Metabolic profiling of human urine by CE-MS using a positively charged capillary coating and comparison with UPLC-MS
Rawi
Ramautar
*ab,
Ekaterina
Nevedomskaya
b,
Oleg A.
Mayboroda
b,
André M.
Deelder
b,
Ian D.
Wilson
c,
Helen G.
Gika
d,
Georgios A.
Theodoridis
d,
Govert W.
Somsen
a and
Gerhardus J.
de Jong
a
aDepartment of Biomedical Analysis, Utrecht University, Sorbonnelaan 16, P.O. Box 80082, 3508 TB Utrecht, The Netherlands. E-mail: R.Ramautar@lumc.nl; Fax: +31 30-253-5180
bBiomolecular Mass Spectrometry Unit, Department of Parasitology, LUMC, Leiden, The Netherlands
cDepartment of Clinical Pharmacology and Drug Metabolism and Pharmacokinetics, AstraZeneca, AlderleyPark, Macclesfield, Cheshire, SK10 4TG, United Kingdom
dChemistry Department, Aristotle University of Thessaloniki, Thessaloniki 54124, Greece
First published on 5th November 2010
Abstract
The potential of capillary electrophoresis time-of-flight mass spectrometry (CE-TOF-MS) using capillaries coated with a triple layer of polybrene–dextran sulfate–polybrene (PB–DS–PB) was evaluated for metabolic profiling of human urine. The method covers various metabolite classes and stable metabolic profiles of urine samples were obtained with favourable migration time repeatability (RSDs <1%). The PB–DS–PB CE-TOF-MS method was used for the analysis of human urine samples from 30 males and 30 females, which had been previously analyzed by reversed-phase UPLC-TOF-MS. Multivariate data analysis of the obtained data provided clear distinction between urine samples from males and females, emphasizing gender differences in metabolic signatures. Nearly all compounds responsible for male–female classification in CE-TOF-MS were different from the classifying compounds in UPLC-TOF-MS. Almost all compounds causing classification in the CE-TOF-MS study were highly polar and did not exhibit retention in the reversed-phase UPLC system. In addition, the CE-TOF-MS classifiers had an m/z value in the range of 50–150, whereas 95% of the classifying features found with UPLC-TOF-MS had an m/z value above 150. The CE-TOF-MS method therefore appears to be highly complementary to the UPLC-TOF-MS method providing classification based on different classes of metabolites.
1. Introduction
The aim of metabolic profiling is to analyze a wide range of endogenous metabolites in a biological sample within a single run.1–4 No single analytical technique is capable of providing a comprehensive view of the endogenous metabolites present in a biological sample. In order to increase the coverage of the metabolome, several analytical techniques have been used in conjunction enabling the detection of metabolites which cannot be detected by the use of only one method.5,6 For instance, hydrophilic interaction chromatography (HILIC) has been used as a complementary method to a reversed-phase LC (RPLC) method in order to improve the coverage of (highly) polar metabolites in rat and human urine.7,8 Recently, it has been demonstrated that the retention of highly polar analytes in RPLC can be improved by using modified C18 stationary phases such as mixed functional RP phases and Synergi C18 materials embedded with polar groups.9,10 It is becoming more and more evident that for the satisfactory coverage of a given metabolome, data from multiple separation techniques should be used.11 Such a strategy, comprising six different analytical methods, has been recently developed for the comprehensive analysis of the microbial metabolome.12A large proportion of the endogenous metabolites present in biological samples are highly polar and ionic and, therefore, capillary electrophoresis (CE) is a very attractive separation technique for the metabolic profiling of such samples, as compounds are separated on the basis of their charge-to-size ratio.13,14 Other features of CE include the relatively fast and highly efficient separations with minimal sample pretreatment. These aspects, together with the small sample requirement, make CE particularly suitable for the analysis of biological samples that are volume-limited.15
Recently, we have developed a CE-TOF-MS method for the analysis of amino acids and related compounds in human urine.16 In this method, CE capillaries were noncovalently coated with a bilayer of polybrene (PB) and poly(vinyl sulfonate) (PVS) providing a considerable electro-osmotic flow (EOF) at low pH, thus facilitating the fast separation of amino acids using a background electrolyte (BGE) of 1 M formic acid (pH 1.8). Although this CE-TOF-MS method can be used for the fast and reproducible profiling of amino acids in human urine samples, the separation window for cationic compounds was limited (ca. 10 min). A longer separation window for cationic compounds can be obtained by using bare fused-silica capillaries at low pH. However, using bare fused-silica capillaries we found considerable migration time variation for amino acids spiked in urine (RSDs of 5–15%).16 Another approach to increase the separation window for cationic compounds is by using a CE-TOF-MS method based on PB–dextran sulfate (DS)–PB coated capillaries. In this set-up, cationic compounds will migrate after the EOF time at low pH conditions and as a result a larger separation window is obtained.
