A printed nanolitre-scale bacterial sensor array
Sahar
Melamed
a,
Laura
Ceriotti
b,
Wilfried
Weigel
c,
François
Rossi
b,
Pascal
Colpo
b and
Shimshon
Belkin
*a
aDepartment of Plant and Environmental Sciences, Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel. E-mail: shimshon@vms.huji.ac.il; Fax: +972 26585559; Tel: +972 26584192
bEuropean Commission, Joint Research Center, TP 203, Via E Fermi, 2749, 21020, Ispra, VA, Italy
cScienion AG, Volmerstr. 7b, 12489 Berlin, Germany
First published on 26th October 2010
Abstract
The last decade has witnessed a significant increase in interest in whole-cell biosensors for diverse applications, as well as a rapid and continuous expansion of array technologies. The combination of these two disciplines has yielded the notion of whole-cell array biosensors. We present a potential manifestation of this idea by describing the printing of a whole-cell bacterial bioreporters array. Exploiting natural bacterial tendency to adhere to positively charged abiotic surfaces, we describe immobilization and patterning of bacterial “spots” in the nanolitre volume range by a non-contact robotic printer. We show that the printed Escherichia coli-based sensor bacteria are immobilized on the surface, and retain their viability and biosensing activity for at least 2 months when kept at 4 °C. Immobilization efficiency was improved by manipulating the bacterial genetics (overproducing curli protein), the growth and the printing media (osmotic stress and osmoprotectants) and by a chemical modification of the inanimate surface (self-assembled layers of 3-aminopropyl-triethoxysilane). We suggest that the methodology presented herein may be applicable to the manufacturing of whole-cell sensor arrays for diverse high throughput applications.
Introduction
The use of genetically engineered microorganisms as accurate and sensitive reporters of their chemical environment is gaining increasing attention. Consequently, numerous reports describe diverse bacterial strains capable of emitting a dose-dependent signal (luminescent, fluorescent, electrochemical or colorimetric) in the presence of either specific target chemicals or global biological effects such as toxicity or genotoxicity.1–6 In contrast to traditional approaches for monitoring chemicals, based on physical or chemical analyses, living bioreporter cells provide data such as bioavailability of chemicals, their effects on living systems, and their synergistic or antagonistic behavior in mixtures.7 Many of these reporters are engineered by fusing a specific promoter, known to be induced by the target chemical or effect, to a reporter system such as the bacterial bioluminescence genesluxCDABE.7–10To turn such strains into biosensors, however, they need to be integrated into a hardware platform containing all the components essential for a sensing device,11 including the necessary electronics and signal acquisition elements. In this paper we describe the successful deposition of an array of live bioluminescent and fluorescent reporter cells onto solid surfaces by printing nanolitre-scale drops of cell suspensions by an adapted commercial non-contact DNA printer.
Immobilization of bacteria on abiotic surfaces can be carried out using diverse approaches. One potential solution involves the direct deposition of the cells onto the surface, exploiting the natural adhesive properties of bacteria. Bacterial adhesion may be divided into (a) an initial, instantaneous and reversible physical phase and (b) a time-dependent irreversible “molecular” phase. The initial adhesion of a bacterium to a solid surface is mediated by non-specific electrostatic or hydrophobic interactions, by hydrogen bonds, and by van der Waals forces. As bacterial cell surfaces are usually negatively charged, they adhere more intensely to positively charged surfaces.12 The attachment can be further optimized by chemical modification of the surface and/or by biological modification of the cells.13–17 A common chemical surface modification may be achieved by the formation of self-assembled layers or monolayers (SAM).18,193-Aminopropyl-triethoxysilane (APTES), for example, can be used to functionalize glass by forming self-assembled layers with positively charged functional amino groups that attract the negatively charged bacteria.20 Initial adhesion is further stabilized by the generation of bonds between individual cells, as well as by excreted exopolysaccharides (EPS) that strengthen the attachment of the bacteria to each other as well as to the surface.21 External cellular structures can interact with the surface, strengthening the interaction and rendering it irreversible; one of these surface structures is curli: thin (2–5 nm diameter), coiled and highly aggregative fimbria that protrude from the cell surface and form an extracellular matrix. Curli are optimally expressed below 30 °C, at low levels of nutrients and during stationary phase.22–25 An Escherichia coli mutant, PHL628, able to adhere spontaneously to inert solid surfaces, was previously described.17 This phenotype was caused by a point mutation in the ompR regulatory protein, leading to overproduction of curli.
When immobilized for whole-cell sensor applications, it is desired that the live bacteria are deposited into specific locations according to the biosensor design; such patterning also allows the deposition of different bioreporter types on the same platform. A recent technique demonstrated for patterning live cells is the cell printing by ink-jets or related non-contact technologies.26–28 In the current study we took cell printing several steps further by using a modified non-contact robotic printer, originally designed for arraying biomolecules, to generate printed arrays of reporter cells.
