Droplet microfluidics—a tool for protein engineering and analysis
Haakan N.
Joensson
and
Helene
Andersson-Svahn
*
Albanova University Center, Royal Institute of Technology (KTH), Stockholm, Sweden. E-mail: helene.andersson-svahn@biotech.kth.se
First published on 7th October 2011
Introduction
Protein engineering is a field that seeks to tailor-make the traits of proteins from natural precursors or de novo designed proteins. Inhabiting the subcellular length scale, and an aqueous environment, the efficient screening and analysis of proteins calls for fluid handling on the micro scale. Microfluidics has come to play an important supporting role in DNA sequencing, with many of the most common DNA sequencing instruments including vital microfluidic components of some form. Furthermore, a number of companies utilizing droplet microfluidics are currently offering products related to DNA sequencing. Microfluidic tools for protein engineering can build on these advances. In this focus article we will further explore the application of droplet microfluidics for protein engineering.Engineered proteins are expected to be increasingly important in industrial biotechnology-based manufacturing of commodities such as fine chemicals and fuels, many of them related to bioenergy and biosustainability as discussed in our last article.1 Recent estimates value the industrial enzymes market in the multi-billion dollar range. The manufacture of therapeutics and implementation of their use offer significant areas for engineered proteins to make an impact. The 2010 biologicals market reached $149 billion, where protein therapeutics accounted for 49% and monoclonal antibodies for 32%. Currently, more than 130 protein therapeutics have been approved for clinical use by the US Food and Drug Agency.2 These proteins range from enzymes and affinity molecules to protein hormones, vaccines and proteins used in diagnostics. Even though not all of these proteins were specifically engineered, the proportion of engineered proteins is large and still rising.
The cost of DNA synthesis (Fig. 1) by conventional means has decreased to a fraction of a dollar per base pair. At the same time novel microfluidic solutions for DNA synthesis have been developed.3 The low costs of DNA synthesis combined with the large number of protein library technologies at hand—with capacities to produce 108–1015 different sequences—has created a need for automated high-throughput technologies to effectively screen and analyze proteins that can cope with the size of these libraries.
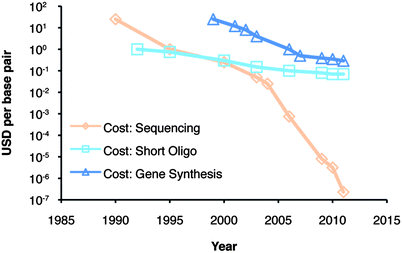 | ||
| Fig. 1 The cost per base pair in US dollars (USD) of DNA synthesis, short oligonucleotide synthesis and DNA sequencing over the last 20 years. Image adapted from http://www.synthesis.cc with permission. | ||
Droplet microfluidics
Droplet microfluidics involves the controlled formation and manipulation of nano- to femtolitre droplets of one fluid phase in another fluid immiscible with the first. Earlier systems exploiting two immiscible fluids for biological experimentation have been utilized for protein analysis prior to the advent of microfluidic circuits for droplet generation, notably in 1961 the Nobel Prize winner Joshua Lederberg used aqueous droplets in oil to study the characteristics of β-galactosidase4 in what has to be one of the earliest examples of a single biomolecule study. Generation of droplets in microfluidic circuits has however enabled the formation of droplets with an extreme monodispersity of size, enabling quantitative assays in droplets. In many cases aqueous droplets are generated in a hydrocarbon or fluorocarbon oil continuous phase containing surfactants to stabilize the interface, allowing the droplets to remain stable over long periods of time. The development of a multitude of functional manipulations of droplets e.g., splitting, fusion, incubation, and most notably active sorting, capable of processing thousands of droplets per second, provides the basis for automated high-throughput handling of fluid packets. Combined with biological assays these manipulations have the potential to serve as a high-throughput screening and experimentation platform.Several companies have been started around the use of droplet microfluidic technologies (or have come to use them), for example RainDance Technologies, QuantaLife, Sphere Fluidics, Emerald Biosciences and GnuBIO. These companies have thus far mainly focused on products and solutions related to DNA sequencing. The first such product brought to market was introduced by RainDance Technologies in 2008. Emerald Biosciences, which markets a protein crystallization screening product using microfluidic formulation and screening in a plug format, is the only company to provide products relating to protein screening or analysis. While commercial efforts have focused around genomics and sequencing, the tools and automation developed are in many respects very well suited for screening and analysis of engineered proteins. Proteins, such as green fluorescent protein, have for example been expressed in vitro in droplet microfluidic devices.5
Droplet microfluidics has been proposed as a screening platform for a plethora of applications such as in chemical synthesis, screening for small molecule drugs, cells or proteins. While all of these applications are of tremendous interest, the droplet microfluidics technology platform with the poignant characteristic that the encapsulation reagents and surfactants used must be compatible with the droplet contents,6 the limited chemical variations and generally large size of biopolymers such as proteins make these a particularly interesting group of molecules to apply these set of screening techniques to, rather than, e.g., a library of small drug molecules, which tend to have more diverse chemical traits.
Protein engineering and screening
Recombinant DNA, DNA synthesis and related technologies have been at the core of the rise of molecular biotechnology over the last 40 years. The expression of foreign or synthetically designed proteins are now commonplace in many different hosts, notably CHO, E. Coli, etc., and are currently some of the fastest growing areas in biotechnology, e.g. biologicals in therapeutic drugs and diagnostics as well as enzymes for industrial use, particularly in the biofuels and bioprocessing industries. Some of the largest grossing of these products involve engineered proteins, i.e. proteins which have been improved starting from an ancestor protein found in nature e.g. engineered insulin analogues. Two general strategies are used in protein engineering; rational design and an evolutionary approach known as directed evolution, although these are to some extent used complementarily to screen sequence space for protein fitness (Fig. 2). | ||
| Fig. 2 The basic experimental workflow for directed evolution. | ||
Directed evolution is an iterative process resembling Darwinian evolution, but where the fitness providing trait can be selected. The technique takes a known sequence as its starting point. Subsequently a sequence library is generated, containing many mutated copies of the starting sequence under an elevated mutational rate. Finally the mutated copies where the desired trait has been most improved are selected and the selected sequences are used as starting material for a new library for continued iteration (Fig. 3). Rational design using in silico methods has so far often been difficult because of incomplete information about protein function, structure and dynamics. In silico modeling can however outperform evolutionary approaches in the speed by which sequences can be screened and offers the possibility of accessing parts of sequence space not readily available to in vitro screening. Even though many rationally designed proteins have thus far, once expressed, shown a very limited function, these may be used as the starting material for subsequent rounds of directed evolution, directly probing the local sequence space for more fit variants.
 | ||
| Fig. 3 Fitness landscapes of proteins where lighter colors indicate a high degree of fitness. (A) The hypothetical fitness landscape for a certain protein trait across sequence space. The two protruding regions indicate two sequence regions where protein fitness is above base level for the required trait. (B) Two protein fitness landscapes. The smoothness of the protein fitness landscape has implications for whether or not a directed evolution approach is likely to be successful. Reprinted by permission from Macmillan Publishers Ltd: Nat. Rev. Mol. Cell Biol. © 2009.13 | ||
Proteins are engineered for traits such as affinity, selectivity, stability or enzymatic activity. For improved affinity, screening of engineered protein libraries expressed on single cells or phages is commonplace, mainly using flow cytometry. This approach does however generally require the protein to be expressed on the cell surface and not in the soluble form which often is the form it is eventually intended to be used. Compartmentalization of single cells or alternative expression systems offer the possibility to select by secreted proteins and screening for traits other than affinity. For example, enzyme variants with improved functionality can be selected by the product of a substrate reaction contained in the compartment along with the DNA sequence information.
Protein libraries
Diverse libraries of DNA sequences, created by error-prone PCR, recombination or other techniques can be expressed as proteins in a number of different formats.7 While no library format can cover the entire sequence space, which for a even a 20 residue polypeptide amounts to 1026 sequences, libraries as large as 1015 members have been constructed by e.g., mRNA or ribosome display. Cell based methods such as bacterial or yeast based display offer a format where production can be scaled up without the need for further cloning. Yeast cells allow post translational modifications which are specific to eukaryotic cells. The libraries of cells produced typically range between 108 to 1010 members. Phage display, which utilizes phage particles incapable of independent proliferation, is a robust system which enables somewhat larger libraries, typically on the order of 1012 sequences. Most of the currently used systems were constructed to confer the linkage between the genotype and protein phenotype. In droplets, or other compartmentalized single cell/clone systems, the compartment can supply that linkage, increasing the flexibility of the assay and alleviating some of the restrictions that a physical linkage places upon some of the non-droplet methods.Droplets as a high-throughput screening tool for engineered proteins
In screening for protein traits besides affinity, generally researchers must still resort to microtiter plate based methods, which when dealing with large libraries require a substantial investment in robotics for microtiter plate handling and additional large scale automated equipment such as colony pickers etc. Recently, Agresti et al.8 demonstrated a droplet based screening platform capable of screening a library of 107 variants of the horseradish peroxidase enzyme in less than 10 h using 150 μl of reagents. This represented a 1000-fold increase in speed and a million-fold decrease in cost compared to performing the same screen in an automated microtiter plate format. Droplet microfluidics has also been used for analysis of protein traits, such as enzyme kinetics.9Even though droplet microfluidics has been combined with several detection techniques, such as mass spectrometry, the main set of techniques used are fluorescence based (Fig. 4). Most droplet sorting applications for screening purposes reported thus far, i.e. by dielectrophoresis,10 have relied on sequential sorting to limit the number of droplets sorted per day to <109. Screening the largest libraries currently available will require an increase in throughput of 6 orders of magnitude. In order for droplet microfluidics to reach throughputs of that magnitude, passive and parallel selection methods are necessary. Examples of passive and parallelizable selection methods include droplet separation by size,11 which has yet to be coupled with selection of protein traits or affinity selection in microfluidic channels using continuous phase microfluidics and acoustic separation.12
![Fluorescence detection of E. coli expressed β-galactosidase by turnover of a fluorogenic substrate in droplets has been used as a basis for fluorescence activated droplet sorting. Image reproduced from [10], Royal Society of Chemistry 2009.](/image/article/2011/LC/c1lc90102h/c1lc90102h-f4.gif) | ||
| Fig. 4 Fluorescence detection of E. coli expressed β-galactosidase by turnover of a fluorogenic substrate in droplets has been used as a basis for fluorescence activated droplet sorting. Image reproduced from [10], Royal Society of Chemistry 2009. | ||
Conclusions
In order to address the challenges that lie ahead in drug development and industrial biotechnology, it is clear that protein engineering and synthetic biology are important technologies which must rely on automated high-throughput workflows. In this focus article, we have highlighted some of the ways in which microfluidics in general and droplet microfluidics in particular is positioned to assist in meeting those challenges. In the near term, droplet microfluidics has the potential to play an important role in the screening and analysis of engineered proteins. In the longer perspective, the integration of droplet microfluidic protein screening and microfluidic synthesis of nucleotides offers attractive routes to automating larger parts of the protein engineering process, nearing the establishment of a test bed for synthetic biology.All opinions are those of the authors and do not reflect the views of Lab on a Chip or the Royal Society of Chemistry.
References
- M. Uhlen and H. Andersson Svahn, Lab Chip, 2011, 11, 3389–3393 RSC.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS.
- C.-C. Lee, T. M. Snyder and S. R. Quake, Nucleic Acids Res., 2010, 38, 2514–2521 CrossRef CAS.
- B. Rotman, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS.
- P. S. Dittrich, M. Jahnz and P. Schwille, ChemBioChem, 2005, 6, 811–814 CrossRef CAS.
- R. C. Wootton and A. J. Demello, Nature, 2010, 464, 839–840 CrossRef CAS.
- M. Baker, Nat. Methods, 2011, 8, 623–626 CrossRef CAS.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS.
- L. Mazutis, J.-C. Baret, P. Treacy, Y. Skhiri, A. F. Araghi, M. Ryckelynck, V. Taly and A. D. Griffiths, Lab Chip, 2009, 9, 2902–2908 RSC.
- J. C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- H. N. Joensson, M. Uhlen and H. A. Svahn, Lab Chip, 2011, 11, 1305–1310 RSC.
- J. Persson, P. Augustsson, T. Laurell and M. Ohlin, FEBS Journal, 2008, 275, 5657–5666 CAS.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |