body of knowledge focused on the temporal secretion of pro- and anti-inflammatory cytokines during the FBR.
Acknowledgements
We thank David Cross of the Department of Poultry Science and Center of Excellence for Poultry Science at the University of Arkansas for preparing the histology slides. Dr John Hahn, DVM, is acknowledged for surgical advice. Carol Rodlun, IACUC Project/Program and Central Animal Laboratory Facility Manager, is acknowledged for assistance in setting up animal studies. Professor Joon Jin Song, Department of Mathematical Sciences, and Director of the J. William Fulbright College Statistics Resource Center, is acknowledged for statistics advice. We thank Professor Jeannine Durdik for constructive comments on the manuscript. NIH EB 001441 and the Arkansas Biosciences Institute are acknowledged for support of this work.References
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS.
- P. Thevenot, W. Hu and L. Tang, Curr. Top. Med. Chem., 2008, 8, 270–280 CrossRef.
- L. Tang and W. Hu, Expert Rev. Med. Devices, 2005, 2, 493–500 Search PubMed.
- D. T. Luttikhuizen, M. C. Harmsen and M. J. Van Luyn, Tissue Eng., 2006, 12, 1955–1970 CrossRef CAS.
- C. L. Baum and C. J. Arpey, Dermatol. Surg., 2005, 31, 674–686 CAS ; discussion 686.
- J. Li, J. Chen and R. Kirsner, Clin. Dermatol., 2007, 25, 9–18 CrossRef.
- T. R. Kyriakides and P. Bornstein, Thromb. Haemostasis, 2003, 90, 986–992 Search PubMed.
- A. Meager, The Molecular Biology of Cytokines, Wiley, Chichester, New York, 1998 Search PubMed.
- D. Rossi and A. Zlotnik, Annu. Rev. Immunol., 2000, 18, 217–242 CrossRef CAS.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS.
- R. Adamson, J. Wound Care, 2009, 18, 349–351 Search PubMed.
- J. M. Daley, S. K. Brancato, A. A. Thomay, J. S. Reichner and J. E. Albina, J. Leukocyte Biol., 2010, 87, 59–67 Search PubMed.
- W. K. Ward, J. Diabetes Sci. Technol., 2008, 2, 768–777 Search PubMed.
- A. A. Sharkawy, B. Klitzman, G. A. Truskey and W. M. Reichert, J. Biomed. Mater. Res., 1998, 40, 586–597 CrossRef CAS.
- A. S. Eriksson, L. M. Bjursten, L. E. Ericson and P. Thomsen, Biomaterials, 1988, 9, 86–90 CrossRef CAS.
- H. R. Ford, R. A. Hoffman, E. J. Wing, D. M. Magee, L. McIntyre and R. L. Simmons, Arch. Surg., 1989, 124, 1422–1428 Search PubMed.
- T. Nedrebo, R. K. Reed, R. Jonsson, A. Berg and H. Wiig, J. Physiol., 2004, 556, 193–202 CAS.
- L. Baldwin and J. A. Hunt, Cytokine, 2008, 41, 217–222 CrossRef CAS.
- X. Wang, M. R. Lennartz, D. J. Loegering and J. A. Stenken, Cytokine, 2008, 43, 15–19 CrossRef CAS.
- A. Rodriguez, H. Meyerson and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2009, 89, 152–159.
- A. J. Rosenbloom, D. M. Sipe and V. W. Weedn, J. Neurosci. Methods, 2005, 148, 147–153 CrossRef CAS.
- J. A. Stenken, M. K. Church, C. A. Gill and G. F. Clough, AAPS J., 2010, 12, 73–78 Search PubMed.
- X. Ao and J. A. Stenken, Methods, 2006, 38, 331–341 CrossRef CAS.
- R. R. Patlolla, R. Mallampati, S. V. Fulzele, R. J. Babu and M. Singh, Toxicol. Lett., 2009, 185, 168–174 CrossRef CAS.
- Y. Morita, S. Oda, T. Sadahiro, M. Nakamura, T. Oshima, S. Otani and H. Hirasawa, Cytokine, 2009, 47, 48–55 CrossRef CAS.
- M. Xiong, Y. Yang, G. Q. Chen and W. H. Zhou, Brain Res. Bull., 2009, 79, 351–357 CrossRef CAS.
- R. Lang, D. Patel, J. J. Morris, R. L. Rutschman and P. J. Murray, J. Immunol., 2002, 169, 2253–2263 CAS.
- X. Wang, M. R. Lennartz, D. J. Loegering and J. A. Stenken, Anal. Chem., 2007, 79, 1816–1824 CrossRef CAS.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29, e45 CrossRef CAS.
- L. A. DiPietro, M. Burdick, Q. E. Low, S. L. Kunkel and R. M. Strieter, J. Clin. Invest., 1998, 101, 1693–1698 CrossRef CAS.
- W. G. Brodbeck, G. Voskerician, N. P. Ziats, Y. Nakayama, T. Matsuda and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2003, 64, 320–329.
- R. J. Schutte, L. Xie, B. Klitzman and W. M. Reichert, Biomaterials, 2009, 30, 160–168 CrossRef CAS.
- L. A. DiPietro, P. J. Polverini, S. M. Rahbe and E. J. Kovacs, Am. J. Pathol., 1995, 146, 868–875 CAS.
- L. A. Dipietro, M. G. Reintjes, Q. E. Low, B. Levi and R. L. Gamelli, Wound Repair Regen., 2001, 9, 28–33 CrossRef CAS.
- C. Gretzer, L. Emanuelsson, E. Liljensten and P. Thomsen, J. Biomater. Sci., Polym. Ed., 2006, 17, 669–687 CrossRef CAS.
- W. G. Brodbeck, Y. Nakayama, T. Matsuda, E. Colton, N. P. Ziats and J. M. Anderson, Cytokine, 2002, 18, 311–319 CrossRef CAS.
- D. A. Rappolee, D. Mark, M. J. Banda and Z. Werb, Science, 1988, 241, 708–712 CrossRef CAS.
- D. A. Rappolee and Z. Werb, Curr. Top. Microbiol. Immunol., 1992, 181, 87–140 Search PubMed.
- J. A. Jones, D. T. Chang, H. Meyerson, E. Colton, I. K. Kwon, T. Matsuda and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2007, 83, 585–596 CrossRef.
- D. T. Chang, E. Colton and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2009, 89, 490–498 CrossRef.
- T. O. Collier and J. M. Anderson, J. Biomed. Mater. Res., 2002, 60, 487–496 CrossRef CAS.
- M. Dadsetan, J. A. Jones, A. Hiltner and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2004, 71, 439–448.
- A. Kinasiewicz, K. Dudzinski, A. Chwojnowski, A. Werynski and J. Kawiak, Transplant Proc., 2007, 39, 2914–2916 CrossRef CAS.
- S. H. Jackman, M. B. Yoak, S. Keerthy and B. L. Beaver, Ann. Clin. Lab. Sci., 2000, 30, 201–207 Search PubMed.
- G. G. Vaday, S. Franitza, H. Schor, I. Hecht, A. Brill, L. Cahalon, R. Hershkoviz and O. Lider, J. Leukocyte Biol., 2001, 69, 885–892 Search PubMed.
- A. M. Ferreira, S. Takagawa, R. Fresco, X. Zhu, J. Varga and L. A. DiPietro, J. Invest. Dermatol., 2006, 126, 1900–1908 CrossRef CAS.
- S. J. Leibovich and R. Ross, Am. J. Pathol., 1975, 78, 71–100 CAS.
- T. Yamamoto, B. Eckes, C. Mauch, K. Hartmann and T. Krieg, J. Immunol., 2000, 164, 6174–6179 CAS.
- E. Engelhardt, A. Toksoy, M. Goebeler, S. Debus, E. B. Brocker and R. Gillitzer, Am. J. Pathol., 1998, 153, 1849–1860 CAS.
- G. J. Randolph and M. B. Furie, J. Immunol., 1995, 155, 3610–3618 CAS.
- M. Romano, M. Sironi, C. Toniatti, N. Polentarutti, P. Fruscella, P. Ghezzi, R. Faggioni, W. Luini, V. van Hinsbergh, S. Sozzani, F. Bussolino, V. Poli, G. Ciliberto and A. Mantovani, Immunity, 1997, 6, 315–325 CrossRef CAS.
- Y. Sato and T. Ohshima, Int. J. Legal Med., 2000, 113, 140–145 CrossRef CAS.
- K. Asadullah, R. Sabat, A. Wiese, W. D. Docke, H. D. Volk and W. Sterry, Arch. Dermatol. Res., 1999, 291, 628–636 CrossRef CAS.
- Y. Sato, T. Ohshima and T. Kondo, Biochem. Biophys. Res. Commun., 1999, 265, 194–199 CrossRef CAS.
- D. F. Fiorentino, A. Zlotnik, T. R. Mosmann, M. Howard and A. O'Garra, J. Immunol., 1991, 147, 3815–3822 CAS.
- P. H. Hart, E. K. Hunt, C. S. Bonder, C. J. Watson and J. J. Finlay-Jones, J. Immunol., 1996, 157, 3672–3680 CAS.
- T. Maier, M. Gueell and L. Serrano, FEBS Lett., 2009, 583, 3966–3973 CrossRef CAS.
- M. K. O'Bryan, O. Gerdprasert, D. J. Nikolic-Paterson, A. Meinhardt, J. A. Muir, L. M. Foulds, D. J. Phillips, D. M. de Kretser and M. P. Hedger, Am. J. Physiol., 2005, 288, R1744–R1755 CAS.
- N. Favre, G. Bordmann and W. Rudin, J. Immunol. Methods, 1997, 204, 57–66 CrossRef CAS.
- D. M. Higgins, R. J. Basaraba, A. C. Hohnbaum, E. J. Lee, D. W. Grainger and M. Gonzalez-Juarrero, Am. J. Pathol., 2009, 175, 161–170 CrossRef CAS.
- J. Sambrook, T. Maniatis and E. F. Fritsch, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 2nd edn, 1989 Search PubMed.
Footnote |
| † Present address: Clinical Research Center, Albert Einstein College of Medicine of Yeshiva University, Bronx, NY 10461, USA. |
| This journal is © The Royal Society of Chemistry 2011 |