One of the major challenges encountered in the development of cell-based devices is the long-term maintenance of the viability and activity of the cells. A large number of preservation and immobilization methods have been investigated towards this goal,34 one of the most effective being the involvement of organic osmoprotectants, either externally added or synthesized by the cells. When a bacterium is grown at a high osmolarity, it synthesizes or accumulates compatible solutes, small organic molecules (e.g., trehalose, glutamate, proline) that increase the internal osmotic pressure and stabilize cell components in response to the increased osmotic stress.35 It has been shown that accumulation of trehalose was correlated with improved survival after periods of dryness,36 and that extracellular compatible solutes protect air-dried bacteria from deleterious effects of desiccation.36,37 In the current work, we applied both modes of action of osmoprotectants to enhance bacterial survival.
A future development envisaged for whole cell biosensors will be the incorporation of an array of live sensor cells, differing in their response characteristics, onto the same platform, in a concept resembling that of an artificial nose or tongue.2,8 In the present manuscript, we present a novel method for generating such a cell array, exploitable for future whole-cell sensor biochips, by printing minute (nanolitre range) spots of bacteria. Immobilization was optimized by chemical modification of the surface (APTES) and by biological modification of the bacterium (curli overproduction). The immobilized reporter bacteria displayed a high degree of survival and responsiveness to toxicants following printing and storage of up to 2 months.
Experimental
Strains and media
Several E. coliK12 strains were used (Table 1). Strain PHL628, a curli overproducing strain, was kindly provided by Prof. Philippe Lejeune.17 Strains SM110, SM111, SM117 and SM118 were constructed in the current work. Strains AG1688,29MG1655,30 W3110,32RFM443,31 and HB10133 were taken from our lab collection. All strains were kept on Luria–Bertani (LB) agar plates at 4 °C or in 50% glycerol at −80 °C and were grown in rich (LB) or minimal (M63) media.38| Strain | Host | Plasmid | Phenotype/genotype | Reference |
|---|---|---|---|---|
| AG1688 | — | — | F′ 128lacI q lac:: Tn5 | 29 |
| MG1655 | — | — | F − l − ilvG-rfb-50 rph-1 | 30 |
| RFM443 | — | — | F − galK2 lac74 rpsL200 | 31 |
| W3110 | — | — | F − λ − rph-1 INV(rrnD, rrnE) | 32 |
| HB101 | — | — | F−mcrB mrr hsdS20 (rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20(SmR) glnV44 λ− | 33 |
| PHL628 | MG1655 | — | malA::kan R, ompR234 | 17 |
| SM110 | PHL628 | pES2:recA::GFPuv | ampR, kanR | Current work |
| SM111 | PHL628 | pRS4:recA::DsRed2 | ampR, kanR | Current work |
| SM117 | PHL628 | pBR2TTS:CP25::luxCDABE | ampR, kanR | Current work |
| SM118 | PHL628 | pBR2TTS:cydA::luxCDABE | ampR, kanR | Current work |
Chemicals
Ectoine and hydroxyectoine were purchased from Bitop AG, Germany. Glycerol was purchased from Frutarom (Israel). All other chemicals were purchased from Sigma-Aldrich Corporation (USA).Genetic engineering of the reporter strains
A plasmid harboring cydA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :luxCDABE fusion was constructed. Primers, designed for the cydA promoter amplification based on its sequence and on available information as to its regulation, were used to amplify the regulatory regions of cydAgene from E. coliMG1655 genome by PCR. Sense primer sequence was 5′ GGCAAGGGTACCGCATCAGG 3′ and the antisense primer sequence was 5′ GCCATACCGAGCTCCAGTGG 3′. The bold bases indicate enzyme restriction sites. After cutting with restriction enzymes KpnI and SacI (New England Biolabs, USA), the PCR products were ligated into a pBR2TTS vector,10 a medium copy number plasmid (T4 DNA ligase, Roche) that harbors Photorhabdus luminescens luxCDABEgenes downstream from a multiple cloning site. The vector was first transformed into E. coliAG1688 and then transferred to E. coliPHL628. All other plasmid vectors, harboring either bioluminescence (luxCDABE) or fluorescence (GFPuv or DSRed) reporter genes, were taken from our lab collection and transformed into E. coliPHL628 (Table 1).
:luxCDABE fusion was constructed. Primers, designed for the cydA promoter amplification based on its sequence and on available information as to its regulation, were used to amplify the regulatory regions of cydAgene from E. coliMG1655 genome by PCR. Sense primer sequence was 5′ GGCAAGGGTACCGCATCAGG 3′ and the antisense primer sequence was 5′ GCCATACCGAGCTCCAGTGG 3′. The bold bases indicate enzyme restriction sites. After cutting with restriction enzymes KpnI and SacI (New England Biolabs, USA), the PCR products were ligated into a pBR2TTS vector,10 a medium copy number plasmid (T4 DNA ligase, Roche) that harbors Photorhabdus luminescens luxCDABEgenes downstream from a multiple cloning site. The vector was first transformed into E. coliAG1688 and then transferred to E. coliPHL628. All other plasmid vectors, harboring either bioluminescence (luxCDABE) or fluorescence (GFPuv or DSRed) reporter genes, were taken from our lab collection and transformed into E. coliPHL628 (Table 1).
Adhesion assay by crystal violet (CV) staining
The degree of surface attachment was assayed by CV staining of adherent cells as described previously,39 with a few modifications. Briefly, bacteria were grown overnight in LB or M63 at 37 °C with agitation, and were diluted ×1/4 in fresh medium. Aliquots of the diluted culture were added to either glass test tubes (13 × 100 mm, Kimble, USA) or to a PVC or a polystyrene 96-well microtiter plate (Falcon 3912, Becton Dickinson, USA or Nunc, Denmark, respectively), and incubated at 30 °C without shaking. After different time periods the non-adhered bacteria were removed by washing with tap water, and 1% (w/v) CV (Sigma Aldrich, USA) was added for 5 min before rinsing again in tap water. To quantify the adhered bacteria, the unrinsed CV was dissolved in acetone–ethanol (20:80), and CV concentration was determined by measuring the OD600 of the sample with a plate reader (Victor2, Wallac, Finland) in the case of PVC/polystyrene plates or by a spectrophotometer (Sarstedt, Germany) in the case of the glass test tubes.
Surface modification
Standard microscope glass slides were coated with APTES in the following manner. The slides were cleaned in acid piranha (H2SO4:H2O2, 7:3) for 15 min, rinsed extensively in water and dried at 100 °C for 15 min. Next, the slides were incubated in 1% APTES in dry toluene for 3 h, rinsed in toluene, and dried at 100 °C for 30 min. The contact angle of the treated surface was between 20° and 30°.
Bacterial deposition using a robotic printer
A non-contact robotic printer (sciFlexarrayer S3, Scienion AG, Germany) with the ability to print liquid volumes as low as 0.1 nl per drop was used for printing the cells. The printer was equipped with a special holder for the piezo dispensing capillaries that allowed an adjustment of the temperature of the printed samples between 20 and 50 °C. The capillaries were coated with a hydrophobic polymer to improve the stability of the print process of the cell-containing samples. Cultures of strains SM110 and SM111 were grown overnight at 30 °C with agitation in M63 medium with the appropriate antibiotics, with or without NaCl (0.5 M) and ectoine (1 mM). Bacteria were precipitated by centrifugation (4000 rpm for 10 min) and suspended in 250 µl of the designated osmoprotectant solution (0.5 M ectoine, 0.5 M hydroxyectoine, 0.5 M lysine, 0.25 M glutamine, 0.5 M glutamate, 0.25 M trehalose, 0.5 M sucrose, 0.24 M glycine betaine, 0.5 M proline, 0.5 M glycerol) or an unamended medium (M63). Sample volumes between 0.5 nl and 2.5 nl, consisting of one or multiple drops, were deposited by the robotic printer on either bare or APTES-coated glass slides. Samples were analyzed by an epi-fluorescence microscope (filter sets 13 and 20, Axivert 135TV, Zeiss, Germany) and by a high resolution SEM (Sirion, FEI, Holland) following Au coating.Activity and induction of arrayed cells
Bioluminescent reporter strains SM117 and SM118, harboring a CP25::luxCDABE and a cydA::luxCDABE fusion, respectively, were grown, treated and spotted as described above, either as 1 spot of 500 nl or in 25 spots of 1 nl volume in each well of a 96-well microtiter plate. The plates were incubated at 4 °C; at time zero and at designated intervals, 100 µl of medium (either LB or M63), with or without a toxicant, were added to at least 2 wells of each treatment. Luminescence was measured by a microtiter plate luminometer (Victor2, Wallac, Finland) at 37 °C at 10 minute intervals for 3 hours. Light emission was quantified by the instrument's arbitrary relative light units (RLU). Responses to the toxicants are reported either as the fold increase in RLU over the non-induced control (response ratio) or as the difference between the induced and non-induced systems (ΔRLU). Where indicated, bacteria were printed in a 96-well microtiter plate, the bottom of which was replaced by APTES-coated glass, allowing better adhesion of the cells. At each time point wells were washed with LB or M63 media prior to the activity test.For long-term experiments, the bacteria were printed using the robotic printer in a 96-well microtiter plate, 25 spots of 1 nl in each well. The plates were kept at −20 °C, 4 °C, or 24 °C with or without desiccation for various time periods. At time zero and at the designated sampling times, LB medium was added to at least 2 wells of each treatment with or without a toxicant and luminescence was measured as before, at 37 °C, at 10 minute intervals for 6 hours.
Results and discussion
Screening and selection of bacterial host strain
Deposition of live bacteria onto a solid platform may be conducted with the cells encapsulated in a polymer, which provides solid support, surface attachment and a hydrated environment; the viscosity of the polymer suspension, however, often hinders non-contact deposition by a nozzled instrument. An alternative mode, adopted in the present study, is to array the cells onto the surface without the assistance of a polymeric matrix, thereby allowing low-volume nozzle-deposition and a much finer patterning resolution. For this to succeed, a bacterial strain had to be selected that would adhere firmly to the solid inanimate substrate by its own power. We have thus screened several E. coli strains for their ability to attach to three types of surfaces: polystyrene, poly[vinyl-chloride] (PVC) and glass. The degree of surface attachment was assayed by crystal violet (CV) staining of the attached cells after the non-adherent cells have been washed off.A comparison was made between five E. coliK12 strains (W3110, MG1655, RFM433, HB101, and PHL628), the latter being a curli-overproducing mutant.17 As can be observed in Fig. 1, the extent of bacterial adhesion greatly varied between strains and substrates. Both strains HB101 and PHL628 exhibited the highest overall degree of attachment, with the latter displaying superior affinity to glass. Consequently, strain PHL628 was selected for the subsequent steps of the study, a decision that was also motivated by the high number of genome mutations characteristic of strain HB101 and the unclear mechanism of its adhesion.33,40,41
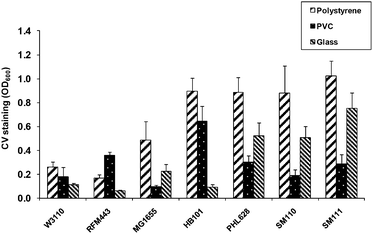 | ||
| Fig. 1 Adhesion of E. coli strains to polystyrene, PVC and glass after 24 h incubation at 30 °C. HB101, W3110 and RFM443 were tested with LB medium while MG1655, PHL628, SM110 and SM111 were tested with M63 medium. Error bars represent standard deviations of at least 3 repeats in the same experiment. | ||
Strain PHL628 was therefore transformed separately with 4 different vectors containing various promoter-reporter gene combinations (Table 1), thus generating 4 different reporter strains, either fluorescent (SM110, SM111) or luminescent (SM117, SM118). As shown for strains SM110 and SM111 (Fig. 1), adhesion characteristics of the PHL628 derivatives were similar to that of their parental strain.
Surface modifications and optimization of cell attachment
As a first step towards the patterning of the envisioned cell arrays, we attempted to optimize the deposition process. An array was formed by printing nanolitre spots of PHL628-based reporter strains, using a non-contact robotic printer. The cells were printed onto modified surfaces and tested for the bacterial adhesion, activity and ability to respond to target chemicals. Microscope glass slides, either non-treated or coated with APTES, recently shown to promote mammalian cell adhesion,42 served as the printed surface.Suspensions of fluorescent strains SM110 and SM111 were printed in volumes of 0.5 nl to 2.5 nl, resulting in spots 50 to 200 µm in diameter (Fig. 2A) showing the expected interdependence of these parameters. The spots were allowed to dry at 30 °C or at 4 °C and the degree of adhesion was tested after 24 hours, after removing non-adhered cells by gentle rinsing. As can be seen from microscopic analysis (Fig. 2), bacteria printed over the APTES coated glass displayed a higher degree of adhesion compared to bacteria printed over non-modified glass (Fig. 2A–C). As discussed above, this may be attributed to the attraction between the positively charged APTES layers and the negatively charged cells. These results are in agreement with the report of Ploux et al. (2007),43 demonstrating that PHL628 cells favorably adhered to positively charged amine functional groups. No correlation was observed between adherence and drop size (data not shown). Fig. 2D displays the tandem deposition of two different fluorescent reporter strains (SM110 and SM111, see Table 1), demonstrating the potential of the printing process to generate an array of unlimited number of strains. It is tempting to envision an array harboring a large number of bacterial reporter strains, each engineered to detect a different class of compounds, for detecting the biological effects of a broad range of compounds in a single device. In view of the stability and firm attachment of the surface-attached bacteria, we believe the approach described herein can be readily adaptable for incorporation into diverse microfluidic platforms.
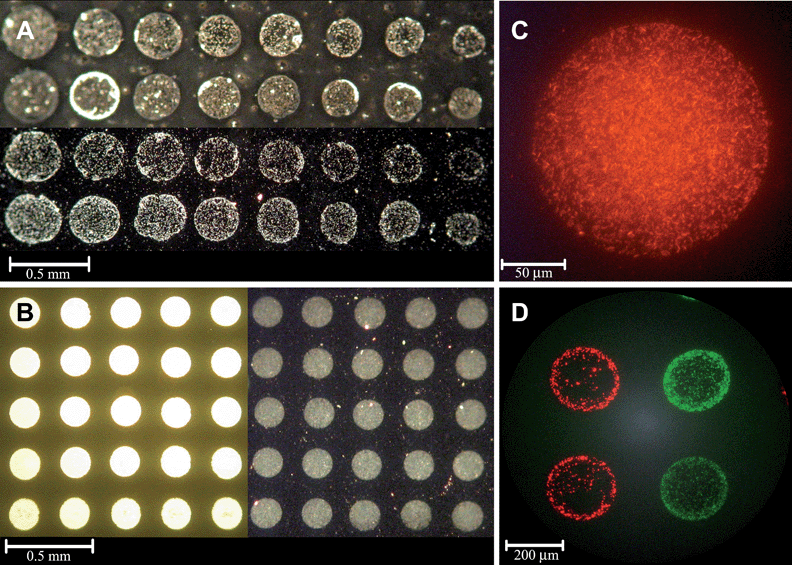 | ||
| Fig. 2 (A) Fluorescent reporter strains SM110 and SM111 printed (0.5–2.5 nl) on a non-modified glass slide, immediately after printing (upper 2 rows) and after 24 h incubation at 30 °C, following removal of non-adhered cells (2 bottom rows) (light microscope). (B) Strain SM111 (1 nl) printed on APTES-coated glass immediately after printing (5 left columns) and after 24 h incubation at 4 °C, following removal of non-adhered cells (5 right columns and (C)). (D) Strains SM110 and SM111 printed (2 nl) on APTES-coated glass (epifluorescence microscope). | ||
Two of the main advantages of non-contact printing are superior spot morphologies and a reduced likelihood for contamination, as the substrate and the printing nozzle are separated. One potential disadvantage is the potential formation of tiny satellite droplets;44 in the experiments described here, however, we have not encountered this problem. Another risk may lie in the high shear rates that the liquid experiences during nozzle passage and surface impact.45,46 A high shear rate, caused by the oscillating wave inside the piezo dispensing capillary and that depends on the viscosity and possible interactions between the cells and the glass surface of the print nozzle, can exert mechanical stress on the cells. This problem was minimized by using hydrophobically coated print nozzles, and by a slight increase of the print head temperature to reduce viscosity. Other parameters that had to be adjusted were the diameter of the orifice of the print nozzle (about 50 µm) and the settings for the applied piezoelectric pulse (pulse amplitude, width and frequency) of the printing device, when printing sub-nanolitre droplets of cell solutions. It was thus essential to test cell viability following the printing process.
As shown in Fig. 3A, residual viability (24 hours post-printing) markedly depended on medium composition. The strain used for this study, SM117, harbors a bacterial bioluminescence operon (luxCDABE) under the control of a synthetic constitutive promoter CP25 (Table 1).47 Strain SM117 thus emits a strong and stable luminescent signal that, in our case, served as a convenient measure of viability. Spots of this strain were printed in a 96-well microtiter plate, one spot of 500 nl in each well, and luminescence was measured before printing, immediately after printing and following 24 hours incubation at 4 °C. When printed in minimal medium M63 (right hand bars), bacterial viability after 24 hours at 4 °C was very low, with only 0.3% of the initial luminescence remaining (Fig. 3A). To at least partially adapt the cells and protect them from the expected stresses, both growth and deposition media were modified. The bacteria were grown with an imposed osmotic stress (0.5 M NaCl) in the presence of 1 mM ectoine, forcing the cells to uptake the compatible solute to correct the osmotic imbalance. It has been previously shown that the expression of both genes involved in ectoine uptake, ompC and proP, increases at high osmolarity.35 In addition, ten known osmoprotectants, including ectoine, were individually added to the deposition medium following the osmotically stressed growth, and their effect on residual bioluminescence was compared. Bacteria pre-grown under osmotic stress but deposited with no additional osmoprotectant fared significantly better (4% residual activity) than cells pre-grown without osmotic stress, presumably due to ectoine accumulation. Osmotically stressed bacteria printed with all the osmoprotectants, except for lysine, clearly showed higher activity than bacteria printed with M63 only (Fig. 3A). In addition, when comparing the luminescence before printing, it seems that the presence of the osmoprotectants in the medium did not affect bacterial activity. It was previously reported that high osmolarity may repress curli expression, leading to a weaker adhesion,48,49 but that the ompR234 mutation in PHL628 counteracts this repression.49 In our case, high osmolarity did not affect the immobilization of the printed bacteria (data not shown).
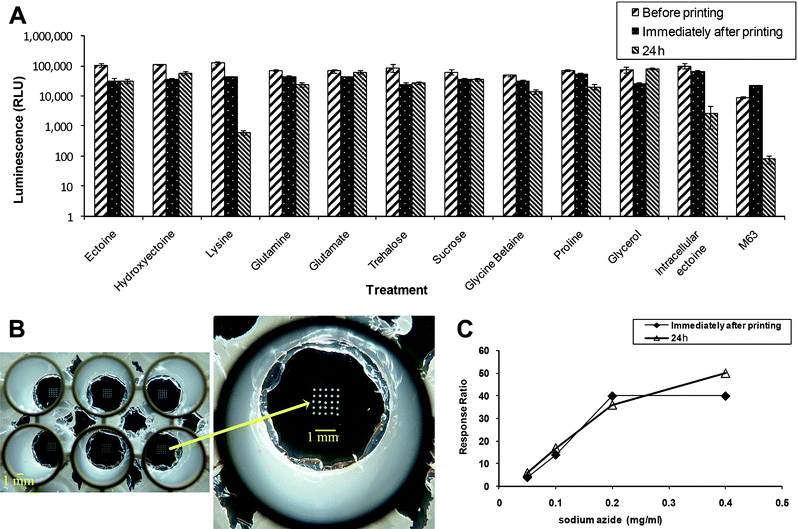 | ||
| Fig. 3 (A) Residual activity of constitutively bioluminescent (CP25:luxCDABE) reporter strain SM117 following desiccation and rehydration. Spots (500 nl) were printed either with no additions (M63), pre-growth under osmotic stress (NaCl 0.5 M) and ectoine (intracellular ectoine), or pre-grown under osmotic stress (NaCl 0.5 M) and ectoine with different osmoprotectants added to the printing medium. Bioluminescence is displayed before printing, immediately after printing, and after incubation for 24 h at 4 °C. Error bars represent standard deviations of 8 repeats in the same experiment. (B) Twenty five spots, 1 nl each, of strain SM118 in ectoine, printed onto the wells of 96-well plate with an APTES coated glass bottom. (C) Response to different concentrations of sodium azide of strain SM118 (cydA:luxCDABE), printed (25 × 1 nl) in ectoine, immediately after the printing and after incubation for 24 h at 4 °C. | ||
In addition to the preservation of bacterial viability, it is also essential to verify that the sensing activity is retained. To test for this we have used strain SM118, which harbors an inducible promoter (cydA) fused to luxCDABE and strongly luminesces in the presence of respiratory inhibitors, such as sodium azide. Twenty five spots of SM118, 2 nl each, were printed on an APTES-coated glass at the bottom of a well of a 96-well microtiter plate (Fig. 3B). Bacteria were exposed to sodium azide immediately after printing and 24 hours following incubation at 4 °C, and luminescence was measured at each time. Fig. 3C demonstrates that the response ratio of these cells (luminescence in the presence of azide divided by luminescence in its absence) remained unaffected following 24 hours at 4 °C.