In the present study we have evaluated the potential of this PB–DS–PB CE-TOF-MS method for metabolic profiling of human urine by first examining the type of metabolite classes that can be analyzed. Subsequently, this method was evaluated by the analysis of urine samples from 30 male and 30 female subjects. Multivariate data analysis of the CE-TOF-MS data was carried out in order to test the ability of the method to discriminate between evident metabolic signatures. The results were compared with the data and metabolic information obtained by reversed-phase UPLC-TOF-MS on the same set of samples. Metabolic profiling of the human urine samples was performed with conventional CE-TOF-MS and UPLC-TOF-MS systems.
2. Materials and methods
2.1 Chemicals and reagents
All the employed chemicals were of analytical grade or higher purity. L-Tyrosine, L-phenylalanine, L-asparagine, L-histidine, formic acid and ammonium hydroxide were purchased from Fluka (Buchs, Switzerland). Creatinine, glutathione, tyramine, proline betaine, hippuric acid, N-methylnicotinamide, polybrene (PB), dextran sulfate (DS) and poly(vinyl sulfonate) (PVS) were from Sigma-Aldrich (Steinheim, Germany). Methanol was obtained from Chromasolv™. Ammonium acetate was purchased from Merck (Darmstadt, Germany).2.2 Coating procedure
The PB–DS–PB coating was prepared by sequentially rinsing the capillary with 10% (m/v) PB solution for 15 min at 350 mbar, deionized water for 5 min at 1380 mbar, 3% (m/v) DS solution for 15 min at 350 mbar, deionized water for 5 min at 1380 mbar, and, finally with a 10% (m/v) PB solution for 15 min at 350 mbar. The capillary was then ready for use with the BGE of choice. Before analysis, capillaries were flushed with BGE for 5 min at 1380 mbar. Between runs, the coated capillaries were flushed with a 1% (m/v) PB solution and BGE, each for 5 min at 1380 mbar.2.3 CE-TOF-MS and UPLC-TOF-MS
CE-TOF-MS experiments were performed on a Beckman Coulter PA 800 instrument (Beckman Coulter, Fullerton, CA, USA) coupled to a microToF mass spectrometer from Bruker (Bruker Daltonics, Bremen, Germany) via an ESI electrospray interface from Agilent (Waldbronn, Germany). Separations occurred in a 100 cm long PB–DS–PB coated capillary with an internal diameter of 50 μm. A BGE of 1 M formic acid (FA) (pH 2.0) was used for the separation of cationic and basic compounds. A sheath liquid composed of methanol/water (1/1, v/v) containing 0.1% FA was infused via a 2.5 mL Hamilton syringe at 4 μL min−1. Samples were hydrodynamically injected by applying a positive pressure of 0.5 psi for 30 s. A voltage of −30 kV (reversed polarity) was applied during separation. The electrospray voltage was −4.5 kV. MS transfer parameters were optimized by direct infusion of an ESI tuning mix (Agilent Technologies, Waldbronn, Germany). Data were acquired over the mass range of 50–1000 m/z with a repetition rate of 1 Hz.The experimental conditions for UPLC-TOF-MS analysis were largely based on previously developed method.6 Briefly, chromatography was performed on an Acquity UPLC system (Waters) with an Acquity C18 BEH column 100 × 2.1 mm, 1.7 μm. The injection volume was 10 μL. A gradient program was applied for analyte elution starting from 100% A (0–2min) and then switching to 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶10 A∶B (2.01 min) followed by linear gradient to 100% B at 6.5 min where it stayed isocratic for column clean-up till 7 min. The flow rate used was 0.2 mL min−1 for the first two min and 0.4 mL min−1 for the rest of the run. Solvents were: 0.1% formic acid in LC-MS quality water (A) and 0.1% formic acid in acetonitrile (B). MS was performed using a Waters Micromass Q-TOF Micro (Milford, MA, USA) operating in positive ion electrospray mode. Full scan data were collected from 80 to 850 m/z over a period of 9 min with a scan time of 0.3 s.
∶10 A∶B (2.01 min) followed by linear gradient to 100% B at 6.5 min where it stayed isocratic for column clean-up till 7 min. The flow rate used was 0.2 mL min−1 for the first two min and 0.4 mL min−1 for the rest of the run. Solvents were: 0.1% formic acid in LC-MS quality water (A) and 0.1% formic acid in acetonitrile (B). MS was performed using a Waters Micromass Q-TOF Micro (Milford, MA, USA) operating in positive ion electrospray mode. Full scan data were collected from 80 to 850 m/z over a period of 9 min with a scan time of 0.3 s.
2.4 Test mixture and urine samples
A test mixture of standard substances was prepared and used to evaluate the CE-TOF-MS method. It comprised tyramine, dopamine, creatinine, hippuric acid, glutathione, proline betaine and amino acids (L-glutamic acid, L-histidine, L-methionine, L-phenylalanine, L-tyrosine). Initially, 1 mg mL−1 stock solutions of each analyte were prepared by dissolving appropriate amounts in water. Subsequently, aliquots of stock solutions were diluted with water in a 1.5 mL glass vial in order to obtain a working solution in which each analyte was present at a 50 μg mL−1 concentration. Stock solutions were kept at −20 °C until usage. Human urine samples were collected and stored at −80 °C prior to usage as described in ref. 6 and 17. Before CE-TOF-MS analysis, urine samples were mixed with BGE (1∶1, v/v) and centrifuged at 10000 g for 5 min. The repeatability of the CE-TOF-MS method was determined by ten consecutive analyses of a pooled human urine sample spiked with 50 μM of each of the test compounds. Prior to UPLC-TOF-MS analysis urine samples were diluted to a final dilution ratio 1∶5 (v/v) with water (containing 0.1% formic acid).
2.5 Data analysis
CE-TOF-MS data were exported to mzXML format using Compass Export (Bruker Daltonics). For the alignment and peak picking XCMS software has been used (Scripps Center, CA, USA) with default parameters except for “missing” and “extra”, both set to 15. The resulting table contained areas for the found peaks across all the samples. These values were normalized to total areas of the corresponding samples to compensate the difference in concentrations between the subjects due to absolute urine volume. Subsequently the table was imported into SIMCA-P+ (Umetrics, Windsor, UK) software for multivariate analysis. Principal component analysis (PCA) and Partial Least Squares Discriminant Analysis (PLS-DA) were performed using Pareto scaling. UPLC-TOF-MS data were processed by MarkerLynx software with the following track peak parameters: peak width at 5% height: 15 s, peak-to-peak baseline noise: 80, intensity threshold: 100, mass window: 0.05 amu, retention time window: 0.2 min, noise elimination level: 6 and mass tolerance: 0.50 amu. Peak list data obtained by MarkerLynx were further processed by SIMCA P version 11 for multivariate data analysis.3. Results and discussion
3.1 Evaluation of the PB–DS–PB CE-TOF-MS method
Metabolic profiling of urine is quite challenging as many chemical classes should be analyzed simultaneously within a single run. A test mixture comprising compounds such as amino acids, organic acids, amines and catecholamines was analyzed first to determine the type of metabolite classes that can be covered with the PB–DS–PB CE-TOF-MS method. When analyzing the test mixture with the PB–DS–PB CE-TOF-MS method using 1 M formic acid (pH 2.0) as BGE, most organic acids co-migrated with the EOF as they are neutral at pH 2.0. Hippuric acid, which is slightly negatively charged at pH 2.0, migrated directly before the EOF time (ca. 9 min), as shown in Fig. 1. Compounds that are positively charged at pH 2.0 such as amino acids, catecholamines and small peptides migrated after the EOF time. As the EOF time was ca. 9 min, the effective separation window for positively charged compounds is ca. 20 min if we consider a maximum separation time of 30 min, which is twice as large as the separation window obtained with the PB–PVS CE-TOF-MS method at low pH.16,18 Moreover, positively charged compounds are separated using a positively charged coated capillary, thereby preventing potential electrostatic adsorption to the capillary wall. Plate numbers ranged from 100000 to 300000 for the test compounds. In general, various metabolite classes can be covered with this method (i.e. organic acids, nucleosides, amino acids, catecholamines, small peptides and amines); however, small multivalent cationic compounds will not be detected due to their strong electrophoretic mobility towards the cathode (i.e., opposite to the direction of the EOF).
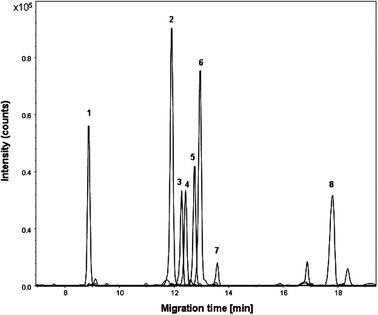 | ||
| Fig. 1 Multiple extracted ion electropherogram obtained during CE-TOF-MS analysis of a pooled human urine sample spiked with test compounds (50 μM). Peaks: 1, hippuric acid; 2, proline betaine; 3, N-methylnicotinamide; 4, L-tyrosine; 5, L-phenylalanine; 6, tyramine; 7, L-asparagine; 8, L-histidine. Conditions: BGE, 1 M formic acid (pH 2.0); sample injection, 0.5 psi for 30 s; separation voltage, −30 kV. | ||
With the PB–DS–PB CE-TOF-MS method, about 500 molecular features (i.e. the number of peaks detected above a certain intensity threshold within the CE run time) were detected in human urine, which was almost twice as many as observed with the PB–PVS CE-TOF-MS method.18 For a reliable comparison of metabolic profiles and to be able to observe small changes in sample composition, migration time stability is of crucial importance. Fig. 2 shows base peak electropherograms of the repeated analysis (n = 10) of a pooled human urine sample. The RSDs for migration times of the test compounds were always smaller than 1%, indicating that stable profiles were obtained (Table 1). The good migration-time repeatability for test compounds in urine samples can be primarily attributed to the use of the PB–DS–PB capillary coating. The RSDs for peak areas of the test compounds were smaller than 10% (Table 1), which is acceptable for metabolomics studies using ESI-MS. In summary, the PB–DS–PB CE-TOF-MS method appears suitable for the profiling of polar metabolite classes in human urine within a single run.
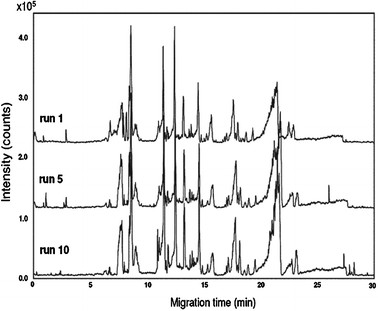 | ||
| Fig. 2 Repeated CE-TOF-MS analyses (n = 10) of a pooled human urine sample. Base peak electropherograms (m/z 50–1000) of the first, middle and last run are shown. Conditions: BGE, 1 M formic acid (pH 2.0); sample injection, 0.5 psi for 30 s; separation voltage, −30 kV. | ||
| Compound | Migration time/min | Migration time RSD (%) | Peak area RSD (%) |
|---|---|---|---|
| Hippuric acid | 8.9 | 0.8 | 7.6 |
| Proline betaine | 11.9 | 0.9 | 8.3 |
| N-Methylnicotinamide | 12.2 | 0.9 | 9.4 |
| L-Tyrosine | 12.4 | 0.9 | 8.5 |
| L-Phenylalanine | 12.8 | 0.8 | 8.3 |
| Tyramine | 13.0 | 0.9 | 9.3 |
| L-Asparagine | 13.7 | 0.7 | 8.4 |
| L-Histidine | 17.8 | 0.9 | 9.7 |
3.2 PB–DS–PB CE-TOF-MS of male and female urine
The PB–DS–PB CE-TOF-MS method was applied to the analysis of urine samples from 30 male and 30 female human subjects, and both unsupervised (PCA) and supervised (PLS–DA) multivariate statistical analysis of the CE-MS data were carried out. The PCA scores plot (data not shown) revealed a tendency for the grouping of the female and male urine samples. Therefore, PLS–DA, which turns the projection so that it reflects not only the variation present in the data, but also maximizes the difference between the sample groups, was applied. The PLS–DA scores plot shows a separation of female and male urine samples (Fig. 3A). The distinction between female and male urine samples is significant in the first PC, reflecting differences in metabolic profiles between urine from female and male subjects. The model parameters for the explained variance and predictive ability were R2 = 0.45 and Q2 = 0.38. To ensure that discrimination in the PLS–DA model was not the result of data overfitting, the model was validated using permutation testing, which helps to assess overfitting by randomly permuting class labels and refitting a new model with the same number of components as the original model. An overfitted model will have similar R2 (goodness of fit) and Q2 (goodness of prediction) values to that of the randomly permuted data. A good and meaningful model will have R2 and Q2 values that are always higher than that of models based on permuted data. Fig. 3B shows that the model parameters for the explained variation, R2, and the predictive capability, Q2, were significantly higher than of the models based on the permuted data, indicating a good model. | ||
| Fig. 3 (A) PLS–DA scores plot of urine samples analyzed by CE-TOF-MS (■ females; △ males). R2 = 0.45; Q2 = 0.38. (B) Cross validation using random permutations, ▲ stand for R2 values (on the left—for actual model, the rest for permuted ones), ■ for Q2 values. | ||
An important output of PLS–DA is the possibility to estimate and rank the influence of individual features on the model with VIP (variable influence on the projection). In theory, all features above threshold (α ≥ 1) are considered to be significant for the given model, but in practice, the threshold depends on the size of the data set. A limited number of samples may result in overfitting of the model and, therefore, only features with α ≥ 1.5 and standard deviation significantly lower than the ranking factor were selected for molecule assignment and identification. This resulted in 27 compounds responsible for discriminating male from female urine samples. The provisional identification of these compounds was performed by using m/z values combined with the migration times of the compounds, and these data were compared with those of the standards and databases, such as the Human Metabolome Database.19 For this purpose, the use of TOF-MS is very practical as the accurate mass measurement obtained for unknown compounds considerably reduces the list of possible candidates. As TOF-MS was used for detection, a number of possible elemental compositions were obtained from the accurate mass of the metabolite peaks. These elemental compositions were matched against available databases using the deduced molecular formula as a search criterion. By using this approach 8 out of the 27 compounds with a VIP ≥ 1.5 could be provisionally identified (Table 2). It was not possible to distinguish between 1- and 3-methylhistidine, as the PB–DS–PB CE-TOF-MS method could not separate these compounds.
| Name | Molecular formula | m/z observed | m/z calculated | Error/mDa | Migr. timeb observed/min | Migr. timeb standards/min |
|---|---|---|---|---|---|---|
| a ND, not determined (no standard available). b Migr. time = migration time. | ||||||
| Methylhistidine | C7H11N3O2 | 170.1080 | 170.0924 | 15.6 | 17.6 | 17.6 |
| Glutamic acid | C5H9NO4 | 148.0850 | 148.0532 | 31.8 | 10.9 | 10.8 |
| Pyroglutamic acid | C5H7NO3 | 130.0630 | 130.0426 | 20.4 | 9.1 | NDa |
| Hypotaurine | C2H7NO2S | 110.0802 | 110.0196 | 60.7 | 17.1 | NDa |
| Threonine | C4H9NO3 | 120.0912 | 120.0582 | 33.0 | 15.2 | 15.1 |
| Methionine | C5H11NO2S | 150.0920 | 150.0511 | 40.9 | 15.7 | 15.7 |
| Methylnicotinamide | C7H9N2O | 138.0728 | 138.0715 | 1.3 | 12.1 | 12.2 |
| Proline betaine | C7H13NO2 | 144.1160 | 144.1019 | 14.1 | 12.0 | 11.9 |
3.3 Comparison with reversed-phase UPLC-TOF-MS
The same set of urine samples had previously been analyzed by a reversed-phase UPLC-TOF-MS method. The separation time was 15 min, which was two times shorter than the separation time in the PB–DS–PB CE-TOF-MS method. With the UPLC-TOF-MS study, PLS–DA also revealed a classification of male and female urine samples (Fig. 4). The fraction of the differences between the groups explained by the PLS–DA model, expressed by the cumulative R2 for the two components, was calculated to be 0.95 for the UPLC-TOF-MS dataset, which was higher than that obtained from the CE-TOF-MS data. The predictive capability of the PLS–DA model (Q2 = 0.80) was also better for the UPLC-MS data than for the CE-TOF-MS data.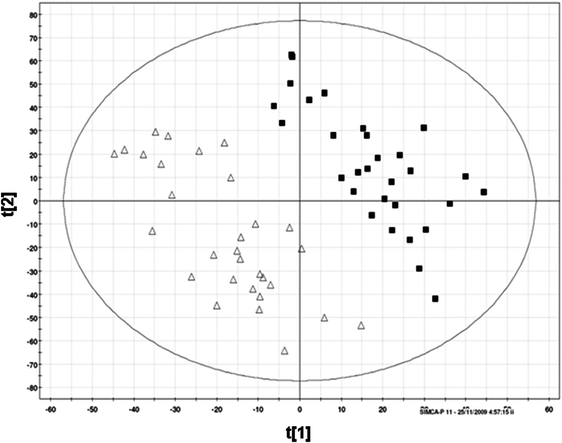 | ||
| Fig. 4 PLS–DA scores plot of urine samples analyzed by UPLC-TOF-MS (■ females; Δ males). R2 = 0.95; Q2 = 0.80. | ||
About 300 features with a VIP ≥ 1.5 were responsible for the separation of female and male urine in the UPLC-TOF-MS study, which is about ten times higher than the number of classifying features obtained with CE-TOF-MS. This could probably be related to the fact that the concentration sensitivity of UPLC-TOF-MS is considerably higher than that of CE-TOF-MS, as much larger injection volumes (10 μL vs. 30 nL for CE) can be applied, and that the reversed-phase UPLC method covers a wider range of compounds than CE. Interestingly, with the exception of glutamic acid, the provisionally identified compounds responsible for the classification of female and male urine samples in the CE-MS study (Table 2) showed no retention in the reversed-phase UPLC system. That is, they eluted with the column dead time as was confirmed by injection of standard compounds. When comparing the lists of m/z values of features responsible for gender classification obtained with UPLC-TOF-MS and CE-TOF-MS, seven m/z values, i.e., 85.03, 105.04, 120.09, 130.06, 131.03, 132.05, and 170.11, appeared in both lists. In the CE-MS study, three of these m/z values were assigned to pyroglutamic acid (m/z 130.06), threonine (m/z 120.09) and methylhistidine (m/z 170.11). However, as these compounds did not exhibit retention in the UPLC-MS system, the respective m/z values found with UPLC-MS originate from different compounds. Another interesting aspect is that almost all features causing classification in the CE-TOF-MS study had an m/z value in the range of 100–150, whereas more than 95% of the classifying features found with UPLC-TOF-MS had an m/z value above 150. Hence, these results indicate that the two separation methods provide complementary metabolic information and, therefore, an improved coverage of urinary metabolites is obtained by using these two techniques in conjunction. This is very important for metabolomics studies where the aim is to obtain maximum information on the endogenous metabolites in body fluids. For the identified metabolites causing the classification of female and male urine quantification is possible using an internal or external calibration approach, however, this was not the goal of the present study.
4. Concluding remarks
In the present study, the suitability of a PB–DS–PB CE-TOF-MS method for metabolic profiling of human urine samples was demonstrated. Profiles covering several metabolite classes in human urine could be recorded in a repeatable way. On the basis of CE-TOF-MS data, urine from female subjects could be distinguished from urine samples obtained from males, and the classifying metabolites reflected the polar fraction of the urinary metabolome. The same set of samples had also been measured by a UPLC-TOF-MS method and samples from males and female subjects were also well resolved. However, the metabolites causing the classification in the CE-TOF-MS study were largely different from the compounds responsible for the classification obtained with UPLC-TOF-MS. Hence, this study has shown that CE-TOF-MS and UPLC-TOF-MS may be considered as complementary techniques for metabolic profiling of human urine as the analysis of highly polar and charged compounds is better attainable with CE-TOF-MS.It will be interesting to compare PB–DS–PB CE-TOF-MS with hydrophilic interaction liquid chromatography (HILIC)-MS for metabolic profiling of human urine. As HILIC-MS is also highly suited for the separation of polar compounds it would be illustrative to establish the complementary character of these systems. Moreover, it would be interesting to compare the CE-MS method with nano-LC-MS for metabolic profiling of small-sized samples as a significant improvement in sensitivity can be achieved through down-scaling of the LC column diameter and by miniaturization of the ESI source (nano-ESI). For this comparison, the sheath–liquid interface for the coupling of CE to MS may be replaced by a sheathless interface in order to make it compatible with nano-ESI-MS.
Abbreviations
| PB–DS–PB | polybrene–dextran sulfate–polybrene |
| PB–PVS | polybrene–poly(vinyl sulfonate) |
| CE-TOF-MS | capillary electrophoresis time-of-flight mass spectrometry |
| EOF | electro-osmotic flow |
| BGE | background electrolyte |
Acknowledgements
H. Gika’s work is funded by a Marie Curie ERG 202132 Reintegration grant from the European Committee. H. Gika acknowledges the provision of laboratory space for computational work from the Laboratory of Forensic Medicine and Toxicology, Medical School Aristotle University Thessaloniki.References
- J. van der Greef, P. Stroobant and R. van der Heijden, Curr. Opin. Chem. Biol., 2004, 8, 559–565 CrossRef CAS.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS.
- J. K. Nicholson, J. Connelly, J. C. Lindon and E. Holmes, Nat. Rev. Drug Discovery, 2002, 1, 153–161 CrossRef CAS.
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155–171 CrossRef CAS.
- E. M. Lenz and I. D. Wilson, J. Proteome Res., 2007, 6, 443–458 CrossRef CAS.
- G. Theodoridis and I. D. Wilson, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 141–142 CrossRef CAS.
- H. G. Gika, G. A. Theodoridis and I. D. Wilson, J. Sep. Sci., 2008, 31, 1598–1608 CrossRef CAS.
- R. Mohamed, E. Varesio, G. Ivosev and L. Burton, et al. , Anal. Chem., 2009, 81, 7677–7694 CrossRef CAS.
- K. T. Myint, K. Aoshima, S. Tanaka, T. Nakamura and Y. Oda, Anal. Chem., 2009, 81, 1121–1129 CrossRef CAS.
- J. Ding, C. M. Sorensen, Q. Zhang and H. Jiang, et al. , Anal. Chem., 2007, 79, 6081–6093 CrossRef CAS.
- J. M. Buscher, D. Czernik, J. C. Ewald, U. Sauer and N. Zamboni, Anal. Chem., 2009, 81, 2135–2143 CrossRef CAS.
- M. J. van der Werf, K. M. Overkamp, B. Muilwijk, L. Coulier and T. Hankemeier, Anal. Biochem., 2007, 370, 17–25 CrossRef CAS.
- S. Ullsten, R. Danielsson, D. Backstrom, P. Sjoberg and J. Bergquist, J. Chromatogr., A, 2006, 1117, 87–93 CrossRef CAS.
- R. Ramautar, G. W. Somsen and G. J. de Jong, Electrophoresis, 2009, 30, 276–291 CrossRef CAS.
- M. R. Monton and T. Soga, J. Chromatogr., A, 2007, 1168, 237–246 CrossRef CAS ; discussion 236.
- R. Ramautar, O. A. Mayboroda, R. J. Derks and C. van Nieuwkoop, et al. , Electrophoresis, 2008, 29, 2714–2722 CrossRef CAS.
- H. G. Gika, G. A. Theodoridis, J. E. Wingate and I. D. Wilson, J. Proteome Res., 2007, 6, 3291–3303 CrossRef CAS.
- R. Ramautar, A. A. van der Plas, E. Nevedomskaya and R. Derks, et al. , J. Proteome Res., 2009, 8, 5559–5567 CrossRef CAS.
- D. S. Wishart, C. Knox and A. C. Guo, et al. , Nucleic Acids Res., 2009, 37, D603–D610 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |