DOI:
10.1039/C005217P
(Paper)
Lab Chip, 2011,
11, 152-162
Received
22nd April 2010
, Accepted 20th August 2010
First published on 18th October 2010
Abstract
We explore the design and operation of an optical-tweezers electrophoresis apparatus to resolve polymer adsorption dynamics onto a single micro-sphere in a micro-fluidic environment. Our model system represents a broader class of micro-fluidic electrophoresis experiments for biosensing and fundamental colloid and surface science diagnostics. We track the adsorption of 100 kDa poly(ethylene oxide) homopolymer onto a colloidal silica sphere that is optically trapped in a crossed parallel-plate micro-channel. The adsorption dynamics are probed on the ∼1 μm particle length scale with ∼1 s temporal resolution. Because the particle electrophoretic mobility and channel electro-osmotic flow are exquisitely sensitive to the polymer layer hydrodynamic thickness, particle dynamics can be complicated by polymer adsorption onto the micro-channel walls. Nevertheless, using experiments and a theoretical model of electro-osmotic flow in channels with non-uniform wall ζ-potentials, we show that such influences can be mitigated by adopting a symmetrical flow configuration. The equilibrium hydrodynamic layer thickness of 100 kDa poly(ethylene oxide) on colloidal silica is ∼10 nm at polymer concentrations ≳10 ppm (weight percent), with the dynamics reflecting polymer solution concentration, flow rate, and polydispersity.
1 Introduction
When a colloidal particle is confined in a micro-fluidic environment by an optical trap, rapid and precise control over the surrounding solvent can be achieved when, for example, introducing an analyte that has specific or non-specific affinity for the colloidal substrate. In optical tweezers electrophoresis (OTE), an oscillatory electric field is applied to the trapped particle, and the scattered light furnishes the electric-field-induced particle dynamics with high spatial and temporal resolution. Such experiments have potential as biosensing platforms and for novel studies of physicochemical interactions. In principle, conventional microelectrophoresis and electrophoretic light scattering techniques can serve similar purposes, but they are often limited by particle sedimentation and entrainment by hydrodynamic flow. Furthermore, bulk mixing of colloidal dispersions and analyte solutions typically yields dynamics that reflect bulk mixing rather than the receptor-ligand interaction potential.
We recently demonstrated OTE as a high resolution, micro-particle velocimetry tool for mapping electro-osmotic flow profiles in micro-channels at frequencies up to 50 kHz.1 In that work, the particles were bare colloidal micro-spheres in salt solutions with fixed concentration. Here we explore OTE for probing the dynamics when a trapped micro-sphere is abruptly exposed to a solution of adsorbing homopolymer. While the fluid flow can be well controlled in this micro-fluidic environment, the high sensitivity of OTE expedites quantitative studies of how electro-osmotic flow and quasi-steady micro-channel hydrodynamics influence polymer adsorption and, thus, the particle dynamics.
Adsorbing polymers are often added to colloidal dispersions to control stability and rheology.2–4 Thick layers generally impart steric stability, with thin layers providing opportunities for polymer bridging. The polymer layer hydrodynamic thickness on colloidal particles has been measured using dynamic light scattering.5,6 However, the adsorption dynamics can be complicated by the mixing hydrodynamics, and instrumental sensitivity is lost when nanometer thick layers develop on micrometer sized particles. The electrophoretic mobility of colloidal particles can be extraordinarily sensitive to the hydrodynamic layer thickness, but, again, bulk mixing of polymer-particle suspensions yields an apparent electrophoretic mobility whose change reflects mixing dynamics and polymer-mediated inter-particle interactions. Similarly to dynamic light scattering, this makes it difficult to directly probe the polymer-particle interaction and, indeed, details of the adsorption dynamics.
Other techniques based on streaming potential in capillary tubes7,8 or the quartz-crystal micro-balance9,10probe polymer adsorption on larger, macroscopic length and time scales. These configurations tend to probe bulk polymer diffusion and/or slow relaxations due to dynamic equilibrium when adsorption and desorption rates are comparable. When adsorption is faster than desorption, due to high binding affinity, for example, the final adsorbed layer structure may reflect polymer conformational dynamics, which can occur on time scales spanning many decades.
In this study, we use single-particle micro-electrophoresis as a tool for probing polymer adsorption dynamics. A custom-built optical-tweezers electrophoresis (OTE) apparatus is integrated with a parallel-plate micro-fluidic channel, and the electrophoretic mobility of a colloidal micro-sphere is tracked after imposing an abrupt local change in polymer concentration. We demonstrate the technique using a 100 kDa poly(ethylene oxide) (PEO) homopolymer adsorbing onto colloidal silica micro-spheres. Similarly to capillary streaming potential instruments, OTE does not directly measure hydrodynamic layer thickness. Nevertheless, electrokinetic theory furnishes a sensitive correspondence between the electrophoretic mobility and hydrodynamic layer thickness.11 Moreover, since the hydrodynamic layer thickness is also known to be a sensitive indicator of polymer conformation,12,13 the experiments can probe polymer conformation dynamics in a manner that has previously not been achieved.
Using optical tweezers, we trap a single micro-sphere in a crossed parallel-plate glass micro-channel, subjecting it to an oscillatory electric field while emersed in steady pressure driven flows of background electrolyte and polymer solution. The harmonic response of the micro-sphere displacement is measured using back-focal-plane interferometry, which furnishes the electrophoretic mobility at ∼1 s time intervals. When flowing PEO solution past the micro-sphere, polymer adsorption lowers the apparent ζ-potential.14 Thus, in principle, positioning the micro-sphere at the conventional point of zero electro-osmotic flow (EOF) furnishes particle mobility measurements in channels with spatially uniform charge or wall ζ-potential. However, when polymer solution is introduced into clean micro-channels, it can adsorb to the walls, producing a time-dependent, non-uniform wall ζ-potential and accompanying (non-conventional) EOF. Similar phenomena have been observed in planar15 and cylindrical16 channels of dissimilar materials.
To understand how the wall ζ-potential dynamics influence the micro-sphere dynamics, we calculate EOF profiles with a stepwise discontinuous wall ζ-potential in a straight parallel-plate channel. This model successfully accounts for anomalous particle mobility measurements when polymer solution enters the crossed micro-channel perpendicular to the electric field. On the other hand, experiments and theory show that particle mobility is not significantly susceptible to wall ζ-potential dynamics when polymer solution enters the crossed channel parallel to the electric field. While non-uniform ζ-potentials have been explored to enhance mixing in micro-fluidic devices,17,18 they are detrimental in capillary electrophoretic separations.19,20
The OTE apparatus is described in section 2 with our protocols for detecting polymer adsorption. Section 3 briefly addresses the theory of optical tweezers velocimetry, and summarizes mathematical models for calculating fluid flow and electric fields in the parallel-plate micro-channels, as well as a simple model to connect particle electrophoretic mobility to polymer hydrodynamic layer thickness. Section 4 presents the results. We show how OTE mobilities are obtained from particle displacement power spectra, and compare measurements of apparent wall ζ-potentials and particle mobilities in the absence and presence of PEO. These data are obtained from apparent particle mobility profiles across the gap of channels with spatially uniform ζ-potentials. We then map out the transverse electric field using the OTE response of a single micro-sphere, and verify the measurements with theoretical calculations. Finally, we report particle mobility time series during PEO adsorption in two micro-channel flow configurations, drawing on theory to explain anomalies due to non-uniform wall ζ-potentials. From the particle mobility time series, we report equilibrium layer thicknesses and the long-time polymer adsorption time scales. Section 5 concludes with a brief summary.
2 Materials and methods
Our custom-built optical tweezers apparatus comprises a steerable high power 1064 nm ND-YVO4 laser (BL-106C, Spectra-Physics) directed into the back-aperture of a Plan-Apochromat 100x NA 1.4 oil-immersion objective (MRY10014, Nikon) mounted on an inverted microscope (TE-2000U, Nikon). The optically trapped particle position is measured with nanometer spatial and millisecond temporal resolution using back-focal-plane interferometry with a quadrant silicon photodiode at 0 V bias (YAG-444-A, Perkin Elmer Optoelectronics or Spot-9dmi, OSI Optoelectronics Inc.). Photodiode signals are converted to x- and y-voltage signals with a dual-axis position-sensing amplifier (501, UDT Instruments), and filtered at the Nyquist frequency with a 4-pole low-pass Bessel-filter (3362, Krohn-Hite) used as input to a 16-bit data-acquisition oscilloscope (CompuScope 1602, Gage Applied Sciences). A schematic of the optical train is shown in Fig. 1(a). The electric field is generated with an arbitrary function generator (AFG310, Tektronix) and amplified using a high-voltage amplifier (601C, Trek). Custom written programs to control the optical tweezers are written in Labview, and programs for data analysis are written in Matlab.
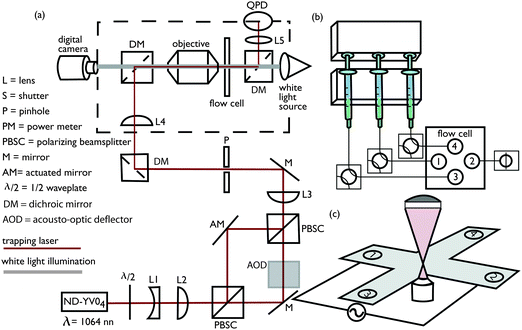 |
| | Fig. 1 Schematic of optical tweezers electrophoresis instrument and accompanying micro-fluidic hardware. (a) Optical tweezers and back-focal plane interferometry. (b) Syringe pump, valves, and connections to micro-channel electrophoresis flow cell (with an asymmetric flow configuration). (c) Crossed parallel-plate micro-channel electrophoresis flow cell. | |
2.3
Polymer adsorption experiments and micro-channel flow configurations
Separate solutions of poly(ethylene oxide) and silica micro-spheres dispersed in 1 mM NaCl electrolyte are initially pumped into the micro-channel with a syringe pump (NE-1600, New Era Pumps). The stock silica spheres are specified by the manufacturer (SS03N, Bangs Labs) as mono-disperse, non-functionalized, and non-porous; they are provided as an aqueous 10 wt% dispersion with particle radius, density, and refractive index, respectively, a = 0.4 μm, ρp = 1.96 g cm−3, and n = 1.45. The silica micro-spheres were diluted to ∼0.1 ppm (weight percent) and injected with 10 mL, 14.5 mm inner diameter syringes, and the poly(ethylene oxide) (PEO) solutions (25322-68-3, Acros Organics), with polymer molecular mass Mp ≈ 100 kDa and polydispersity index Mw/Mn ≈ 2.8 (GPC Viscotek TDA 301, Cirrus Inc.), were injected with 3 mL, 8.66 mm inner diameter syringes. Before adsorption, steady flow rates Q1 ≈ 2.8 μL min−1 for micro-sphere dispersions, and Q2 ≈ 1 μL min−1 for polymer solutions were allowed to develop, and, thus, the micro-sphere dispersion to polymer solution flow ratio s = Q1/Q2 ≈ 3. Flow separation occurs within the channel intersection, with the PEO solution front located l ≈ 1 mm from the intersection mid-point where an initially bare micro-sphere is optically trapped. Next, to initiate polymer adsorption, the micro-sphere dispersion flow is stopped (Q1 = 0) and the polymer solution flow rate increased to Q2 ≈ 4 μL min−1, producing an average fluid velocity U ≈ 100 μm s−1 at the trapped micro-sphere (s = 0). Experiments conducted with various polymer solution flow rates indicate that the subsequent change in particle electrophoretic mobility reflects the polymer adsorption dynamics when U≳50 μm s−1. In this work, continuous position time series for optically trapped micro-spheres were sampled at 1 kHz while applying an electric field with frequency (unless stated otherwise) fE = 50 Hz. Position time series were divided into equal ∼1 s segments to resolve temporal changes in the electrophoretic mobility. Prior to polymer adsorption, the micro-sphere position fluctuations21 were sampled at 20 kHz to obtain the optical trap corner frequency fc and the voltage-position sensitivity β, both of which are required for absolute mobility measurements. We adopted two flow protocols for polymer adsorption experiments. For the asymmetric flow configuration shown in Fig. 1(b), port (3) introduces polymer solution, port (4) introduces micro-spheres, port (2) is open to waste, and port (1) is blocked. For the symmetric flow configuration, port (1) introduces micro-spheres, port (2) introduces polymer solution, and ports (3) and (4) are open to waste. Calculated gap-averaged velocity streamlines are shown in Fig. 2(b) and (c), respectively, for the symmetric and asymmetric flows established before initiating polymer adsorption (left panels) and during polymer adsorption (right panels).
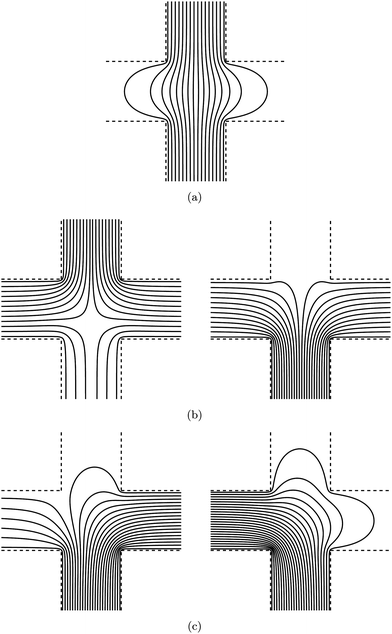 |
| | Fig. 2 Streamlines of ∇ψ from solutions of ∇2ψ = 0 in a two-dimensional infinitely extending cross with channel width aspect ratio r = 1. Electric-field lines (a), and gap-averaged velocity streamlines for symmetric (b) and asymmetric (c) flow configurations, both with electrolyte to polymer solution flow ratio s = 3 (left) and s = 0 (right), which correspond, respectively, to the flows before and during polymer adsorption onto the trapped micro-sphere. In (b), polymer solution enters from the bottom and electrolyte solution (left panel) enters from the top, whereas in (c) polymer enters from the left and electrolyte (left panel only) enters from the right. | |
3. Theoretical background
We combine optical tweezers and micro-electrophoresis,22–29 to measure the electrophoretic mobility of a single micro-sphere in a dynamic micro-fluidic environment. Briefly, the application of an oscillatory electric field E(t) = E0eiωEt sets the fluid in motion with an electro-osmotic velocity u(t) = ûeiωEt. In addition, the electrical force FE = ![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) EeiωEt acting on the particle contributes to the oscillatory particle displacement X =
EeiωEt acting on the particle contributes to the oscillatory particle displacement X = ![[X with combining circumflex]](https://www.rsc.org/images/entities/i_char_0058_0302.gif) eiωEt. Newton's second law, including inertial, electrical, optical, and hydrodynamic drag forces30–32 gives
eiωEt. Newton's second law, including inertial, electrical, optical, and hydrodynamic drag forces30–32 gives| | 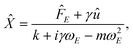 | (1) |
where m is the particle mass, γ is the frequency dependent drag coefficient (which, in general, includes electro-viscosity and inertia), k is the (stationary) optical trap stiffness, and 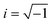 . To calibrate the optical tweezers, we adopt the steady Stokes drag approximation γ = γ0 = 6πηa, which, according to numerically exact solutions of the standard electrokinetic model,33 is correct to within 1% at frequencies f ≲ 1 kHz when κa ≈ 40 and ζ ≤ 3kBT/e. Here, κ is the reciprocal Debye length (κ−1 ≈ 10 nm with 1 mM NaCl electrolyte), ζ is the well-known particle ζ-potential, kBT is the thermal energy, and e is the fundamental charge.
. To calibrate the optical tweezers, we adopt the steady Stokes drag approximation γ = γ0 = 6πηa, which, according to numerically exact solutions of the standard electrokinetic model,33 is correct to within 1% at frequencies f ≲ 1 kHz when κa ≈ 40 and ζ ≤ 3kBT/e. Here, κ is the reciprocal Debye length (κ−1 ≈ 10 nm with 1 mM NaCl electrolyte), ζ is the well-known particle ζ-potential, kBT is the thermal energy, and e is the fundamental charge.
Because the voltage from the quadrant photo-diode detector is proportional to the particle displacement X, the one-sided voltage power spectrum
| |  | (2) |
furnishes the voltage-position sensitivity
β and trap stiffness
k, where
fc =
k/(2π
γ0) is the trap corner frequency, and
δ is the Dirac delta function.
The power spectrum is normalized by requiring that the first (Lorentzian) term yields ∫∞0〈P(f)〉df = kBTβ2/k. The power contributing to the Dirac delta function at f = fE furnishes the apparent electrophoretic mobility
| | 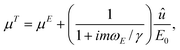 | (3) |
which, at low frequencies (
ωE ≪
γ/
m), is the sum of the intrinsic particle mobility
μE and the EOF mobility
û/
E0. Note that
| | 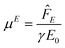 | (4) |
is the mobility reported from conventional micro-
electrophoresis,
i.e., in a stationary fluid without an optical trap.
In our OTE experiments, the changing apparent mobility is directly measured. For discretely sampled particle positions, the spacing between frequency bins fj and fj+1 is Δf = fj+1 − fj = 1/ts, where ts is the sampling time. With δ(f − fE) → tsδfj,fE, f → fj, and using eqn (3) and (4) with fj ≪ γ/m, the discrete power spectrum P(f) → P(fj) from eqn (2) is
| | 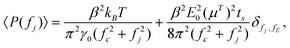 | (5) |
where
δfj,fE is the Kronecker delta; and
ts is chosen so that
fEts is an integer, which prevents leakage from the discrete delta function into adjacent frequency bins.
The Kronecker delta term furnishes the particle mobility at the forcing frequency fj = fE. All points P(fj) in the first (Lorentzian) term of eqn (5) have exponential probability distributions, and are uncorrelated.30 Thus, the precision of (μT)2 can be estimated from the ratio of the delta function and Lorentzian terms in eqn (5) at fj = fE. Accordingly, the mobility amplitude signal-to-noise ratio
| | 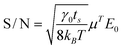 | (6) |
is similar to the result of Galneder
et al.23 for particle
ζ-potentials. Errors in
μT depend, in part, on errors in measuring
β and
fc. Standard least-squares fitting of
eqn (5) for all
fj ≠
fE to an experimentally measured power
spectrum, using the statistical methods described by Flyvbjerg
et al.,
30 furnishes the standard deviations
σfc and
σβ of
fc and
β, respectively. From the delta function term in
eqn (5), the standard deviation
σμT is obtained from a first-order Taylor expansion, assuming that
β and
fc are uncorrelated, giving
| | 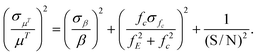 | (7) |
3.2 Hydrodynamics in micro-electrophoresis flow channels
The following sub-sections detail mathematical models to quantify the electric field, pressure-driven flow, and electro-osmotic flow in parallel-plate micro-channels with H ≪ W and Reynolds number Re = UHρ/η ≪ 1. Here, U is the average fluid velocity, and ρ and η are, respectively, the fluid density and shear viscosity. Note that the channel gap position z spans the distance between the top and bottom glass plate surfaces at ± H/2 from the gap center. Electric-field lines and representative pressure-driven gap-averaged velocity streamlines predicted by these models are shown in Fig. 2. Note that the electric-field lines in panel (a) and the gap-averaged velocity streamlines in panels (b) and (c) are all conveniently calculated (numerically and analytically) as the streamlines of the gradient of a scalar that satisfies Laplace's equation.
3.2.1 Electric field.
The electric field E = −∇ϕ, where ϕ(x, y) is the electrostatic potential. In the bulk fluid, i.e., beyond the diffuse double layers at channel walls where the charge vanishes, ϕ satisfies Laplace's equationwith boundary conditionat the electrodes, and| | ∇ϕ·![[n with combining circumflex]](https://www.rsc.org/images/entities/i_char_006e_0302.gif) = 0 = 0 | (10) |
on all other (electrically insulating) boundaries. Here, V is the applied voltage, and is an outward unit normal in the xy-plane. Fig. 2(a) highlights the flaring of electric-field lines that occurs at the central location where OTE is performed. Further quantitative analysis and comparison of this theory with experiment are undertaken below.
3.2.2 Pressure driven flow.
For steady pressure-driven flow in parallel-plate channels, the fluid velocity u is governed by the Hele-Shaw equations| | 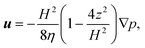 | (11) |
where the pressure p(x, y) satisfies Laplace's equation
Note that the gap-averaged velocity
| | 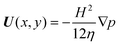 | (13) |
furnishes the boundary condition
| | 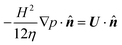 | (14) |
for the pressure. Streamlines of
U are shown in
Fig. 2(b) and (c), respectively, for
symmetric and
asymmetric flow configurations. The flow ratios
s = 3 (left panels) and
s = 0 (right panels), respectively, correspond to the pressure-driven electrolyte and
polymer solution flows that prevail prior to (left panels) and after (right panels) initiating
polymer adsorption onto the trapped micro-sphere.
3.2.3 Electro-osmotic flow.
Electro-osmotic flow is driven by the electrical body force of the electric field E acting on the free charge within diffuse double layers at the charged interfaces. In the bulk fluid, where the free charge vanishes, the fluid velocity u at low Reynolds numbers satisfies the unsteady Stokes equations| | 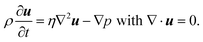 | (15) |
When κH ≫ 1,2 the bulk fluid appears to slip along the walls with velocitywhere Ê = E/|E| and u± = −εε0ζ±η−1|E|. Here, εε0 is the fluid (water) dielectric constant, and ζ± denote apparent wall ζ-potentials at z = ± H/2 (top and bottom).
For example, in a long straight channel with longitudinal oscillatory electric field E = E0eiωtÊ, the fluid velocity and pressure gradient have the forms u = û(z)eiωtÊ and ∇p = ![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif) xeiωtÊ. Furthermore, with blocked ends and equal wall ζ-potentials (u+ = u− = uEO), the pressure gradient is specified by demanding zero net flow, giving
xeiωtÊ. Furthermore, with blocked ends and equal wall ζ-potentials (u+ = u− = uEO), the pressure gradient is specified by demanding zero net flow, giving
| | 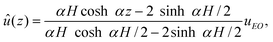 | (17) |
where
α2 =
iωρ/
η is the squared reciprocal viscous diffusion length. Note that a blocked channel with steady EOF has the familiar parabolic velocity profile
| | 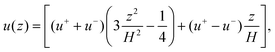 | (18) |
which has points of zero EOF at
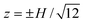
when
u+ =
u− =
uEO. Note that
eqn (18) is also a good quasi-steady approximation of
eqn (17), since deviations |
u(
z)| − |
û(
z)| are at most a few percent of
uEO at dimensionless frequencies |
αH| ≤ 3.
3.3
Polymer layer hydrodynamic layer thickness
An approximate expression relating the hydrodynamic layer thickness Lh of an uncharged, ion-penetrable polymer layer to the particle electrophoretic mobility is available for κa ≫ 1 with Lh/![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) B ≫ 1, where B is Brinkman screening length of the adsorbed polymer layer. The bare particle ζ-potential ζ0 is related to the apparent polymer-coated particle ζ-potential by tanh[eζ/(4kBT)] = tanh[eζ0/(4kBT)]e−κLh.34,35 Thus, substituting the well known Smoluchowski mobility μE = εε0ζ/η, which is generally accurate when κa≳50, gives
B ≫ 1, where B is Brinkman screening length of the adsorbed polymer layer. The bare particle ζ-potential ζ0 is related to the apparent polymer-coated particle ζ-potential by tanh[eζ/(4kBT)] = tanh[eζ0/(4kBT)]e−κLh.34,35 Thus, substituting the well known Smoluchowski mobility μE = εε0ζ/η, which is generally accurate when κa≳50, gives| | 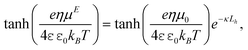 | (19) |
which is consistent with Ohshima's analytical approximations for uniform uncharged layers36 Here, the polymer layer is hydrodynamically impenetrable, i.e., Lh/B ≫ 1, so the layer thickness is interpreted as the hydrodynamic thickness. More accurate solutions of the electrokinetic model for soft colloidal spheres are available for uniform14,36 and nonuniform layers with the influences of polarization and relaxation.11,37 However, for 100 kDa PEO adsorbing on silica micro-spheres, we estimate 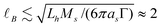 nm with Lh ≈ 10 nm, mass coverage Γ ≈ 0.65 mg m−2,38 and PEO statistical segment molecular mass Ms ≈ 71 g mol−1 and Stokes radius as = 0.175 Å.39
nm with Lh ≈ 10 nm, mass coverage Γ ≈ 0.65 mg m−2,38 and PEO statistical segment molecular mass Ms ≈ 71 g mol−1 and Stokes radius as = 0.175 Å.39
4. Results and discussion
4.1
OTE with bare and PEO-coated silica micro-spheres and glass micro-channel walls
A representative OTE voltage power spectrum is shown in Fig. 3(a) for a silica micro-sphere prior to polymer adsorption. The height of the delta peak at fE = 50 Hz decreases with polymer adsorption. For example, after ∼20 min exposure to a 100 kDa PEO solution with concentration c ≈ 50 ppm, the electrophoretic mobility decreased from μE ≈ 3.05 to 1.36 μm cm V−1s−1. A Lorentzian fit to the power spectra at frequencies in the range 40–800 Hz furnished trap corner frequencies fc ≈ 278 ± 6 and 280 ± 6 Hz, and voltage-position sensitivity β ≈ 503 ± 9 and 498 ± 9 nm V−1 for the bare and PEO-coated particles, respectively. The upper frequency limit is at least twice the trap corner frequency, and ∼5 times lower than the ≈ 4 kHz parasitic photo-diode low-pass-filter corner frequency. PEO adsorption has a negligible influence on the passive particle fluctuations, as evidenced by Lorentzian fits, which indicate a change in particle size that is at most a few percent of the nominal particle diameter. Accordingly, calibration requires the acquisition of one power spectrum before or after each adsorption experiment.
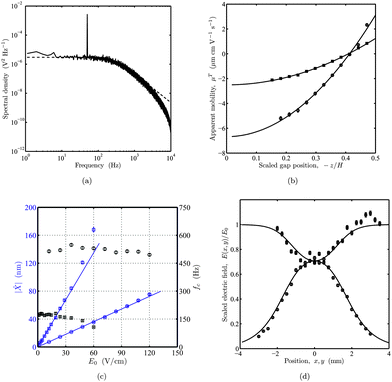 |
| | Fig. 3 Instrumental aspects of optical tweezers electrophoresis. (a) Representative power spectrum of the voltage fluctuations from an optically trapped silica micro-sphere (prior to PEO adsorption). The oscillatory electric field produces the delta peak at frequency fE = 50 Hz with an amplitude that is proportional to the square root of the apparent electrophoretic mobility. (b) Apparent mobility μT of a bare (circles) silica micro-sphere, and another silica micro-sphere after exposure to a 100 kDa PEO solution with concentration c ≈ 50 ppm (squares) versus scaled channel gap position – z/H. Mobilities and accompanying errors bars are from eqn (3) and (7), respectively. Lines are least-squares fits of eqn (18) with the quasi-steady EOF mobility û/E0 from eqn (3). The apparent mobility equals the intrinsic mobility μE at z/H ≈ ± 0.289. See the main text for details. (c) Electric-field-induced particle displacement amplitude || (blue) and trap corner frequency fc (black) at fE = 50 Hz versus electric-field amplitude E0. Lines are linear weighted least-squares fits. Nominal laser powers are ∼40 mW (squares) and ∼120 mW (circles). (d) Scaled electric field E(x, y)/E0versus positions x and y along the central axes of a crossed parallel-plate micro-channel with channel width W = 3 mm (width aspect ratio r = 1). Symbols are experimental data obtained by scaling the apparent mobility with E0 = V/L as the only fitting parameter. Circles and squares correspond to x = 0 and y = 0, respectively, with the electric field directed along the x-axis. Lines are from an analytical solution of Laplace's equation. | |
To establish the degree to which polymer adsorbs to the particle and micro-channels walls, we obtained profiles of the apparent particle mobility across the gap of a closed parallel-plate micro-channel. These experiments adopted an electric field that drives quasi-steady electro-osmotic flow at a frequency fE = 30 Hz. Apparent electrophoretic mobilities of micro-spheres before and after the introduction of a 100 kDa PEO solution with concentration c ≈ 50 ppm are shown in Fig. 3(b) as a function of the channel gap position. Least-squares fits of eqn (18) to the profiles furnish intrinsic particle mobilities μE ≈ − 3.41 and −1.26 μm cm V−1s−1, and wall-slip mobilities u±/E0 ≈ 6.53 and 2.49 μm cm V−1s−1. Note that the negative particle mobilities and positive wall-slip velocities both correspond to surfaces bearing negative charge. Unfortunately, the profiles with and without adsorbed polymer could not be obtained for the same silica micro-sphere. Nevertheless, assuming the two particles have comparable surface charge densities, it is clear that polymer adsorbs to the particle and channel walls, decreasing the magnitude of the respective ζ-potentials by factors of 2.7 and 2.6. Accordingly, eqn (19) infers respective polymer layer thicknesses Lh ≈ 9.0 and 7.6 nm on the colloidal silica and glass.
4.2 Instrumental aspects of OTE in parallel-plate channels
The temporal resolution of our instrument is limited by the amplitude and frequency of the electric-field-induced particle displacements. Under quasi-steady conditions, particle displacement amplitude || in the absence of an optical trap is O(μEE0/fE), suggesting that increasing fE demands an accompanying increase in E0. However, displacements || ≲ 100 nm must be maintained to guarantee position detection and an optical trapping force that are linear with respect to the particle displacement. To establish instrumental limitations and sensitivity, || (blue symbols) and fc (black symbols) are plotted as a function of E0 in Fig. 3(c) at two laser powers. Note that || is obtained from power spectra according to eqn (5) using P(fE) ≈ β2||2ts/2, and error bars are from eqn (7) with σ/ ≈ σμT/μT. The data with low laser power (40 mW, squares) indicate electric-field-induced excursions ≳100 nm from the trap center when E0≳50 V cm−1. At such large displacements, the weaker optical restoring force (evidenced by the diminishing corner frequency fc) yields displacements that have a positive deviation from the low-amplitude linear response. At a higher laser power (120 mW, circles), the micro-spheres experience small (≲ 100 nm) displacements at electric field strengths up to at least 120 V cm−1. Note also that the approximately constant values of fc attest to the linear dependence of the optical trapping force with respect to electric-field-induced excursions ||.
Quantitative determination of the absolute electrophoretic mobility requires accurate knowledge of the undisturbed electric field at the particle position. As demonstrated in Fig. 2(a), flaring of the electric field lines at the intersection of a crossed channel yields an electric field that is significantly lower than the value E0 = V/L obtained by dividing the applied voltage V by the electrode separation L. We measured the electrophoretic mobility at various positions traversing the channel intersection, in the directions x parallel and y perpendicular to the applied electric field. Since the apparent mobility μT is independent of x and y, spatial variations in the particle displacements reflect variations in the electric field strength E(x, y) when mobilities are measured at a constant gap position z. The scaled electric field E(x, y)/E0 along the central axes of a crossed channel with either x = 0 or y = 0 is shown in Fig. 3(d). Here, the principal electric field is directed along the x-axis with the particle trapped at a constant gap position 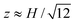 . The electric field strength is inferred from the apparent electrophoretic mobility of a micro-sphere at fE = 30 Hz. Again, error bars are from eqn (7). The lines in Fig. 3(d) are an analytical solution of Laplaces's equation obtained from a Schwarz-Christoffel transformation for a crossed channel, with width aspect ratio r = 1 [see Fig. 2(a)], furnishing
. The electric field strength is inferred from the apparent electrophoretic mobility of a micro-sphere at fE = 30 Hz. Again, error bars are from eqn (7). The lines in Fig. 3(d) are an analytical solution of Laplaces's equation obtained from a Schwarz-Christoffel transformation for a crossed channel, with width aspect ratio r = 1 [see Fig. 2(a)], furnishing 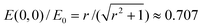 . Since || ∝ E(x,y), we adopted E0 = V/L as the single fitting parameter to scale the experimental data. This clearly provides an excellent correspondence between theory and experiment and, accordingly, we adopted an electric-field strength E(0, 0) ≈ 0.707V/L to compute all subsequent particle electrophoretic mobilities obtained with this channel geometry.
. Since || ∝ E(x,y), we adopted E0 = V/L as the single fitting parameter to scale the experimental data. This clearly provides an excellent correspondence between theory and experiment and, accordingly, we adopted an electric-field strength E(0, 0) ≈ 0.707V/L to compute all subsequent particle electrophoretic mobilities obtained with this channel geometry.
4.3 EOF in closed parallel-plate channels with discontinuous wall-slip velocity
Polymer adsorption onto the micro-channel walls produces spatially varying wall ζ-potentials and EOF that modulate the apparent particle electrophoretic mobility. To help understand these influences, we consider two simple models for straight channels that each have a stepwise discontinuity in the wall ζ-potential. In the first model, there is a transverse discontinuity, whereas in the second there is a longitudinal discontinuity. In both models, the channel co-ordinates (x, y, z) span the rectangular domain − L/2 ≤ x ≤ L/2 (length), − W/2 ≤ y ≤ W/2 (width), and − H/2 ≤ z ≤ H/2 (height), where L ≫ W ≫ H, and the uniform electric field E is directed along the x-axis.
In the experiments, the height H ≈ 140 μm is much larger than the Debye-length κ−1 ≈ 10 nm, so we adopt the foregoing (Helmholtz-Smoluchowski) wall-slip boundary conditions. With forcing frequency fE = 50 Hz, frequency-dependent EOF profiles from eqn (17) are practically the same as the steady parabolic profiles from eqn (18), which, in turn, were demonstrated in Fig. 3(b) to be in good agreement with experiments. Thus, the following theoretical calculations are for quasi-steady EOF, with the bulk EOF velocity having cartesian components u = (u,v,w).
4.3.1 Jump discontinuity in wall-slip velocity across channel width.
We consider a wall-slip velocity discontinuity at a (transverse) position y = y0 across a straight channel with width W. This mimics the quasi-steady migration of a polymer adsorption front across the channel when the polymer adsorption time τa is much shorter than the characteristic migration time τm ∼ W/U ≈ 30 s. This is the case when the polymer concentration c is high enough to yield τa≲ τm with τa ∼ Γ2/(Dc2) for diffusion-limited adsorption, e.g., τa ≈ 2 s when c ≈ 150 ppm. Under these conditions, the accompanying effective wall ζ-potential discontinuity migrates across the channel, with a velocity that reflects the polymer solution flow rate. This situation represents our OTE experiments when c≳100 ppm. Interestingly, when c is low enough to yield τa≳τm, convection limits the diffusion length to 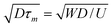 , and, hence, Fick's law and scaling analysis suggest that
, and, hence, Fick's law and scaling analysis suggest that 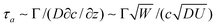 . Thus, at very low polymer concentrations, an initially non-uniformly coated channel is expected to become uniformly coated on a much longer time scale. This situation is representative of our OTE experiments when c≲ 10 ppm. Under these conditions, the position of the wall ζ-potential discontinuity is expected to remain fixed, but with an amplitude that vanishes as the initially uncoated side of the channel attains the same ζ-potential as the initially coated side.
. Thus, at very low polymer concentrations, an initially non-uniformly coated channel is expected to become uniformly coated on a much longer time scale. This situation is representative of our OTE experiments when c≲ 10 ppm. Under these conditions, the position of the wall ζ-potential discontinuity is expected to remain fixed, but with an amplitude that vanishes as the initially uncoated side of the channel attains the same ζ-potential as the initially coated side.
The unidirectional flow in the x-direction satisfies the Stokes equations, which simplify to
| | 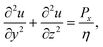 | (20) |
where
u =
u(
y,
z). The wall-slip boundary conditions are
| | 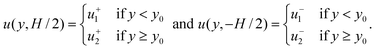 | (21) |
We solved eqn (20) for u = u1 + u2 by superposing the solutions u1 and u2 of two simpler problems. In the first problem, u1 is the electric-field driven flow that satisfies the boundary conditions of eqn (21) with Px = 0, while in the second problem u2 is the well-known pressure-driven flow with no-slip boundary conditions. Superposing u1 and u2 to ensure zero net flow specifies Px ≠ 0, as required for a closed channel.
A Schwarz-Christoffel transformation of the complex upper half plane into an infinite strip gives
| | 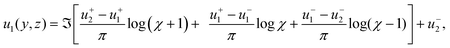 | (22) |
where
| | 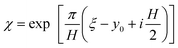 | (23) |
is the conformal map with
ξ =
y +
iz. As expected,
u1 → (
u+1 −
u−1)
z/
H + (
u+1 +
u−1)/2 (simple shear) when
y −
y0≲
H and, similarly,
u1 → (
u+2 −
u−2)
z/
H + (
u+2 +
u−2)/2 when
y –
y0≳
H. Note that the conformal map furnishes an exact solution for
u1 when |
y −
y0|≲
H.
Since
| | 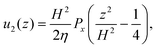 | (24) |
superposition gives
| | 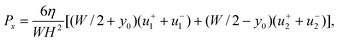 | (25) |
so, finally,
| | 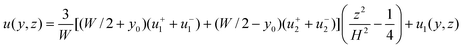 | (26) |
with
u1(
y,
z) given by
eqn (22).
A representative solution with u1+ = u1− = 0 and u2+ = u2− = u0 is examined in Fig. 4. Iso-contours of u(y, z) are plotted in panel (a) with y0 = 0 (channel center line). This is the instantaneous quasi-steady EOF due to one half of the channel (y > y0 = 0) having slip velocity u0 and the other half (y < y0 = 0) having zero slip. As expected, the iso-contours reflect uniform parabolic profiles when |y − y0|≳H. Panel (b) shows velocity profiles u(0, z) (vertical profiles at the channel center) as a function of y0 with W = 10H. The solid line indicates a slowly varying and significant contribution to u at 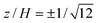 where OTE adsorption experiments are typically performed. Note that increasing y0 mimics a polymer concentration front migrating across the channel, perpendicular to the principal electric field. Accordingly, the channel is bare when y0 = −5H = −W/2, and completely coated when y0 = 5H = W/2. This general behavior is expected in our OTE experiments when adopting the asymmetric flow configuration described in section 2.3.
where OTE adsorption experiments are typically performed. Note that increasing y0 mimics a polymer concentration front migrating across the channel, perpendicular to the principal electric field. Accordingly, the channel is bare when y0 = −5H = −W/2, and completely coated when y0 = 5H = W/2. This general behavior is expected in our OTE experiments when adopting the asymmetric flow configuration described in section 2.3.
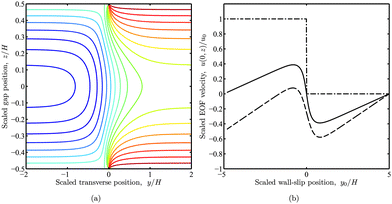 |
| | Fig. 4 Fluid velocity u(y, z) in a straight parallel-plate channel with a transverse wall-slip discontinuity at y = y0 and zero net flow. The wall-slip velocities are u(y, ± H/2) = 0 when y < y0, and u(y, ± H/2) = u0 when y > y0. (a) Iso-contours of u(y, z) with y0 = 0 starting at u = u0 (red) and decreasing in increments of 0.1u0. (b) u(0, z) versus y0 scaled with the channel height H for positions across the gap 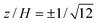 (conventional point of zero EOF, solid), 0 (center, dashed), and ±0.5 (wall, dash-dotted) with W = 10H. (conventional point of zero EOF, solid), 0 (center, dashed), and ±0.5 (wall, dash-dotted) with W = 10H. | |
4.3.2 Jump discontinuity in wall-slip velocity along channel length.
This model integrates a wall-slip velocity discontinuity at a (longitudinal) position x = x0 along a straight channel with length L to mimic the quasi-steady migration of a polymer adsorption front along the length of a straight channel when the polymer adsorption time τa ≪ τm ∼ H/U≈1 s. However, when the PEO concentration c≲ 150 ppm, τa≳τm. In this case, we consider a model where the wall-slip velocity varies linearly with respect to x over a length scale H. In both cases, p = p(x, z) and u = [u(x,z),0,w(x,z)]. At the top and bottom walls, w(x, z = ± H/2) = 0,| | 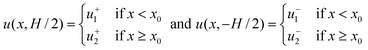 | (27) |
and u → [u(z),0,0] [given by eqn (18)] as x → ±∞. Introducing a stream function ψ(x, z), where ∇ψ = [−w, 0, u], guarantees that ∇·u = 0, and the steady momentum equation becomeswhich we solve using a standard finite-difference methodology.
The bulk fluid velocity with wall-slip velocities u1+ = u1− = 0 and u2+ = u2− = u0 are shown in Fig. 5(a) with x0 = 0. As expected, the flow establishes a parabolic profile when x≳H, and vanishes when x≲ H. Fig. 5(b) shows u(0, z) and w(0, z) versus x0 at several gap positions z. Here, increasing x0 mimics the longitudinal migration of a polymer concentration front parallel to the applied electric field. Interestingly, near the wall-slip velocity discontinuity, w(0, z) is non-zero at 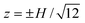 (dotted line). However, at the conventional point of zero EOF (solid lines), Fig. 4(b) and 5(b), respectively, show that the EOF is finite over length-scales comparable to the channel width W and height H. Note that the small non-zero EOF contribution (solid line) in Fig. 5(b) decreases by an order of magnitude when the wall-slip velocity varies linearly over a distance H, as seen in Fig. 5(d). Here, a linearly varying wall-slip velocity produces negligible EOF at the conventional point of zero EOF (solid line), suggesting, again, that OTE experiments with the symmetric flow configuration are less susceptible to the adverse influences of polymer adsorption onto the channel walls.
(dotted line). However, at the conventional point of zero EOF (solid lines), Fig. 4(b) and 5(b), respectively, show that the EOF is finite over length-scales comparable to the channel width W and height H. Note that the small non-zero EOF contribution (solid line) in Fig. 5(b) decreases by an order of magnitude when the wall-slip velocity varies linearly over a distance H, as seen in Fig. 5(d). Here, a linearly varying wall-slip velocity produces negligible EOF at the conventional point of zero EOF (solid line), suggesting, again, that OTE experiments with the symmetric flow configuration are less susceptible to the adverse influences of polymer adsorption onto the channel walls.
![Fluid velocity vectors (left panels) in straight parallel-plate channels with zero net flow and non-uniform wall-slip velocity with x0 = 0. Velocity components (right panels) u(0, z)/u0versus wall-slip position x0/H ≠ 0 for channel gap positions (solid), 0 (dashed), and ± 0.5 (dash-dotted). The dotted line is , i.e., the vertical component of the velocity at the conventional position of zero EOF. (a) and (b) have discontinuous wall-slip velocity: u(x, ± H/2) = 0 when x < x0, and u(x, ± H/2) = u0 when x > x0. (c) and (d) have a linearly varying wall-slip velocity: u(x, ± H/2) = 0 when x < x0 − H/2, u(x, ± H/2) = u0 when x > x0 + H/2, and u(x, ± H/2) = [(x − x0)/H + 1/2]u0 when x0 − H/2 < x < x0 + H/2.](/image/article/2011/LC/c005217p/c005217p-f5.gif) |
| | Fig. 5 Fluid velocity vectors (left panels) in straight parallel-plate channels with zero net flow and non-uniform wall-slip velocity with x0 = 0. Velocity components (right panels) u(0, z)/u0versus wall-slip position x0/H ≠ 0 for channel gap positions 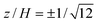 (solid), 0 (dashed), and ± 0.5 (dash-dotted). The dotted line is (solid), 0 (dashed), and ± 0.5 (dash-dotted). The dotted line is 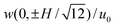 , i.e., the vertical component of the velocity at the conventional position of zero EOF. (a) and (b) have discontinuous wall-slip velocity: u(x, ± H/2) = 0 when x < x0, and u(x, ± H/2) = u0 when x > x0. (c) and (d) have a linearly varying wall-slip velocity: u(x, ± H/2) = 0 when x < x0 − H/2, u(x, ± H/2) = u0 when x > x0 + H/2, and u(x, ± H/2) = [(x − x0)/H + 1/2]u0 when x0 − H/2 < x < x0 + H/2. , i.e., the vertical component of the velocity at the conventional position of zero EOF. (a) and (b) have discontinuous wall-slip velocity: u(x, ± H/2) = 0 when x < x0, and u(x, ± H/2) = u0 when x > x0. (c) and (d) have a linearly varying wall-slip velocity: u(x, ± H/2) = 0 when x < x0 − H/2, u(x, ± H/2) = u0 when x > x0 + H/2, and u(x, ± H/2) = [(x − x0)/H + 1/2]u0 when x0 − H/2 < x < x0 + H/2. | |
Apparent electrophoretic mobility μT time-series for bare micro-spheres abruptly exposed to 100 kDa PEO solutions with concentrations c ≈ 2–150 ppm are shown in Fig. 6(a). These OTE experiments were conducted with a micro-sphere trapped at the center of a crossed parallel-plate micro-channel, at the conventional point of zero EOF. The left and right panels compare, respectively, similar experiments with the asymmetric and symmetric flow configurations. The most striking difference is that the asymmetric configuration yields an ostensible, non-monotonic change in the mobility that is characterized by an initial “hump” at the two highest polymer concentrations c ≈ 50 and 150 ppm. As explored in previous sections, PEO adsorption reduces the wall-slip velocity and particle electrophoretic mobility. The theoretical calculations in section 4.3 revealed that a migrating polymer solution front produces a spatially varying wall-slip velocity that has a much weaker influence on the electro-osmotic flow velocity at the conventional point of zero EOF when the front migrates (along the electric field) in a manner that is representative of the symmetric flow configuration. For example, μT in the left panel when c ≈ 50 (blue) and 150 ppm (black) resembles the superposition of the corresponding time series on the right and the model EOF profile in Fig. 4(b) (solid line). Note also that the asymmetric flow configuration (left panel) of Fig. 6 yields bare particle apparent mobilities |μT0| < |μE0|. This is expected when the micro-sphere and channel walls are both negatively or positively charged, and less than half of the channel is polymer coated, i.e., y0 < 0, and is verified by the numerical calculations presented in Fig. 4(b) (solid line). Here, the EOF velocity 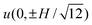 is positive when y0 < 0, thus adding a (diminishing) contribution [by eqn (3)] to the apparent mobility of a negatively charged micro-sphere.
is positive when y0 < 0, thus adding a (diminishing) contribution [by eqn (3)] to the apparent mobility of a negatively charged micro-sphere.
 |
| | Fig. 6 Representative electrophoretic mobility time series from OTE. (a) Apparent mobility μTversus time t for bare silica micro-spheres abruptly exposed to 100 kDa PEO solutions at t = 0 s with concentrations c ≈ 2 (magenta), 6 (green), 16 (red), 50 (blue) and 150 ppm (black). PEO solutions flow perpendicular (left) and parallel (right) to the electric field with flow rate Q1 ≈ 4 μL min−1. (b) Fractional decrease in (apparent) mobility 1 − μT/μT0versus time t for bare micro-spheres with (apparent) mobility μT0 when t < 0 s and PEO pre-adsorption onto the channel walls. See the main text for details. At t = 0 s, each particle is abruptly exposed to a 100 kDa PEO solution with concentration c ≈ 150 ppm flowing perpendicular to the electric field. In the left panel, Q1 ≈ 1 (green), 2 (red), 4 (blue), and 8 μL min−1 (black). The right panel compares data with (black) and without (red) polymer pre-adsorption onto the channel walls: Q1 ≈ 4 μL min−1. | |
The left panel of Fig. 6(b) compares mobility time series—now plotted as the fractional decrease in mobility 1 − μT/μT0—with c ≈ 150 ppm for several PEO solution flow rates Q1 = 1–8 μL min−1. As expected, increasing the polymer solution flow rate in this asymmetric flow configuration increases the initial anomalous dynamics, i.e., the breadth of the initial “hump” decreases with increasing flow rate. For these experiments, we conducted pre-adsorption to partially mitigate anomalous EOF effects due to non-uniform polymer adsorption onto the channel walls. Here, before trapping a micro-sphere, we flowed only polymer solution through the channel for ∼30 min to fully coat the channel walls. Next, we turned on the electrolyte flow (containing micro-spheres) and trapped a bare particle ∼10 min later, just before initiating a standard polymer adsorption experiment. The right panel of Fig. 6(b) compares time series with (black) and without (red) pre-adsorption. Here, the initial “hump”. These clearly demonstrate the advantage of irreversibly coating the channel walls to maintain a uniform and constant effective wall ζ-potential.
4.5 Poly(ethylene oxide) layer thickness development on silica micro-spheres
The apparent mobility time series in Fig. 6(a) are replotted in Fig. 7 as the fractional decrease in electrophoretic mobility. Recall, an initially bare silica micro-sphere is abruptly exposed to 100 kDa PEO from solutions with concentrations c ≈ 2–150 ppm in the asymmetric (a) and symmetric (b) flow configurations. As polymer adsorbs to the particles and, indeed, the channel walls, the mobility varies rapidly within the first ten seconds, with an approximately exponential relaxation to equilibrium occurring on a time scale of several minutes.
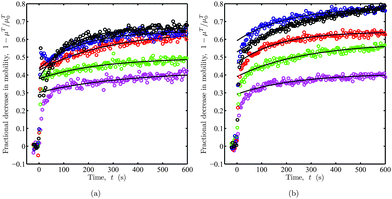 |
| | Fig. 7
OTE fractional decrease in (apparent) electrophoretic mobility 1 – μT/μT0. Replotted from raw mobility data in Fig. 6(a) with asymmetric (left) and symmetric (right) flow configurations. Lines are least-squares fits of eqn (29) for t≳50 s. The fitting parameters are listed in Table 1. | |
Experiments with mono-dispersed PEO solutions (to be reported elsewhere), suggest that the latter time scale is due to thermodynamic exchange of small adsorbed polymer coils for larger ones. We fitted the first-order relaxation model
| | | μT = μT∞(1 + A0e−t/τ) | (29) |
to the mobility time series with
t≳50 s to furnish a first-order time constant
τ and equilibrium mobility
μT∞. These parameters and the accompanying equilibrium layer thicknesses
Lh from
eqn (19) are listed in
Table 1. For PEO concentrations
c≳16 ppm,
Lh ∼ 10 nm is comparable to the thicknesses obtained by others from dynamic light scattering with
silica nano-particles,
40–42 and streaming potentials with
quartz capillaries.
7
|
c (ppm) |
Flow configuration |
μ
T
0 (μm cm V−1s−1) |
μ
T
∞ (μm cm V−1s−1) |
τ (s) |
L
h
(nm) |
| 150 |
asymmetric |
3.48 |
1.17 |
117 |
10.3 |
| 150 |
symmetric |
3.80 |
0.72 |
299 |
15.9 |
| 50 |
asymmetric |
3.27 |
1.16 |
164 |
9.9 |
| 50 |
symmetric |
3.49 |
0.76 |
203 |
14.6 |
| 16 |
asymmetric |
2.87 |
1.01 |
288 |
10.0 |
| 16 |
symmetric |
2.98 |
1.06 |
167 |
9.9 |
| 6 |
asymmetric |
3.22 |
1.62 |
249 |
6.4 |
| 6 |
symmetric |
3.13 |
1.31 |
277 |
8.3 |
| 2 |
asymmetric |
3.12 |
1.80 |
351 |
5.1 |
| 2 |
symmetric |
3.54 |
2.10 |
236 |
4.8 |
Our OTE instrument also reveals information concerning the kinetics of polymer layer development at short times, i.e., within the first several seconds. When c≳16 ppm, layer development appears to be limited by polymer conformation dynamics. With c≲16 ppm, the growth rate is proportional to c, as expected when the adsorption is limited by bulk polymer diffusion. More quantitative kinetic data can be obtained by fitting a two-time scale model with fast τ1 and slow τ2 exponential relaxation time constants. Nevertheless, we find that τ2 ≈ τ, where τ (listed in Table 1) is the time constant from fitting eqn (29) to the data when t≳50 s. Note that, with the asymmetric flow configuration, τ generally decreases with increasing polymer concentration c, but remains fairly constant with the symmetric configuration. For the asymmetric flows, τ is perturbed by wall-adsorption dynamics and anomalous EOF. For example with c ≈ 2 ppm, the wall-adsorption time τa ∼ 500 s effectively increases τ.
The equilibrium layer thickness is often lower with the asymmetric flow configuration (flow perpendicular to the electric field). This reflects the channel being partially coated in the asymmetric flow configuration before adsorption onto the micro-sphere with |μT0|≲ |μE0|. For symmetric flows, τ seem to reflects a single, slow relaxation time corresponding to conformational changes in the layer structure and, possibly, thermodynamic exchange due to polydispersity. Furthermore, the amplitude of this relaxation also increases with polymer concentration. Thus, when the initially bare surface is rapidly coated with low molecular weight polymer, there may ultimately be a higher fraction of the higher molecular weight chains that end up as loops and tails, thereby increasing the final hydrodynamic layer thickness.
The distinct increase in layer thickness with the bulk polymer concentration, and evidence of a plateau at high polymer concentrations (c≳16 ppm) might be due to thermodynamic exchange or irreversible conformational dynamics. For example, with adsorption from low concentration solutions, thin layers might prevail due to the longer diffusion-limited, short-time dynamics providing more time for chains to organize compactly on the surface, e.g., with a higher portion of the chains forming trains. On the other hand, this might reflect the statistical thermodynamics of adsorption isotherms with saturation, where the chemical potential of the adsorbed flexible chains varies with the chemical potential/concentration of the free chains in solution. Evidently, flow rate, polymer concentration, and polydispersity all impact the layer kinetics and ‘equilibrium’ structure. Such details are beyond the scope of this work, but will be explored in future studies to be undertaken with the OTE instrument.
5. Summary
We adopted optical tweezers electrophoresis (OTE) to measure the apparent electrophoretic mobilities of silica micro-spheres in a crossed parallel-plate micro-channel during the adsorption of an industrial grade 100 kDa poly(ethylene oxide). The experiments were conducted with low-frequency (50 kHz) oscillatory electric fields, furnishing time series with ∼1 s resolution. Equilibrium layer thicknesses were ∼10 nm with bulk PEO concentrations ≳16 ppm (weight percent).
This study highlights the importance of understanding electro-kinetic phenomena at the micro-channel walls for correctly interpreting dynamics of the intrinsic micro-particle mobility and accompanying polymer adsorption. We presented the proof of concept for a novel diagnostic tool that can probe polymer dynamics on the micrometer length scale. Such a small length scale furnishes kinetic data reflecting the dynamics of developing polymer layers that are much more laterally uniform at short times than can be achieved with millimeter-scaled geometries, such as in capillary streaming potential or quartz-crystal micro-balance instruments.
Simple EOF models were advanced to support our hypothesis that anomalous micro-channel EOF prevails when introducing adsorbing polymer solution. Transient adsorption onto the channel walls adversely influences EOF when the polymer solution is introduced into a channel perpendicular to the electric field (asymmetric flow configuration). However, when the polymer solution is introduced in a symmetric manner, i.e., flowing parallel to the electric field, our model indicates that anomalous EOF influences are significantly reduced, meaning that that EOF velocity at the conventional point of zero EOF is small compared to the intrinsic particle electrophoretic velocity.
These differences are evident when comparing the qualitative forms of the short-time electrophoretic mobility time series. Interestingly, too, the flow configuration also impacts the long-time relaxation and equilibrium electrophoretic mobility. Our investigations suggest that a symmetric flow configuration is preferable for achieving an electrophoretic mobility time series that reflects—as closely as possible—the intrinsic mobility of the micro-sphere during abrupt exposure to a quasi-step change in polymer solution concentration.
6Acknowledgements
R.J.H. gratefully acknowledges support from the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chairs program; and J.A.vH. thanks the Department of Chemical Engineering, McGill University for generous financial support through a Eugene Ulmer Lamothe graduate scholarship.
References
- J. A. van Heiningen, A. Mohammadi and R. J. Hill, Lab Chip, 2010, 10, 1907–1921 RSC.
-
R. J. Hunter, Foundations of Colloid and Interface Science, Oxford University Press, 2nd edn, 2001 Search PubMed.
-
T. G. M. van de Ven, Colloidal Hydrodynamics, Academic Press, 1989 Search PubMed.
-
G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman and Hall, 1988 Search PubMed.
- L. Couture and T. G. M. van de Ven, Colloids Surf., 1990, 54, 245–260.
- J. A. De Witt and T. G. M. van de Ven, Langmuir, 1992, 8, 788–793 CrossRef CAS.
- N. V. Churaev, I. Sergeeva, V. D. Sobolev, Z. M. Zorin and E. K. Gasanov, Colloids Surf., A, 1993, 76, 23–32 CrossRef CAS.
- J. C. Dijt, M. A. Cohen Stuart and G. J. Fleer, Macromolecules, 1992, 25, 5416–5423 CrossRef CAS.
- M. Rodahl, F. Höök, C. Fredriksson, C. A. Keller, A. Krozer, P. Brzezinski, M. Voinova and B. Kasemo, Faraday Discuss., 1997, 107, 229–246 RSC.
- D. Johannsmann, K. Mathauer, G. Wegner and W. Knoll, Phys. Rev. B: Condens. Matter, 1992, 46, 7808–7815 CrossRef.
- R. J. Hill and D. A. Saville, Colloids Surf., A, 2005, 267, 31–49 CrossRef CAS.
- M. A. Cohen Stuart and H. Tamai, Langmuir, 1988, 4, 1184–1188 CrossRef CAS.
- E. G. M. Pelssers, M. A. Cohen Stuart and G. J. Fleer, J. Chem. Soc., Faraday Trans., 1990, 86, 1355–1361 RSC.
- H. Ohshima, J. Colloid Interface Sci., 2002, 252, 119–125 CrossRef CAS.
- M. Ichiyanagi, S. Sasaki, Y. Sato and H.K., J. Micromech. Microeng., 2009, 19, 045021 CrossRef.
- A. E. Herr, J. I. Molho, J. G. Santiago, M. G. Mungal and T. W. Kenny, Anal. Chem., 2000, 72, 1053–1057 CrossRef CAS.
- J. L. Lin, K. H. Lee and G. B. Lee, Electrophoresis, 2005, 26, 4605–4615 CrossRef CAS.
- C. C. Chang and R. J. Yang, Phys. Fluids, 2009, 21, 052004 Search PubMed.
- J. K. Towns and F. E. Regnier, Anal. Chem., 1992, 64, 2473–2478 CrossRef CAS.
- S. Ghosal, Anal. Chem., 2002, 74, 771–775 CrossRef CAS.
- K. C. Neuman and S. M. Block, Rev. Sci. Instrum., 2004, 75, 2787–2809 CrossRef CAS.
- V. Kahl, A. Gansen, R. Galneder and J. O. Rädler, Rev. Sci. Instrum., 2009, 80, 073704 CrossRef CAS.
- R. Galneder, V. Kahl, A. Arbuzova, M. Rebecchi, J. O. Rädler and S. McLaughlin, Biophys. J., 2001, 80, 2298–2309 CrossRef CAS.
- N. Garbow, M. Evers and T. Palberg, Colloids Surf., A, 2001, 195, 227–241 CrossRef CAS.
- I. Semenov, O. Otto, G. Stober, P. Papadopoulos, U. F. Keyser and F. Kremer, J. Colloid Interface Sci., 2009, 337, 260–264 CrossRef CAS.
- O. Otto, C. Gutsche, F. Kremer and U. F. Keyser, Rev. Sci. Instrum., 2008, 79, 023710 CrossRef.
- G. S. Roberts, T. A. Wood, W. J. Frith and P. Bartlett, J. Chem. Phys., 2007, 126, 194503 CrossRef.
- F. Strubbe, F. Beunis and K. Neyts, J. Colloid Interface Sci., 2006, 301, 302–309 CrossRef CAS.
- F. Strubbe, F. Beunis and K. Neyts, Phys. Rev. Lett., 2008, 100, 218301 CrossRef.
- K. Berg-Sørensen and H. Flyvbjerg, Rev. Sci. Instrum., 2004, 75, 594–612 CrossRef CAS.
- D. Bedeaux and P. Mazur, Physica, 1974, 76, 247–258 CrossRef.
- S. F. Tolić-Nørrelykke, E. Schäffer, J. Howard, F. S. Pavone, F. Jülicher and H. Flyvbjerg, Rev. Sci. Instrum., 2006, 77, 103101 CrossRef.
- R. J. Hill, D. A. Saville and W. B. Russel, J. Colloid Interface Sci., 2003, 268, 230–245 CrossRef CAS.
- G. J. Fleer, L. K. Koopal and J. Lyklema, Colloid. Polym. Sci., 1972, 250, 689–702 CrossRef CAS.
- M. A. Cohen Stuart and J. W. Mulder, Colloids Surf., 1985, 15, 49–55 CrossRef.
- H. Ohshima, Adv. Colloid Interface Sci., 1995, 62, 189–235 CrossRef CAS.
- R. J. Hill, D. A. Saville and W. B. Russel, J. Colloid Interface Sci., 2003, 258, 56–74 CrossRef CAS.
- J. C. Dijt, M. A. Cohen Stuart, J. E. Hofman and G. J. Fleer, Colloids Surf., 1990, 51, 141–158 CrossRef CAS.
- R. J. Hill, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 051406 CrossRef.
- E. Killmann and P. Sapuntzijs, Colloids Surf., A, 1994, 86, 229–238 CrossRef CAS.
- B. Wind and E. Killmann, Colloid Polym. Sci., 1998, 276, 903–912 CrossRef CAS.
- G. P. Van der Beek and M. A. Cohen Stuart, J. Phys., 1988, 49, 1449–1454 CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.