Fig. 4 demonstrates how the addition of an external compatible solute (in this case, trehalose) forms a “blanket” that surrounds the bacteria, partially separating them from their environment, in comparison with the bacteria printed in a non-amended medium. Trehalose was reported to protect the outer phase of the membrane and may also enter the cells by passive diffusion across the cell membrane. It was suggested that trehalose binds to the phospholipid head groups by hydrogen bonds, replacing the hydrogen bonded water molecules, and maintaining the proper liquid-crystal state of the membrane. In the absence of trehalose or another compatible solute, van der Waals forces between the hydrophobic “tails” of the phospholipids increase, changing the membrane state, and causing a deleterious effect.36,50,51 As shown in this paper, this seeming isolation did not hinder the ability of the cells to sense their environment.
 | ||
| Fig. 4 Strain SM110, printed (2 nl spot) on APTES coated glass (A, C, and D) and on non-modified glass (B), in the absence (A and B) or presence (C and D) of trehalose in the printing medium (high resolution SEM). | ||
Reporter activity of printed bacteria
As already mentioned above, strain SM118 harbors a cydA:luxCDABE fusion, and is strongly induced by sodium azide. This compound is a cytochrome inhibitor;52 it is used in hospitals and laboratories as a biocide and is also found as the source of nitrogen gas used in automobile airbags.53
Strain SM118 was printed as an array of 25 spots of 1 nl each, in a 96-well plate, in the presence of 6 different osmoprotectants or in minimal medium M63. Bacteria were exposed to sodium azide (0.2 mg ml−1) immediately after printing and 24 hours later, following a gentle rinsing to remove non-adhered cells, and bioluminescence was recorded for 3 hours. As may be observed in Fig. 5, glycerol, sucrose, ectoine, trehalose and hydroxyectoine had a significant protective effect on the inducible activity, whereas glutamine failed to provide any protection. The response speed under all conditions was similar to that of non-desiccated cells (data not shown).
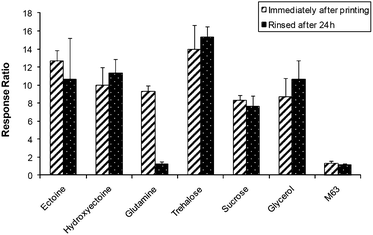 | ||
| Fig. 5 Induction by sodium azide (0.2 mg ml−1) of strain SM118 (cydA:luxCDABE) printed (25 × 1 nl) in different osmoprotectants, immediately after the printing and 24 h later. Error bars represent standard deviations of 3 independent experiments. | ||
Long term maintenance of printed arrays
Clearly, a future biosensor should be “ready for action” after long term storage. We tested the printed bacteria for their ability to remain active and responsive following storage periods of up to 2 months. Based on the results described above, we printed strain SM118 (cydA:luxCDABE), amended with sucrose, trehalose or glycerol in an array of 25 spots, 1 nl each, in the wells of 96-well plates. The plates were stored dry at −20 °C, 4 °C and 24 °C, and at intervals (immediately after printing, one week, one month and two months later) sodium azide (0.2 mg ml−1) was added and luminescence was monitored. As shown in Fig. 6, 4 °C appeared to be the best temperature of those tested, and sucrose provided the best long term protection. Activity after 2 months at 4 °C in the presence of sucrose was over 40% of that at time zero, and response kinetics and dose-dependency were practically unchanged (not shown). Quite clearly, cooling to −20 °C was detrimental; it is unclear at this time whether yet lower temperatures would improve preservation. No significant improvement in residual activity was obtained by storage in desiccated conditions (not shown). While long-term preservation results to date are very encouraging, further work is needed to optimize long term storage conditions of the spotted cells.
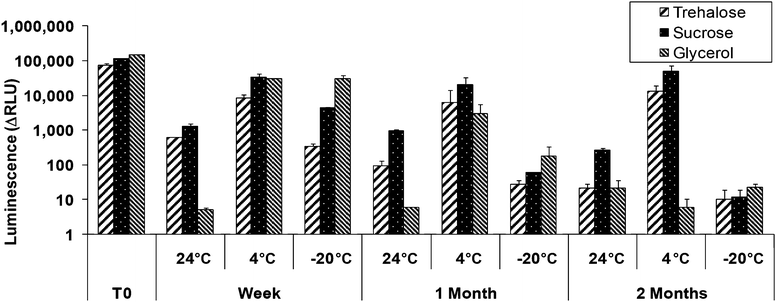 | ||
| Fig. 6 Induction by sodium azide (0.2 mg ml−1) of strain SM118 (cydA:luxCDABE) printed (25 × 1 nl) in trehalose, glutamate or sucrose and incubated at different temperature. Error bars represent standard deviations of duplicates in the same experiment. | ||
Conclusions
The results presented in this paper provide a proof of the concept for the feasibility of constructing live cell arrays made out of “spots” of nanolitre-scale live bacterial bioreporters. Using both bioluminescent and fluorescent toxicant-sensitive genetically engineered bacterial sensor strains, it was demonstrated that they can be spotted, arrayed, preserved and maintain their sensor activities for prolonged periods. For this purpose, we have used non-contact deposition onto a chemically modified surface, and manipulated growth and deposition media for optimal maintenance of activity. In doing so, as far as we are aware, we described the smallest cell printing volume to date by non-contact printing technology, enabling minimization of the printed area and a higher density array. Minimizing the area needed for bacterial deposition could be useful for incorporation into a single-use array biochip, to be applied either in portable field sensors or in laboratory high-throughput devices. While additional research is clearly needed in order to optimize the printing method and further improve bacterial storage, we nevertheless believe that we have progressed several steps forward on the route to functional live cell array biochips. In the course of the study, we have also described a novel specific reporter for the detection of respiratory inhibitors. Sodium azide, a chemical with a constantly increasing world distribution,53 served as the model toxicant. The sensor's response was rapid (20 minutes after exposure) and dose-dependent, and could be maintained for at least 2 months at 4 °C.Acknowledgements
Research was supported by EU 6th framework project Toxichip (project no. 027900) (http://www.toxichip.org/), by US–Israel Binational Research and Development Fund (BARD), contract US-3864-06, and by the Israeli Ministry of Science Infrastructure Program. We are grateful to Prof. Philippe Lejeune of Institut National des Sciences Appliquées de Lyon for the generous donation of E. coli strain PHL628 and to Dr Sharon Yagur-Kroll for her help with the genetic engineering of the reporter strains. Research was also partially supported by National Research Foundation of Korea Grant funded by the Korean Government (NRF-2010-220-D00019).References
- A. Biran, S. Yagur-Kroll, R. Pedahzur, S. Buchinger, G. Reifferscheid, H. Ben-Yoav, Y. Shacham-Diamand and S. Belkin, Microb. Biotechnol., 2009, 3, 412–427 Search PubMed.
- T. Elad, J. H. Lee, S. Belkin and M. B. Gu, Microb. Biotechnol., 2008, 1, 137–148 Search PubMed.
- A. Mulchandani, I. Kaneva and W. Chen, Anal. Chem., 1998, 70, 5042–5046 CrossRef CAS.
- A. Schmidt, C. Standfu-Gabisch and U. Bilitewski, Biosens. Bioelectron., 1996, 11, 1139–1145 CrossRef CAS.
- R. S. Shetty, S. K. Deo, P. Shah, Y. Sun, B. P. Rosen and S. Daunert, Anal. Bioanal. Chem., 2003, 376, 11–17 CAS.
- K. Yagi, Appl. Microbiol. Biotechnol., 2007, 73, 1251–1258 CrossRef CAS.
- S. Belkin, Curr. Opin. Microbiol., 2003, 6, 206–212 CrossRef CAS.
- J. H. Lee, C. H. Youn, B. C. Kim and M. B. Gu, Biosens. Bioelectron., 2007, 22, 2223–2229 CrossRef CAS.
- R. J. Mitchell and M. B. Gu, Biosens. Bioelectron., 2004, 19, 977–985 CrossRef CAS.
- S. Yagur-Kroll, B. Bilic and S. Belkin, Microb. Biotechnol., 2009, 3, 300–310 Search PubMed.
- M. Nic, J. Jirat and B. Kosata, in IUPAC Compendium of Chemical Technology, ed. A. Jenkins, A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 2006 Search PubMed.
- Y. H. An and R. J. Friedman, J. Biomed. Mater. Res., 1998, 43, 338–348 CrossRef CAS.
- M. D. Disney and P. H. Seeberger, Chem. Biol., 2004, 11, 1701–1707 CrossRef CAS.
- R. Muller, S. Ruhl, K. A. Hiller, G. Schmalz and H. Schweikl, J. Biomed. Mater. Res., Part A, 2007, 84, 817–827.
- S. Rozhok, Z. Fan, D. Nyamjav, C. Liu, C. A. Mirkin and R. C. Holz, Langmuir, 2006, 22, 11251–11254 CrossRef CAS.
- M. A. Schembri, G. Christiansen and P. Klemm, Mol. Microbiol., 2001, 41, 1419–1430 CrossRef CAS.
- O. Vidal, R. Longin, C. Prigent-Combaret, C. Dorel, M. Hooreman and P. Lejeune, J. Bacteriol., 1998, 180, 2442–2449 CAS.
- E. Ruckenstein and Z. F. Li, Adv. Colloid Interface Sci., 2005, 113, 43–63 CrossRef CAS.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef CAS.
- A. R. Ferhan, L. Guo and D.-H. Kim, Langmuir, 2010, 26, 12433–12442 CrossRef CAS.
- P. N. Danese, L. A. Pratt and R. Kolter, J. Bacteriol., 2000, 182, 3593–3596 CrossRef CAS.
- P. K. Brown, C. M. Dozois, C. A. Nickerson, A. Zuppardo, J. Terlonge and R. Curtiss, Mol. Microbiol., 2001, 41, 349–363 CrossRef CAS.
- A. Olsén, A. Arnqvist, M. Hammar and S. Normark, Infect. Agents Dis., 1993, 2, 272–274 Search PubMed.
- A. Olsén, A. Jonsson and S. Normark, Nature, 1989, 338, 652–655 CrossRef CAS.
- C. Prigent-Combaret, G. Prensier, T. T. L. Thi, O. Vidal, P. Lejeune and C. Dorel, Environ. Microbiol., 2000, 2, 450–464 CrossRef CAS.
- R. R. Bradley, M. O. Christina, A. B. Jason, Y. Daniel and J. S. Barry, Biotechnol. J., 2006, 1, 930–948 Search PubMed.
- P. Calvert, Science, 2007, 318, 208–209 CrossRef CAS.
- M. C. Flickinger, J. L. Schottel, D. R. Bond, A. Aksan and L. E. Scriven, Biotechnol. Prog., 2008, 23, 2–17.
- J. C. Hu, R. T. Sauer, N. E. Newell and B. Tidor, Protein Sci., 1993, 2, 1072–1084 CrossRef CAS.
- F. R. Blattner, G. Plunkett, III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau and Y. Shao, Science, 1997, 277, 1453–1462 CrossRef CAS.
- R. Menzel, Anal. Biochem., 1989, 181, 40–50 CAS.
- B. J. Bachmann, Microbiol. Mol. Biol. Rev., 1972, 36, 525–557 CAS.
- H. W. Boyer and D. Roulland-dussoix, J. Mol. Biol., 1969, 41, 459–472 CrossRef CAS.
- J. Bjerketorp, S. Hakansson, S. Belkin and J. K. Jansson, Curr. Opin. Biotechnol., 2006, 17, 43–49 CrossRef CAS.
- B. Kempf and E. Bremer, Arch. Microbiol., 1998, 170, 319–330 CrossRef CAS.
- D. T. Welsh and R. A. Herbert, FEMS Microbiol. Lett., 1999, 174, 57–63 CrossRef CAS.
- M. Manzanera, S. Vilchez and A. Tunnacliffe, FEMS Microbiol. Lett., 2004, 233, 347–352 CrossRef CAS.
- J. H. Miller, A Short Course in Bacterial Genetics: A Laboratory Manual for Escherichia coli and Related Bacteria, Cold Spring Harbor laboratory, NY, 1992 Search PubMed.
- L. A. Pratt and R. Kolter, Mol. Microbiol., 1998, 30, 285–293 CrossRef CAS.
- S. Lacks and B. Greenberg, J. Mol. Biol., 1977, 114, 153–168 CrossRef CAS.
- S. A. Rothen, M. Sauer, B. Sonnleitner and B. Witholt, Biotechnol. Bioeng., 1998, 58, 92–100 CrossRef CAS.
- T. Sordel, F. Kermarec-Marcel, S. Garnier-Raveaud, N. Glade, F. Sauter-Starace, C. Pudda, M. Borella, M. Plissonnier, F. Chatelain, F. Bruckert and N. Picollet-D'hahan, Biomaterials, 2007, 28, 1572–1584 CrossRef CAS.
- L. Ploux, S. Beckendorff, M. Nardin and S. Neunlist, Colloids Surf., B: Biointerfaces, 2007, 57, 174–181 CrossRef CAS.
- F.-G. Tseng, C.-J. Kim and C.-M. Ho, J. Microelectromech. Syst., 2002, 11, 427–436 CrossRef.
- L. R. Allain, D. N. Stratis-Cullum and T. Vo-Dinh, Anal. Chim. Acta, 2004, 518, 77–85 CrossRef CAS.
- T. Okamoto, T. Suzuki and N. Yamamoto, Nat. Biotechnol., 2000, 18, 438–441 CrossRef CAS.
- P. R. Jensen and K. Hammer, Appl. Environ. Microbiol., 1998, 64, 82–87 CAS.
- G. Jubelin, A. Vianney, C. Beloin, J.-M. Ghigo, J.-C. Lazzaroni, P. Lejeune and C. Dorel, J. Bacteriol., 2005, 187, 2038–2049 CrossRef CAS.
- C. Prigent-Combaret, E. Brombacher, O. Vidal, A. Ambert, P. Lejeune, P. Landini and C. Dorel, J. Bacteriol., 2001, 183, 7213–7223 CrossRef CAS.
- S. B. Leslie, E. Israeli, B. Lighthart, J. H. Crowe and L. M. Crowe, Appl. Environ. Microbiol., 1995, 61, 3592–3597 CAS.
- M. Potts, Microbiol. Rev., 1994, 58, 755–805 CAS.
- H. C. Lichstein and M. H. Soule, J. Bacteriol., 1944, 47, 221–230 CAS.
- E. A. Betterton, Crit. Rev. Environ. Sci. Technol., 2003, 33, 423–458 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |