High-content screening of drug-induced cardiotoxicity using quantitative single cell imaging cytometry on microfluidic device†
Min Jung
Kim
,
Su Chul
Lee
,
Sukdeb
Pal
,
Eunyoung
Han
and
Joon Myong
Song
*
Research Institute of Pharmaceutical Sciences and College of Pharmacy, Seoul National University, Seoul, 151-742, South Korea. E-mail: jmsong@snu.ac.kr; Fax: +82-2-871-2238; Tel: +82-2-880-7841
First published on 8th November 2010
Abstract
Drug-induced cardiotoxicity or cytotoxicity followed by cell death in cardiac muscle is one of the major concerns in drug development. Herein, we report a high-content quantitative multicolor single cell imaging tool for automatic screening of drug-induced cardiotoxicity in an intact cell. A tunable multicolor imaging system coupled with a miniaturized sample platform was destined to elucidate drug-induced cardiotoxicity via simultaneous quantitative monitoring of intracellular sodium ion concentration, potassium ion channel permeability and apoptosis/necrosis in H9c2(2–1) cell line. Cells were treated with cisapride (a human ether-à-go-go-related gene (hERG) channel blocker), digoxin (Na+/K+-pump blocker), camptothecin (anticancer agent) and a newly synthesized anti-cancer drug candidate (SH-03). Decrease in potassium channel permeability in cisapride-treated cells indicated that it can also inhibit the trafficking of the hERG channel. Digoxin treatment resulted in an increase of intracellular [Na+]. However, it did not affect potassium channel permeability. Camptothecin and SH-03 did not show any cytotoxic effect at normal use (≤300 nM and 10 μM, respectively). This result clearly indicates the potential of SH-03 as a new anticancer drug candidate. The developed method was also used to correlate the cell death pathway with alterations in intracellular [Na+]. The developed protocol can directly depict and quantitate targeted cellular responses, subsequently enabling an automated, easy to operate tool that is applicable to drug-induced cytotoxicity monitoring with special reference to next generation drug discovery screening. This multicolor imaging based system has great potential as a complementary system to the conventional patch clamp technique and flow cytometric measurement for the screening of drug cardiotoxicity.
Introduction
Drug-induced, undesired biological side effects introduce a new level of complexity to safe therapeutic applications limiting drug usage and sometimes results in withdrawal from the market.1 In most cases, the side effects of drugs are mainly associated with the malfunctioning of important organs including heart, liver and kidney. Especially, drug-induced cardiac dysfunction leads to morbidity and high mortality.2 Cardiac abnormality is essentially associated with the changes in ion channel activities, myocyte structure, extracellular matrix (ECM) structure, and neurohumoral system.3 It also results in protein abnormalities related to Ca2+ and abnormalities of the signaling system. Among these factors, the change in ion channel activity has been recognized as the major cause of drug-induced cardiotoxicity. Therefore, high-content screening of drug-induced changes in ion channel activity is expected to play a key role in drug discovery and development. High-content screening is a drug discovery method that uses images of living cells as the basic unit for molecule discovery.The surface electrocardiogram (ECG) provides information on the electrical events including atrial/ventricular depolarization and ventricular repolarization within the heart. In ECG, QT interval that indicates ventricular depolarization (i.e., a decrease in the electrical potential across a membrane) and repolarization (i.e., recovery of the resting potential) represents the duration of the ventricular action potential and includes QRS interval which reflects activation time of both ventricles. QT interval is measured from the onset of the Q wave to the end of the T wave. Numerous overlapping ionic currents contribute to determine the morphology and duration of ventricular APD. Depolarization of the ventricles is initiated by the rapid entry of Na+ through selective sodium channels. This is followed by a rapid repolarization through transiently activating and inactivating outward potassium channels, and subsequently by a plateau phase, mainly determined by the entry of calcium ions through L-type calcium channels. During repolarization the negative transmembrane potential is recovered by the inactivation of calcium channels and the increase in net outward potassium currents carried mainly by the slow and rapid components of the delayed rectifier potassium channels. Inwardly-rectifying potassium channels also contribute to the repolarization. The regulatory factors including Na+/K+-pump restore intracellular ion concentrations to the original state. Thus, the changes in normal ion channel activities result in prolongation of QT interval that induces Torsades de pointes (TdP) and even sudden cell death. In the majority of cases, drugs that prolong the QT interval preferentially inhibit IKr, the rapid component of the delayed rectifier potassium current, or hERG, the gene that encodes for the α-subunit of IKr channels. The fact that all hERG blockers cause TdP has not been well established and a direct link between QT prolongation and arrhythmogenesis is still unclear. However, until now all drugs that have been removed from the marketplace due to TdP have been shown to be hERG blockers that delay repolarization causing QT prolongation.1,4,5
Although in the majority of cases drug-induced prolongation of the QT interval is associated with the inhibition of hERG, the opposing correlate that inhibition of the hERG channel causes a long QT interval is still not conclusively proven. Besides, cardiotoxicity can also be generated by changes in ion pump activities as well as by cardiomyocyte cell death.6 Therefore, the early identification of the risk of drugs should be considered as integrated activities of multiple ion channels at a molecular level and cell proliferation and death at a single cell level.
There are several techniques that are commonly used for evaluation of drug-induced cardiotoxicity, e.g., the patch clamp technique using hERG transfected cells or isolated cardiomyocytes,7Rb+ efflux assay,8 microelectrode assay using Purkinje fibers9 or guinea pig papillary muscle,10 and in vivo electrocardiography.11 Among these, the patch clamp technique is probably the most widely used tool for screening of drug-induced cardiotoxicity that allows monitoring the effect of a single drug only on a single target ion channel at a time. Albeit this technique offers high accuracy, it does not allow for contemporaneous observation of multiple ion channel activities. Thus the comprehensive analysis of cellular response with respect to interactions of different ion channel classes or organelles in a cell is restrained. Moreover, patch clamp technique does not provide high-content results. Therefore, development of a high-content screening technology for simultaneous monitoring of ion channel activities, and hence drug-induced cardiotoxicity is a timely research.
Our group has already developed a high-content quantitative hyperspectral imaging system that via single-cell monitoring can directly depict and quantitate targeted cellular moieties with special reference to next generation drug discovery screening.12–14 In this study, a single cell multicolor imaging system was exuberantly coupled with a simple microfluidic system for quantitative analytical observation of multivariate cellular responses related to ion channel activity and intracellular ion concentrations. Microfluidic systems provide suitable miniaturization for achieving sufficient cell confluence for reliable monitoring and quantification based on cellular assays at the single cell level. A tunable multicolor imaging system, based on acousto-optic tunable filter (AOTF), was set up for synchronous monitoring of different ion channels and ion concentrations viahyperspectral imaging. Analysis using miniaturized platforms often requires automated high-content quantification of fluorescent cell images, obtained under different spectral conditions such as fluorescent intensity, excitation efficiency, focal depth, and optical magnification. The hyperspectral system utilizes the approach of employing region selection to slightly defocused, background-nullified and threshold images that provides uniform threshold distribution over the objects (cells),14 which is absolutely necessary for automated high-content quantitative analysis in lab-on-a-chip devices. Herein, using multiple fluorescent probes we demonstrated the simultaneous quantitative monitoring of intracellular Na+ concentration, potassium ion channel permeability and apoptosis/necrosis in drug treated-cardiomyocyte on the microfluidic system. Compared to conventional 96 or 12 wells the microfluidic system is very advantageous for achievement of reduced sample consumption and less cell contamination in execution of high-content screening. Due to the reduced cell surface coverage on the microfluidic system, the amount of samples that can be used is reduced and exposure to the external environment to influence the cell contamination is diminished. In addition cells detached from the conventional well plate form multilayers generally. On the other hand, cells in the microfluidic platform are apt to form monolayers due to a much smaller cell culture volume. This leads to a great enhancement in cell detection efficiency at the single cell level. Drug-induced changes in the permeability of K+ channel and intracellular Na+ concentration were simultaneously detected using two fluorophores in the microfluidic devices. This multispectral and multicolor imaging also allowed efficient discrimination of simultaneous cellular events (i.e., apoptosis and necrosis) triggered by anticancer drugs by facilitating the observation of an entire emission spectrum of a third fluorophore at an individual wavelength. This enabled us to correlate the cell death pathways with changes in the intracellular Na+.
The conventional methods including the patch-clamp technique have a limit to detect complex changes. On the contrary, the developed single cell multicolor imaging assay could simultaneously provide information on cell death and changes in ion channel activities and intracellular ion concentrations, thereby considerably reducing the measurement time. Since individual cellular response to a particular stimulus is likely to be different, approximately 300–400 numbers of cells in the microchannel were simultaneously detected to increase the statistical confidence which cannot be offered by the patch-clamp technique. In addition, drug-induced cardiotoxicity can be quantitatively analyzed and the change in intracellular ion concentration can be measured at a single cell level on the microfluidic system. Considering the need for fast drug screening in the early stage of drug discovery, implementation of high-content quantitative analytical approaches to image-based cellular assays will add new dimensions to identification of a lead compound. In this context the developed method can pave the way for high-content drug-induced cardiotoxicity screening.
Experimental
Chemicals
FluxOR™ thallium detection kit, streptavidin-coated quantum dot 525/625, and CoroNa™ Red sodium indicator were purchased from Invitrogen (Eugene, OR). Propidium Iodide (PI), annexin V binding buffer (10X concentrate), and annexin V-biotin were obtained from BD Pharmingen™ (San Jose, CA). Camptothecin, cisapride, and digoxin were purchased from Sigma (St. Louis, MO).Synthesis of SH-03
SH-03 {(7S,7aR,13aS)-9,10-dimethoxy-3,3-dimethyl-7,7a,13,13a-tetrahydro-3H-chromeno [3,4-b]pyrano[2,3-h]chromen-7-ol} was synthesized as described previously.12,15 Briefly, NaBH4 (5.8 mg, 0.15 mmol) was added to a solution of deguelin (20 mg, 0.051 mmol) in methanol (2 mL) at 0 °C. The reaction mixture was stirred for 5 min and then the reaction was quenched with water. The mixture was extracted with Et2O and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was further purified by flash column chromatography on silica gel (EtOAc : n-hexane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford 20 mg (99%) of SH-03 as a colorless oil.
:1) to afford 20 mg (99%) of SH-03 as a colorless oil.
Cell culture
The H9c2(2–1) cell line originating from rat cardiac myocyte, was obtained from Korean Cell Line Bank (KCLB, Seoul, Korea) for cardiotoxicity screening of drugs. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY), and 60 μg mL−1penicillin and 100 μg mL−1streptomycin in the microfluidic platform. The commercially available microfluidic device (ibid.; Munich, DE and Verona, WI) was used as cell culture platform. The microfluidic system was made of plastic with the highest optical quality. The microfluidic platform contains six microchannels. The individual microchannel has dimensions of 3.8 mm (width) × 17 mm (length) × 400 μm (height) as shown in Fig. 1. Cell growth area was 0.6 cm2 channel−1. Inside the microchannel was not coated with collagen or poly-L-lysin that has been generally used for cell culture. The surface of the microchannel was scratched so that an environment for cell growth was formed. The total volume of the microchannel was 30 μL. There were holes at both ends of the microchannel. The cell culture medium was introduced into the microchannel through these holes. 30 μL H9c2(2–1) cell solution with 5 × 105cell mL−1 concentration was introduced into a microchannel and incubated in 5% CO2 incubator at 37 °C when the cells were attached on the surface of the microchannel. After the cell attachment, 60 μL cell culture medium was added into the microchannel. Cells were grown under standard conditions, i.e., at 37 °C in a humidified incubator containing 5% CO2 (US AutoFlowt, NuAire, Plymouth, MN, USA).![(A) Optical set up of the multicolor imaging system. Here, 1. Ar-ion laser, 2. Microfluidic platform (6 channels, channel dimension: 3.8 mm × 17 mm × 0.4 mm), 3. Microscope objective lens (40×), 4. Beam splitter, 5. Prism, 6. AOTF, 7. CCD camera. (B) Outline of the assay schemes used for simultaneous monitoring alteration of K+ channel permeability and intracellular [Na+]. H9c2(2–1) rat cardiomyocyte cells were treated with commercially available dyes for detection ofK+ channel permeability and intracellular Na+ concentration. If K+ channels are not blocked, thallium ions (Tl+) flow into the cell through the K+ channel and combine with the Tl+ sensitive dyes to produce a fluorophore (λmax 525 nm). Non-fluorescent Na+-indicator dye freely penetrates the plasma membrane and upon binding to Na+ becomes fluorescent (λmax 579 nm). Blockade of the Na+/K+ pump increases the intracellular Na+ concentration and in turn the fluorescence intensity of the Na+ sensitive dye.](/image/article/2011/LC/c0lc00110d/c0lc00110d-f1.gif) | ||
| Fig. 1 (A) Optical set up of the multicolor imaging system. Here, 1. Ar-ion laser, 2. Microfluidic platform (6 channels, channel dimension: 3.8 mm × 17 mm × 0.4 mm), 3. Microscope objective lens (40×), 4. Beam splitter, 5. Prism, 6. AOTF, 7. CCD camera. (B) Outline of the assay schemes used for simultaneous monitoring alteration of K+ channel permeability and intracellular [Na+]. H9c2(2–1) rat cardiomyocyte cells were treated with commercially available dyes for detection ofK+ channel permeability and intracellular Na+ concentration. If K+ channels are not blocked, thallium ions (Tl+) flow into the cell through the K+ channel and combine with the Tl+ sensitive dyes to produce a fluorophore (λmax 525 nm). Non-fluorescent Na+-indicator dye freely penetrates the plasma membrane and upon binding to Na+ becomes fluorescent (λmax 579 nm). Blockade of the Na+/K+ pump increases the intracellular Na+ concentration and in turn the fluorescence intensity of the Na+ sensitive dye. | ||
Quantitative single cell multicolor imaging cytometer with microfluidic platform and data acquisition
High-content, quantitative single cell multicolor imaging system was set up as described previously.14Fig. 1A depicts the schematic diagram of the single cell multicolor imaging system. A laser beam (488 nm) passed through an interference filter was reflected by a dichromatic mirror and focused onto the microfluidic channel using a microscope objective lens (Olympus 71, Melvile, NY) to excite the cells inside the channel. Fluorescence emission from the cells was collected using the same microscope objective lens and passed through the dichromatic mirror, and directed at a right angle by a prism to the detection window of an acousto-optic tunable filter (AOTF) (Brimrose, TEAF10-0.45-0.7-S). A built-in bi-refringent crystal of the AOTF permits the transmission of the diffracted beam with desired Bragg angle (θ) at a particular wavelength. The fluorescence of the diffracted beam emerging from the AOTF was finally detected by a charge-coupled device (CCD) camera (CoolSNAP cf mono, photometrics, A05F871008). A long pass filter was placed in front of the CCD camera to eliminate the laser scattering. The exposure time to CCD camera was set to 1 s. The fluorescence single cell images were obtained at a particular wavelength as a function of AOTF frequency sweeping. The AOTF was operated over the spectral region ranging from 500 to 630 nm with a 3.75-nm interval at a scanning rate of 1 wavelength s−1. The acquired cellular images were processed and analyzed using MetaMorph (Version 7.1.3.0, Molecular Devices).Drug treatment
To screen the cardiotoxicity of the drugs (cisapride, digoxin, camptothecin and SH-03) cells on the microchannels were treated with different concentrations of each drug. Prior to the drug treatment the cells were cultured overnight to achieve log phase growth. Drug-induced cardiotoxicity was tested when the cell confluency reached 80%. Stock solutions of cisapride (20 mM), digoxin (10 mM), camptothecin (2.8 mM), and SH-03 (25 mM) were prepared in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO). Immediately before the experiment, drug solutions were diluted with DMEM containing 10% FBS to the working concentrations and subsequently the cells were treated with varying concentrations of cisapride (12.5 to 200 nM), digoxin (0.1 to 10 nM), camptothecin (75 to 600 nM). The cells were incubated with a particular drug solution under standard conditions (see above) for different time intervals; cisapride for 12 h; digoxin for 24 h; camptothecin for 6 h; SH-03 for 12 h. Three independent experiments were performed for each particular drug concentration. For each particular drug concentration three independent experiments were performed. The final DMSO concentration in the medium was carefully kept below 0.1% to nullify its toxicity. Cell lines not exposed to drugs, but supplemented with the same concentration of DMSO, and grown under the same conditions as drug-treated cells, were treated as controls.Potassium channel assay
The permeability of potassium channels was monitored by the FluxOR™ potassium channel assay as outlined in Fig. 1B. Once inside the cell, the nonfluorescent AM ester form of the FluxOR™ dye is cleaved by endogenous esterases into a thallium-sensitive fluorescent indicator. FluxOR™ loading buffer was made from Hank's Balanced Saline Solution (HBSS; Invitrogen, Eugene, OR) buffered with 20 mM HEPES and pH adjusted with NaOH to 7.4. Powerload™ concentrate and water-soluble probenecid were used as directed by the kit to enhance the dye solubility and retention, respectively. After drug-treatment the cells were washed with 1X PBS and then incubated with 500 μL Tl+ sensitive dye solution (1:16000 dilutions in HBSS buffer) for 90 min at room temperature. The concentration of the dye was pre-optimized as the highest concentration of the dye that did not allow auto cellular emission (i.e., in absence of Ti+). After that the cell plates were washed once with dye-free assay buffer, before adding a final volume of 400 μL assay buffer containing water-soluble probenecid. Subsequently, the cells were additionally treated with the stimulus buffer (100 μL) containing Tl+ for 90 s, and imaged using the single cellular multicolor imaging system.
Intracellular Na+ concentration
Change in intracellular Na+ concentration was monitored by measuring the fluorescence emission of cells treated with CoroNa™ Red, a sodium-sensitive fluorescence probe (Fig. 1B). Stock solution of CoroNa™ Red (1 mM) was prepared in DMSO. The stock solution was then diluted with 1X PBS to produce a 1 μM working solution. The cells were incubated with 500 μL of CoroNa™ Red solution (1 μM) for 15 min at 37 °C under 5% CO2. After incubation the cells were washed twice with 1X PBS and detached from the culture plate by accutase treatment. The isolated cells were resuspended in 10 μL of 1X PBS and analyzed by the single cellular multicolor imaging system.Apoptotic/necrotic cell staining
Drug-treated cells were washed twice with 1X PBS and detached from the surface of the cell culture plate by incubating with accutase (Thermo Electron Corporation, CO, USA) at 37 °C for 10 min. The detached cells were collected, washed with cold 1X PBS (4 °C), and centrifuged at 220 rcf for 3 min at 4 °C. Subsequently, the cell pellets were resuspended in 100 μL of calcium-enriched binding buffer (BD Biosciences, San Jose, CA) and reacted with 5 μL of annexin V-biotin for 15 min. Annexin V, a human protein with a molecular weight of 36 kDa, has high affinity for phosphatidylserine (PS) externalized on the cell membrane during early apoptosis. The unbound annexin V-biotin was washed out and the cells were redispersed in binding buffer (100 μL) and incubated with 10 nM streptavidin-conjugated quantum dot and/or 10 μL of PI solution. Quantum dot 625 (λmax = 625 nm) was used when cells were simultaneously treated with quantum dot and CoroNa™ red (λmax = 579 nm) and Tl+ sensitive dye (λmax = 524 nm), while quantum dot 525 (λmax = 525 nm) was used when cells were simultaneously subjected to quantum dot, CoroNa™ red and PI (λmax = 619 nm) staining. After 15 min incubation in the dark, the cells were collected by centrifugation (220 rcf, 3 min) and washed twice with 1X PBS. The cell pellets were resuspended in 1X PBS (10 μL) and were subjected to multicolor image analysis.While quantum dot positive (+) cells were considered apoptotic cells, PI (+) cells were identified as necrotic cells. Control cell population was considered to be both PI and quantum dot negative.
Statistical analysis
All measurements were triplicated (n = 3). One-way analysis of variance (ANOVA) and Duncan's multiple range tests were used to determine the significance of differences among drug treatments using SAS (Version 9.1, Statistical Analysis System Software, Cary, NC, USA). Differences were statistically considered to be significant at p < 0.05.Results
High-content screening (HCS) using multiple fluorophores
A new approach to evaluate drug-induced cardiotoxicity was established through the single cell multicolor imaging system allowing simultaneous monitoring of intracellular Na+, the permeability of K+ channel, and apoptotic cell death. The scheme utilizes the simultaneous use of multiple fluorophore having the same excitation but necessarily widely different emission maximums to mitigate the spectral interference in the respective fluorescent images. Therefore our first task was to confirm that the fluorescent images of the cells simultaneously tagged or loaded with different fluorophores used in this study do not overlap with each other. Fig. 2 shows the hyperspectral images of cardiomyocyte cells subjected to the FluxOR™ potassium channel assay, CoroNa™ Red assay and tagging with annexin V-biotin–streptavidin-conjugated QD-625 post-6 h camptothecin (300 nM) treatment. The emission maximums of thallium-sensitive fluorescent indicator (from FluxOR™ dye), sodium-sensitive fluorescence CoroNa™ Red, and streptavidin coated QD-625 were 524, 575, and 625 nm, respectively. Cellular images correspond to the ion channel permeability, intracellular Na+ level and apoptotic cellular events as a function of the specific reactions of cellular components with the used fluorophores. The interaction of the dyes with Tl+ ions or intracellular Na+ or PS was monitored at different cellular emission wavelengths through coincident acousto-optic tunable filter (AOTF) scanning (scan rate 3.75 nm s−1) and CCD (exposure time 1 s) imaging in the spectral region ranging from 500 nm to 630 nm. The fluorescence images of the cells acquired at the emission maximum of each fluorophore were found not to overlap. This coincident operation enabled us to simultaneously monitor a series of cellular responses at the second level to gain an insight into the effect of drugs on cardiomyocyte cells. | ||
| Fig. 2 Hyperspectral images of H9c2(2–1) cells subjected to the FluxOR™ potassium channel assay, CoroNa Red assay and tagging with annexin V-biotin-streptavidin-conjugated QD-625 post-6 h camptothecin (300 nM) treatment. Images were obtained over a spectral region ranging from 500–630 nm with 3.75 nm interval at a scan rate of one wavelength per second. The CCD exposure time was set to 1 s. Images with arrow marks were taken at the respective emission maximum of the fluorophores used. | ||
Effect of cisapride and digoxin on the permeability of K+ channel and Na+ concentration
Cisapride has been known as an hERG channel blocker. On the other hand digoxin has been reported to block the Na+/K+ pump. Their side effects on cardiomyocytes have also been shown using the patch clamp technique.16,17 In previous studies more emphasis was given to monitoring the drug-induced changes occurring in a single ion channel. However, a particular class of ion channels (e.g., Na+/Ca2+ exchanger) in a single cell can also be affected by the activity of ion channels of another class (e.g., Na+/K+ pump). As a new and initial trial to evaluate the comprehensive effects of drugs on activities of several ion channels and intracellular ions, alteration in permeability of K+ channel and intracellular Na+ amount in cisapride or digoxin-treated H9c2(2–1) cells were simultaneously monitored at single cell level using K+ and Na+ indicators. Fig. 3(A, B) shows the corresponding hyperspectral images of the cells treated with varied concentrations of cisapride (0, 12.5, 25, 50, 100 and 200 nM) and digoxin (0, 0.1, 1, 10 and 100 nM). Post drug treatment cells were subjected to both CoroNa™ Red assay and FluxOR™ potassium channel assay. The Y axis in Fig. 3C and D indicates the number percentage (P%) of corresponding fluorescent single cells. P was calculated according to the following equation:| P (%) = (P1/P2) × 100 |
 | ||
| Fig. 3 Effects of cisapride and digoxin on K+ channels and Na+/K+-pumps in H9c2(2–1) cells. Hyperspectral images depict the change in permeability of the K+ channel and intracellular ion concentration after treatment with various cisapride (A) and digoxin (B) doses. (C–D) Quantification of drug-induced cellular responses in H9c2(2–1) cells. All experiments were triplicated and error bars represent the mean ± SD. *P < 0.05 vs. control, n = 3. | ||
Up to a cisapride concentration ≤25 nM the number percentage of fluorescent single cells indicating the permeability of K+ channel remained almost identical to that in the control. However, the permeability of the K+ channel was found to reduce at higher drug concentrations (>25 nM) (Fig. 3A and 3C). The results indicate that the K+ channel permeability in the cardiomyocyte remained unaffected up to a cisapride treatment dose of 25 nM, but was reduced as the concentration of the drug was increased above 25 nM. The permeability of the K+ channel was blocked in proportion to an increment of the treatment dose over the tested drug concentration range (25–200 nM). On the other hand, irrespective of the cisapride concentration the amount of intracellular Na+ remained almost same as that in the control cells. These results clearly suggest that cisapride specifically blocked the K+ channel and did not influence the activity of the Na+/K+ pump.
The IC50 of cisapride as a hERG channel blocker determined using the patch clamp technique was calculated to be 6.5 or 44.5 ± 10.6 nM,16 while the IC50 of cisapride as a K+ channel blocker was independently determined to be 57.0 ± 6.6 nM (Fig. 3C) by the high-content multicolor single cell imaging cytometry. The two values are similar within experimental error, confirming the reliability of measurement of the K+ channel permeability using high-content single cell imaging cytometry.
Fig. 3B and 3D represent the effect of digoxin on the permeability of the K+ channel and intracellular Na+ ion. At lower drug concentrations (≤0.1 nM) no significant change in the permeability of the K+ channel and intracellular Na+ concentration was observed. However, the number percentage of Na+-indicator positive fluorescent single cells increased gradually with further increase in drug concentration (≥1 nM). At a digoxin concentration as high as 100 nM a marked increase in the cell number having increased intracellular Na+ was observed. On the contrary, digoxin did not show any significant effect as a K+ channel block over the tested concentration range (0.1 to 100 nM). The permeability of K+ channel in the treated cells remained almost the same as in the control over the entire drug range tested.
Anticancer drug-induced cardiotoxicity
Prolonged anticancer drug treatment frequently leads to cardiotoxic problems in patients. Anthracyclines anticancer drugs are some of the most effective treatments against a wide range of cancer than any other class of chemotherapy agents ever developed.18 However, they are notorious for causing cardiotoxicity. This cardiotoxicity may be caused by many factors, such as inhibition of the ryanodine receptor of the sarcoplasmic reticulum in the heart muscle cells, or formation of free radicals in the heart including ion channel block. However, there are also other anticancer drugs that do no induce serious cardiotoxicity. As a model drug of that category, camptothecin was chosen to investigate its effects on the permeability of the K+ channel, intracellular Na+ concentration, and cell viability of cardiomyocyte H9c2(2–1) cell lines. Cells were treated with a varied dose of camptothecin (0, 75, 150, 300 and 600 nM) for 6 h. Fig. 4A shows the simultaneous detection of the permeability of the K+ channel, intracellular Na+, and apoptosis in cardiomyocyte single cells post-camptothecin treatment. The number percentage of single cells emitting fluorescence corresponding to permeability of K+ channel and intracellular Na+ ion did not show significant alteration up to a dose of 300 nM of camptothecin. A further twofold increase in camptothecin concentration (600 nM) resulted in a nearly 15% increase in the permeability of the K+ channel as well as in the intracellular Na+ concentration (Fig. 4B). In addition, the percentage of apoptotic cells was significantly increased at higher doses (600 nM) of the drug; the percentage of apoptotic cells was nearly 3.5 times that of the control. The increase in concentration of intracellular Na+ and permeability of the K+ channel at higher dose was thought to arise from camptothecin-induced cytotoxicity rather than from the permeability change of the ion channel.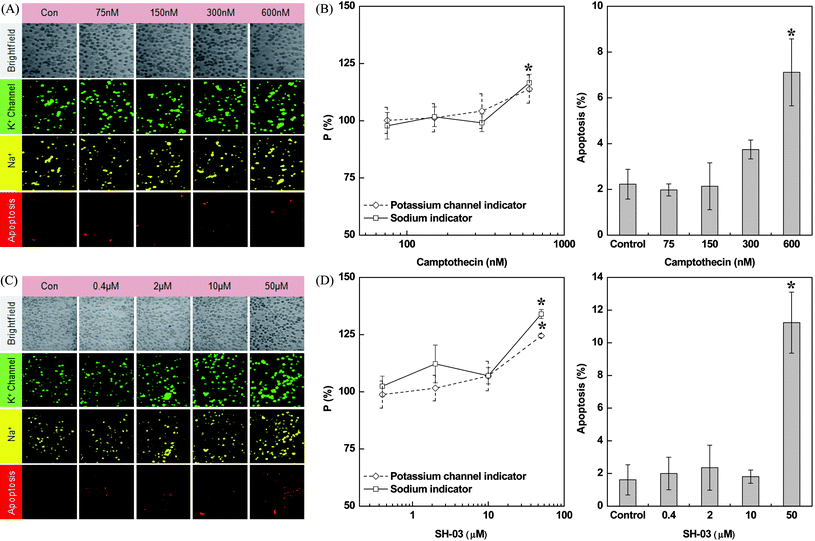 | ||
| Fig. 4 The effect of camptothecin and SH-03 on the permeability of potassium channels, sodium ion concentrations, and apoptosis in the H9c2(2–1) cells. Image (A) and occurrence rate (B) of camptothecin-induced changes in H9c2(2–1) cells after 6 h of drug treatment. Image (C) and occurrence rate (D) of SH-03-induced changes in H9c2(2–1) cells after 12 h of drug treatment. All experiments were triplicated and error bars represent the mean ± SD. *P < 0.05 vs. control, n = 3. | ||
The new anticancer drug candidate SH-03 is a deguelin derivative. Deguelin isolated from Mundulea sericea (Leguminosae) and has specific therapeutic effects on lung cancer cells but is usually noncytotoxic to normal cells.19 Deguelin suppresses angiogenesis as well as tumor growth and induces cell death in cancer cells. Nevertheless, several side effects including cardiotoxicity, respiratory inhibition, and blockage of neural transduction can be generated from high-dose deguelin. To widen the scope of the therapeutic applications of deguelin with diminished adverse effects, SH-03 was synthesized as described in previous studies.14,15Fig. 4C shows the fluorescent images of SH-03-treated cardiomyocyte that were subjected to multiple fluorophore tagging/loading for simultaneous detection of apoptosis, permeability of the K+ channel, and intracellular Na+. At a drug concentration less than 10 μM, the K+ channel activity and intracellular Na+ level in SH-03-treated cardiomyocyte were found to be similar to that in controls. This result demonstrates that SH-03 does not induce significant alteration in activities of K+ channel and Na+/K+-pump at normal use. Also, at that drug concentration no apoptosis was induced. It is noteworthy that 10 μM of SH-03 is a much higher concentration compared to 0.3 μM of camptothecin that did not cause serious cardiotoxicity. However, at a 5-fold higher concentration of SH-03 (50 μM) permeability of the K+ channel as well as the intracellular Na+ level were increased by 25% and 35%, respectively, compared to the control. The percentage of apoptotic cells was approximately six times greater than the controls.
Intracellular Na+ level in apoptotic and necrotic cells
Apoptosis and necrosis exert different effects on ion flow.20Apoptosis results in cell shrinkage and secretion of intracellular medium including Na+ out of the cell. Consequently the intracellular concentration of the sodium ion is decreased.21 On the other hand, necrotic cell death induces damage to the cellular membrane causing cell swelling due to transmission of extracellular solution in to the cell.20 Thus, the sodium ion concentration in necrotic cells is increased. Nevertheless, the phenomenon of ion flow observed in apoptotic and necrotic cells is not clearly understood yet, and more so the phenomenon of ion flow arising from apoptosis or necrosis is still in dispute. Fig. 5 shows drug-induced cell death and alterations in the intracellular Na+ level in H9c2(2–1) cells treated with 1 μM of camptothecin. Intracellular Na+, apoptosis, and necrosis were visualized using a sodium-sensitive fluorescence probe (λem = 579 nm), QD 525 (λem = 525 nm), and PI (λem = 619 nm). To correlate the change of the intracellular Na+ level with apoptosis and necrosis, the Na+-indicator positive fluorescent cellular image was superimposed on the fluorescence image of apoptotic cells (annexin V-QD) or necrotic cells (PI). The cells were an increase of intracellular Na+ concentration was observed were perfectly superimposed on the necrotic cells rather than the apoptotic cells, clearly indicating the increase of intracellular Na+ concentration caused by disruption of the cellular membrane due to necrosis. | ||
| Fig. 5 (A) Optical micrograph (fluorescent mode) of rat cardiomyocyte showing the correlation between drug-induced cell death pathways and sodium ion concentration. Green (apoptosis) and yellow (sodium ion) images are rarely overlapped, while yellow and red (necrosis) images are overlapped completely. (B) Colocalization of markers for apoptosis and necrosis with sodium ion marker. | ||
Discussion
One of the main reasons of drug-induced cardiotoxicity is the blockade of ion channels. The hERG encodes the α-subunit of the rapid delayed rectifier current IKr in the heart. The hERG cardiac potassium channel contributes to terminal repolarization in human cardiomyocytes. Direct blocking of hERG/IKr channels produces acquired long QT syndrome (acLQTS) characterized by drug-induced QT prolongation and TdP arrhythmias. The cardiotoxicity associated with an unintended hERG block has been a major concern in successful drug development and has prompted pharmaceutical companies to screen developmental compounds for hERG blockade and made hERG a major target in drug safety programs. More recently, several therapeutic compounds have been identified that reduce hERG/IKr currents not by direct blocking but by inhibition of hERG/IKr trafficking to the cell surface. Channel trafficking is integral to functional hERG channel biogenesis and surface expression. Its disruption leads to a reduction in number of functional channels at the cell membrane and is an important mechanism by which a number of hERG mutants contribute to the congenital long QT syndrome.22–26Cisapride, known as a hERG channel blocker, strongly binds to two residues (Tyr 652 and Phe 656) located in the S6 domain.27 Insight into the effect of cisapride on cardiomyocyte was achieved using quantitative high content cellular imaging cytometry. In the present study, both the K+ ion channel permeability and intracellular ion level were examined to study the response of cardiac cells to cisapride treatment (Fig. 3A, C). Our data provide an important insight into the terms of identifying the inhibition of hERG channel trafficking (decreasing permeability of K+ channel) by cisapride (Fig. 3C). The IC50 value obtained using the high content cellular imaging was compared with that determined in previous studies using the patch clamp technique. The comparable IC50 values by reiterant HCS assay could provide accurate information of drug-induced cardiotoxicity in the microfluidic platform.
The Na+/K+-ATPase (Na+/K+-pump) plays critical roles in maintaining ion homeostasis.28 Directly blocking of the Na+/K+-pump may lead to cardiotoxicity and apoptosis. Failure of the Na+/K+-pump results in depletion of intracellular K+, accumulation of intracellular Na+, and, consequently, leads to membrane depolarization and increases in intracellular free Ca2+ ([Ca2+]i) due to activation of voltage-gated Ca2+ channels and a reversed operation of the Na+/Ca2+ exchanger. The inhibition of the Na+/K+-pump causes elevated intracellular Na+ levels by diminishing the rate of Na+ influx through the Na+/Ca2+ exchanger. The increasing amount of intracellular Na+ in turn inhibits Ca2+ efflux through the Na+/Ca2+ exchanger. While the permeability of the K+ channel in digoxin-treated cells was identical to the normal condition, the amount of intracellular Na+ was significantly elevated post-digoxin treatment (Fig. 3B, D). We elucidated that digoxin-induced cardiomyocyte toxicity preferentially resulted due to the blocking of Na+/K+-pump as evidenced by the increased intracellular Na+ concentration (Fig. 3B, D). No significant change in K+ channel permeability (Fig. 3D) apparently rules out the possibility of hERG channel blocking or inhibition of channel trafficking.
Since long-term anticancer drug treatment frequently leads to cardiotoxic problems in patients, we also screened the cardiotoxicity of an established (camptothecin) and a potential anticancer drug (SH-03) by monitoring the ion channel permeability, intracellular ion level and cell death pathways (Fig. 4). SH-03 is a rotenoid-containing deguelin analog. Deguelin, isolated from the African plant Mundulea sericea, is known for its antiangiogenic effect and its apoptotic effects in a variety of cell types.29 Despite the potential anticancer activity of deguelin in vivo and in vitro, this agent showed toxic effects in rats.30 Long-term or high-dose deguelin treatment might cause Parkinson's disease-like syndrome in rats.31 To alleviate these constraints of high toxicity and low efficacy of deguelin we synthesized new deguelin derivatives with less toxicity and higher efficacy. Recently, we reported the isolation, synthesis and preliminary structure activity relationship study of SH-03.31SH-03 is effective in activating intracellular hypoxia-inducible factor 1 subunit α (HIF-1α), heat shock protein-90 (Hsp90), the mammalian target of rapamycin (mTOR) and signal transducers and activators of transcription (STAT) proteins in malignant human bronchial epithelium (HBE) and non-small cell lung cancer (NSCLC) cell lines.15 It also exerts an antibacterial effect, specifically on microorganisms belonging to the genus Helicobacter (e.g., Helicobacter pylori).32 Recently, we also showed the anticancer activity and the dynamics of the caspase-mediated apoptotic cascade induced by SH-03 in human leukemia (HL-60) cells.12 However, the effect of SH-03 on cardiomyocyte cells has not been studied earlier and hence, remains unclear. Given the fact that many drugs having remarkable medicinal effects have been removed from the marketplace due to cardiotoxic effects (like TdP), cardiotoxic risk factors for human safety of new chemical entities have to be assayed in vitro with in vivo to minimize the possibility of drug failure during clinical trial. The cell-based cardiotoxicity test can contribute to the cost effective drug discovery process.
Herein, we for the first time screened its cardiotoxicity in cardiomyocyte cells using a new drug-screening platform, and revealed its dose dependent effect on the ion channel permeability, intracellular ion level and induction of apoptosis using high-content cellular imaging cytometry.
Our results (Fig. 4) suggest that both camptothecin and SH-03 do not induce any significant cardiotoxicity at normal use. Like camptothecin, SH-03 did not induce apoptotic cell death or alteration in the permeability of K+ channel and intracellular Na+ level at a concentration ≤10 μM. However, higher concentrations of the drugs do lead to toxicity in the cardiomyocytes. The permeability of the K+ channel and intracellular Na+ level were elevated at a much higher concentration (50 μM) of SH-03. It should be noted that in the case of SH-03-treated cells the toxicity appeared at a much higher concentration (50 μM) compared to the camptothecin-treated cells (0.6 μM).
Death of a single cardiomyocyte cell can proceed with a serial cell death due to the transfer of ions from apoptotic and necrotic cells to normal cardiomyocytes through connection. Because cardiomyocytes have a weak reproduction and proliferation activity, dead cardiomyocytes are not replaced by new cells and this aggravates heart dysfunction. Therefore, drug-induced cardiomyocyte cell death pathways demand serious consideration. Generally, apoptotic cells undergo cell shrinkage as well as variation in the intracellular ion level. An apoptotic cell is usually characterized by lower concentrations of K+ and higher concentrations of H+. However, controversial results about the change in intracellular Na+ levels after apoptosis have caused a dispute. Some studies asserted the increase in intracellular Na+ in apoptotic cells. Bortner et al. (2001) demonstrated that the amount of intracellular Na+ was increased in apoptotic Jurkat T-cells.33 Etoposide-induced apoptosis in the prostate cancer cell line PC3 were shown to accompany increased intracellular [Na+] and [Mg2+] and lower [K+] and [Cl−] (Salidoet al. 2001).34 Elevation of intracellular Na+ concentration was also featured in hypoxia and veratridine-induced neuronal apoptosis.35,36 On the other hand, a number of studies revealed a decrease in intracellular Na+ in apoptotic cells. Many drugs including anisomycin, dexamethasone, thapsigargin and staurosporine induced apoptosis of Jurkat cells and murine S49 Neo cells37 and intracellular [K+] and [Na+] were found to decrease in apoptotic cells. Staurosporine and etoposide were reported to decrease intracellular Na+ and K+ levels in apoptotic U937 cells.38 Necrotic cells are characterized by cell swelling and increased intracellular [Na+], [Ca2+], and [Mg2+]. From the above discussion while it is clear that apoptosis and necrosis exert different effects on ion flow that need to be understood in detail, whether a drug-induced apoptotic process may result in increased intracellular Na+ level in the cardiomyocyte is still an open question and has so far not been directly investigated.
The high content multicolor single cell imaging provides direct visualization to monitor a series of cellular responses at the second level to gain insight into the drug-induced apoptosis and necrosis with respect to the alteration of intracellular Na+ ion level. A higher concentration of camptothecin (1 μM) treatment induced both apoptotic and necrotic cell death modalities in cardiomyocytes (Fig. 5). The images of the sodium sensitive CoroNa™ red positive cells were perfectly superimposed onto the necrotic cells rather than the apoptotic cells, clearly indicating the increase of intracellular Na+ concentration caused by disruption of cellular membrane due to necrosis.
Conclusion
In this study, a drug-induced cardiotoxic effect was estimated by visualizing the alteration in permeability of the ion channel, intracellular ion level, and also induction of cell death (apoptosis or necrosis) using high-content single cellular imaging coupled with a miniaturized sample platform. This developed cellular imaging cytometry is expected to facilitate quantitative multivariate cellular analysis and eliminate false positive errors as it provides high spectral resolution among the fluorophores as shown in Fig. 2. The developed method can be diversely used for many researches. The integration of ion channel activity is related to many processes, such as heartbeat, muscle contraction, hormone secretion and pain perception. Because of the existence of ion channels in most cell types and its roles in a number of biological processes, including ion transportation, regulation of potential difference in plasma membrane, and cell signaling, evaluation of drug-induced toxicity by monitoring ion channel activity can also be applied to cells constituting other organs. However, it should be noted that in this study drug-induced cardiotoxicity was investigated at a particular cellular state. Considering the affinity of the drug to the ion channel is cellular state dependent, applicability and sensitivity of this assay for monitoring the drug interactions with ion channels have to be tested in other cellular states. High content cellular imaging cytometry can be utilized as an analytical tool for automated drug screening targeting specific cellular entities or events.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2010-0017903, 2010-0029775) and a grant of the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea. (A100096).References
- M. C. Sanguinetti and M. Tristani-Firouzi, Nature, 2006, 440, 463–469 CrossRef CAS.
- A. M. Katz, Circ. Heart Failure, 2008, 1, 63–71 Search PubMed.
- M. R. Zile and D. L. Brutsaert, Circulation, 2002, 105, 1503–1508 CrossRef.
- B. Fermini and A. A. Fossa, Nat. Rev. Drug Discovery, 2003, 2, 439–447 CrossRef CAS.
- S. Nattel and L. Carlsson, Innovative approaches to anti-arrhythmic drug therapy, Nat. Rev. Drug Discovery, 2006, 5, 1034–1049 CrossRef CAS.
- O. J. Arola, A. Saraste, K. Pulkki, M. Kallajoki, M. Parvinen and L. M. Voipio-Pulkki, Cancer Res., 2000, 60, 1789–1792 CAS.
- M. Traebert, B. Dumotier, L. Meister, P. Hoffmann, M. Dominguez-Estevez and W. Suter, Eur. J. Pharmacol., 2004, 484, 41–48 CrossRef CAS.
- M. Roy, R. Dumaine and A. M. Brown, Circulation, 1996, 94, 817–823 CAS.
- G. A. Gintant, J. T. Limberis, J. S. McDermott, C. D. Wegner and B. F. Cox, J. Cardiovasc. Pharmacol., 2001, 37, 607–618 CrossRef CAS.
- S. Rajamani, C. Studenik, R. Lemmens-Gruber and P. Heistracher, Br. J. Pharmacol., 2000, 129, 843–852 CrossRef CAS.
- I. Gussak, J. Litwin, R. Kleiman, S. Grisanti and J. Morganroth, J. Electrocardiol., 2004, 37, 19–24 CrossRef.
- P. K. Naoghare, H. A. Ki, S. M. Paek, Y. K. Tak, Y. G. Suh, S. G. Kim, K. H. Lee and J. M. Song, Integr. Biol., 2010, 2, 46–57 RSC.
- H. A. Ki, P. K. Naoghare, B. K. Oh, J. W. Choi and J. M. Song, Anal. Biochem., 2009, 388, 23–27 CrossRef CAS.
- P. K. Naoghare, M. J. Kim and J. M. Song, Anal. Chem., 2008, 80, 5407–5417 CrossRef CAS.
- W. Y. Kim, D. J. Chang, B. Hennessy, H. J. Kang, J. Yoo, S. H. Han, Y. S. Kim, H. J. Park, S. Y. Seo, G. Mills, K. W. Kim, W. K. Hong, Y. G. Suh and H. Y. Lee, Cancer. Prev. Res. (Phila Pa), 2008, 1, 577–587 Search PubMed.
- D. Rampe, M. L. Roy, A. Dennis and A. M. Brown, FEBS Lett., 1997, 417, 28–32 CrossRef CAS.
- T. Haruna, M. Horie, I. Kouchi, R. Nawada, K. Tsuchiya, M. Akao, H. Otani, T. Murakami and S. Sasayama, Circulation, 1998, 98, 2905–2910 CAS.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS.
- J. K. Woo, D. S. Choi, H. T. Tran, B. E. Gilbert, W. K. Hong and H. Y. Lee, Cancer. Prev. Res. (Phila Pa), 2009, 2, 361–369 Search PubMed.
- M. Bhatia, Apoptosis versus necrosis in acute pancreatitis, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 286, G189–196 CrossRef CAS.
- C. D. Bortner and J. A. Cidlowski, J. Biol. Chem., 2003, 278, 39176–39184 CrossRef CAS.
- B. P. Delisle, B. D. Anson, S. Rajamani and C. T. January, Circ. Res., 2004, 94, 1418–1428 CrossRef CAS.
- E. Ficker, Y. A. Kuryshev, A. T. Dennis, C. Obejero-Paz, L. Wang, P. Hawryluk, B. A. Wible and A. M. Brown, Mol. Pharmacol., 2004, 66, 33–44 CrossRef CAS.
- J. S. Cordes, Z. Sun, D. B. Lloyd, J. A. Bradley, A. C. Opsahl, M. W. Tengowski, X. Chen and J. Zhou, Br. J. Pharmacol., 2005, 145, 15–23 CrossRef CAS.
- Y. A. Kuryshev, E. Ficker, L. Wang, P. Hawryluk, A. T. Dennis, B. A. Wible, A. M. Brown, J. Kang, X. L. Chen, K. Sawamura, W. Reynolds and D. Rampe, J. Pharmacol. Exp. Ther., 2005, 312, 316–323 CAS.
- B. A. Wible, P. Hawryluk, E. Ficker, Y. A. Kuryshev, G. Kirsch and A. M. Brown, J. Pharmacol. Toxicol. Methods, 2005, 52, 136–145 CrossRef CAS.
- J. Chen, G. Seebohm and M. C. Sanguinetti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12461–12466 CrossRef CAS.
- X. Q. Wang, A. Y. Xiao, C. Sheline, K. Hyrc, A. Yang, M. P. Goldberg, D. W. Choi and S. P. Yu, J. Cell Sci., 2003, 116, 2099–2110 CrossRef CAS.
- C. Gerhäuser, W. Mar, S. K. Lee, N. Suh, Y. Luo, J. Kosmeder, L. Luyengi, H. H. Fong, A. D. Kinghorn, R. M. Moriarty, R. G. Metha, A. Constantinou, R. C. Moon and J. M. Pezzuto, Nat. Med., 1995, 1, 260–266 CrossRef CAS.
- S. H. Oh, J. K. Woo, Y. D. Yazici, J. N. Myers, W. Y. Kim, Q. Jin, S. S. Hong, H. J. Park, Y. G. Suh, K. W. Kim, W. K. Hong and H. Y. Lee, J. Natl. Cancer Inst., 2007, 99, 949–961 CrossRef CAS.
- P. Caboni, T. B. Sherer, N. Zhang, G. Taylor, H. M. Na, J. T. Greenamyre and J. E. Casida, Chem. Res. Toxicol., 2004, 17, 1540–1548 CrossRef CAS.
- T. Junko and C. Noriko, PCT WO 9815269, 1998.
- C. D. Bortner, M. Gomez-Angelates and J. A. Cidlowski, J. Biol. Chem., 2001, 276, 4304–4314 CrossRef CAS.
- M. Salido, J. Vilches, A. Lopez and G. M. Roomans, Cell Biol. Int., 2001, 25, 499–508 CrossRef CAS.
- F. Petrat, T. Li, N. Dehne, H. deGroot and U. Rauen, Life Sci., 2006, 79, 1606–1615 CrossRef CAS.
- J. Jordan, M. F. Galindo, C. Gonzalez-Garcia and V. Cena, Neuroscience, 2003, 122, 707–715 CrossRef CAS.
- C. D. Bortner, F. M. Hughes Jr and J. A. Cidlowski, J. Biol. Chem., 1997, 272, 32436–32442 CrossRef CAS.
- V. Yurinskaya, T. Goryachaya, I. Guzhova, A. Moshkov, Y. Rozanov, G. Sakuta, A. Shirokova, E. Shumilina, I. Vassilieva, F. Lang and A. Vereninov, Cell. Physiol. Biochem., 2005, 16, 155–162 CrossRef CAS.
Footnote |
| † Published as part of a LOC themed issue dedicated to Korean Research: Guest Editors: Professor Je-Kyun Park and Kahp-Yang Suh. |
| This journal is © The Royal Society of Chemistry 2011 |