Ion mobility mass spectrometry: an elegant alternative focusing on speciation studies†
Gustavo de
Souza Pessôa
ab,
Eduardo Jorge
Pilau
bc,
Fábio Cesar
Gozzo
bc and
Marco Aurelio
Zezzi Arruda
*ab
aGroup of Spectrometry, Sample Preparation and Mechanization (GEPAM), Institute of Chemistry, University of Campinas, UNICAMP, PO Box 6154. 13084-862, Campinas, SP, Brazil. E-mail: zezzi@iqm.unicamp.br
bNational Institute of Science and Technology for Bioanalytics, Institute of Chemistry, University of Campinas, 13084-862, Campinas, SP, Brazil
cDalton Mass Spectrometry Group, Institute of Chemistry, University of Campinas, UNICAMP, PO Box 615413084-862, Campinas, SP, Brazil
First published on 1st December 2010
Abstract
This work is proposed to demonstrate the Traveling-Wave Ion Mobility Specrometry (TWIMS) coupled to Mass Spectrometry (MS) as an alternative technique for speciation analysis between metals/metalloids and biomolecules. Mobilities of bovine carbonic anhydrase bound to Ba2+, Cu2+, Pb2+, Zn2+, Cr3+, Cr6+, Se4+ and Se6+ were estimated. The metal belonging to the bovine carbonic anhydrase structure, commonly found in the commercially available enzyme, was removed by filtration, using centrifugal filter devices. Then, some metals/metalloids were added to 10.0 mmol L−1 ammonium acetate at pH = 6.8 enzyme solution. Experiments were carried out by direct insertion of the sample at 10 μL min−1 flow rate into the ESI source of the instrument. Carbonic anhydrase mobility varied according to the metal bound in its structure, following the order: Zn2+ < Cu2+ < Ba2+ < Pb2+. Metals with higher affinity by the enzyme, such as Zn2+ and Cu2+ had lower mobility, suggesting a higher structural modification, binding itself to the enzyme metallic site. Considering metals with different oxidation states, the enzyme mobility followed the order: Se4+ < Cr6+ < Se6+ < Cr3+.
Introduction
Although necessary, data related to the total concentration of a given analyte are not enough to provide an accurate understanding about its behavior relating to a studied system.1,2 This fact put in evidence the importance of studies/applications involving chemical speciation.3 Inorganic speciation is associated with different oxidation states or isotopic rates,4,5 organic speciation allows to distinguish isomeric ions or proteins isoforms.6–9 Numerous papers are published on metalloid species or organo-metal species in biological fluids.1,2,10–12 In this context, organic, metallo-organic, biomolecules and metal-biomolecules should be considered in speciation analysis.13From this point of view, chemical speciation presents nowadays a transdisciplinar status, creating a perfect atmosphere for involving different expertise in the same work. This enables studies relating to neurological disorders and cardiovascular diseases, which are present in the literature and refer to different forms of the analytes and chemical speciation.10,11 Since the target, in terms of analytes, has been changing, new analytical strategies/instrumentation for carrying out speciation studies need to be explored. This way, hyphenated techniques attaining this task are currently present in the literature, being the coupling between chromatography (i.e. HPLC) and inorganic mass spectrometry (i.e. ICP-MS), one of the most common in terms of hyphenation.4,12
As organic mass spectrometry is capable of providing accurate information related to organo-metallic compounds, it is supposed that it can be successfully applied to speciation studies involving both inorganic and organic species. However, applications in such context are not frequently seen in the literature.14,15 Travelling wave ion mobility spectrometry coupled to mass spectrometry (TWIMS-MS) can be an elegant alternative as a means of increasing selectivity in elemental speciation, at atmospheric pressure, without chromatographic separation.16,17 This technique has been used for protein conformation evaluation in different conditions, being metallothioneins,18 hemoglobin,19 cytochrome c,19 calmodulin,19,20 ubiquitin21 and lysozyme,21 exemplified. Additionally, an electrospray ionization source is capable of producing intact gas-phase ions from non-covalent complexes directly from solution. Thus, direct elemental speciation is more easily attained through soft ionization techniques such as ESI-TWIMS-MS, something that could be more difficult when considering other ionization sources.22
Ion mobility allows the separation of protein bound to a variety of metal ions, being able to separate similar species, to which the differences refer to different oxidation states of the same metal only. The use of TWIMS-MS in speciation analysis of metals associated with biomolecules should be emphasized and encouraged. This recent technique has been presented as a powerful tool, allowing differentiation of ions by shape and size, besides mass and charge.8,9 Modified peptides such as phosphorylated ones can be easily detected and analyzed,23,24 and compounds presenting the same m/z ratio but with different cross-sections can be separated inside the instrument.6,7 Therefore, this potential provides an efficient method for analyzing modified peptides or proteins with same sequences but with modifications in different residues. TWIMS-MS have been applied to assist data analysis obtained in protein cross-linking experiments, more specifically, isomeric dead-end species formed from peptides containing two possible cross-linker reactive sites submitted to ion mobility separation.25
Since ion mobility has an unexplored potential for speciation analysis when dealing with biomolecules, especially taking into account the metal incorporation evaluation, the main goal of this work was to apply this technique to speciation studies, using the bovine carbonic anhydrase (BCA) as proof-of-concept. The ion mobility of Ba, Pb, Zn and Cu in their divalent forms at different concentrations bound to this enzyme were investigated, this technique was also used to explore inorganic speciation of Se4+/Se6+ and Cr3+/Cr6+ in BCA.
Instrumental
Reagents and solutions
All reagents were purchased from Sigma®, including bovine carbonic anhydrase. Ba2+, Pb2+, Zn2+ and Cu2+ solutions were prepared by dilution of their acetate salts in water. Used solutions (85.0 μmol L−1) were prepared by appropriate dilutions from the stock solution. Se4+, Se6+, Cr3+ and Cr6+ solutions were prepared by diluting sodium selenite, sodium selenate, chromium(III) chloride and potassium dichromate in water. Used solutions (85.0 μmol L−1) were prepared in water by appropriate dilutions from the stock solution.Preparation of apo-carbonic anhydrase
Previous studies have shown that commercial BCA contains zinc in its structure, which could interfere with the goal of this study.26 Therefore, zinc removal was carried out before the MS analysis. The chelating agent ortho-phenanthroline (OP), was used as previously reported.27 A solution of OP was added to 11 μmol L−1 of BCA to a final concentration of 1 mmol L−1, both diluted in 50.0 mmol L−1 acetic acetate buffer, pH = 5.0, being this solution kept at 4 °C for 120 h. Then, the buffer was changed to 10.0 mmol L−1 ammonium acetate using centrifugal filter devices (10000 Da molecular weight cutoff, Microcon and Amicon Ultra; Millipore Corporation)28 to maintain ideal conditions in terms of pH for ionization and metal binding. The final protein concentration for measurements was 8.5 μmol L−1 in 10.0 mmol L−1 ammonium acetate, pH = 6.8.Mass spectrometry and TWIMS-MS analysis
Carbonic anhydrase samples were prepared in ammonium acetate buffer (10.0 mmol L−1, pH 6.8) and submitted to MS analysis. TWIMS-MS analysis were performed on a Waters Synapt HDMS (Waters Co., Manchester, UK) instrument. Experiments were carried out by direct infusion of sample at a 10 μL min−1 flow rate into the ESI source of the instrument, with capillary and cone voltages fixed at 3.0 kV and 20 V, respectively. The ion source block and nitrogen desolvation gas temperature were set to 100 and 200 °C, respectively. The Trap and Transfer T-Wave sections were operated at ca. 2.3 × 10−2 mbar of nitrogen, and the ion mobility T-Wave at 0.3 mbar of nitrogen. The Mobility T-Wave was operated with a velocity of 300 m s−1 and the pulse height optimized between 7 and 10 V. A detailed description of the Synapt HDMS has been given elsewhere;29 briefly, the instrument has a quadrupole/ion mobility/orthogonal acceleration time-of-flight (Oa-ToF) geometry. The ion mobility stage contains three stacked-ring ion guides (SRIG), namely Trap, IMS and Transfer. Ions transmitted through the quadrupole are accumulated at the Trap cell, periodically injected into the IMS cell and separated by mobility, after which they are transferred to the Oa-ToF analyzer by the Transfer cell. TWIMS-MS spectra are acquired by synchronizing the release of ions to the IMS cell with acquisition start at the Oa-ToF. The instrument was externally calibrated using phosphoric acid oligomers, and ranging the m/z from 100 to 2000.Visualization of ion mobility data was done as follows. First, a mass spectrum was obtained by combining over the full arrival time distribution (ATD) for each sample. Then, individual ATDs were extracted from ions with +10 charge state corresponding to the apo and cation-bound forms of the enzyme, which allows observation of distinct ion mobility distributions and drift times.
Results and discussion
Preparation of apo-carbonic anhydrase
By employing native mass spectrometry solutions conditions, in this case, at 10 mmol L−1 ammonium acetate, carbonic anhydrase presented three charge states (+10, +11 and +12), at m/z range from 2400 to 3100, as shown in Fig. 1. This result confirms the observations already presented by Lomeli.28 A comparison of mass spectra acquired before and after treatment of BCA with OP (Fig. 1) demonstrates the loss of zinc bound to the protein structure after treatment with OP (m/z 2639, Fig. 1b) when compared to untreated protein (m/z 2645, Fig. 1a), with a mass shift of ca 63.9 Da. Moreover, this difference becomes more evident when exact masses of proteins with and without treatment are compared, as show in Fig. 2. The deconvoluted spectrum of anhydrase without treatment indicates an exact mass of 29085 Da (Fig. 2a), while the enzyme obtained after treatment shows an exact mass of 29021 Da (Fig. 2b), showing a mass difference of 64.0 Da between both, which suggests the metal loss. | ||
| Fig. 1 Carbonic anhydrase mass spectra (a) without treatment and (b) after the treatment with OP. | ||
 | ||
| Fig. 2 Deconvoluted mass spectra of the enzyme (a) without treatment (29085 Da) and (b) after the treatment with OP (29021 Da). | ||
Evaluation of metal addition to carbonic anhydrase by MS
MS has been used to prove metal binding to protein structures.30–32 In the specific case of BCA, the metal plays an important role in biological activity, since the lack of metal inhibits the enzymatic activity.33 Nevertheless, its presence is responsible for biological function of carbonic anhydrase, i.e., catalyze the reversible hydration of carbon dioxide. The metal binding was indicated by the arrows in the mass spectra (Fig. 3). The peaks in the 2924 and 2917 m/z ratio were attributed to the addition of Pb2+ and Ba2+ as shown in Fig. 3b and 3c, respectively. For metal cations such as Zn2+ and Cu2+, the binding of three cations to each molecule of BCA was observed, as shown in Fig. 3d and 3e, respectively. | ||
| Fig. 3 BCA mass spectra at 8.5 μmol L−1(a) in the apo-form, and in the presence of: (b) Pb2+, (c) Ba2+, (d) Zn2+and (e) Cu2+ metals, at 85.0 μmol L−1. | ||
The range of metal concentrations employed was tested between 8.5 and 425 μmol L−1. Binding of zinc and copper with the protein structure was observed at the lowest and highest concentrations, showing a strong affinity between these metals and BCA.34 The same results were not observed for barium and lead, which, at higher concentrations, showed a significant alteration in the protein structure, since the resulting mass spectrum resembled the one of a denatured protein (data not shown). Metal concentrations of 85.0 μmol L−1 yielded mass spectra with peaks corresponding to metal binding to the protein, without alteration of its structure. In fact, the formation of a larger number of species in the presence of metals that have a major affinity to the enzyme, such as Zn2+ and Cu2+, was observed. For metals with lower affinity, such as Pb2+ and Ba2+, only a single metal ion bound per protein molecule was observed.
Metal presence was also evidenced in the enzyme structure, through deconvolution of mass spectrum. The values shown in Table 1 presented the difference in exact mass between the holo and apo enzyme, which is observed when the enzyme is or not submitted to the presence of metals, respectively, and these results do not refer to a specific metallic isotope. The mass difference between anhydrase with and without barium bound has resulted in 137 Da shift. Other mass differences, such as 206, 65 and 64, suggest the addition of Ba2+, Cu2+ and Zn2+, respectively.
Metal affinity is determined by interactions between metal ions and the enzyme binding site, following the Irving-Williams series.34 In the present work, lead presents the smallest dissociation constant with the enzyme (pKD = 9.1).35 The highest affinities of BCA were reported for zinc and copper (pKD = 12 and pKD = 13, respectively).34 The dissociation constant for barium determined by affinity measurements was not published up to this date. In biological systems, the structure of BCA has been shown to bind with Zn2+, even in the presence of a variety of potential cellular interferents, such as Mg2+, Ca2+, Fe2+ and Cu2+.34 Copper could potentially compete with zinc for binding to the enzyme in biological systems.36 In fact, in the present work, BCA has been shown to bind the same number of zinc and copper cations per protein molecule, since its affinity for both metals is similar.34 Thus, the advantage of soft ionization electrospray was demonstrated, since this source was able to maintain those non-covalent ion-molecule interactions.
The binding of Cr3+ and Cr6+ to the protein structure can also be observed in the spectra, as shown in Fig. 4. In the absence of chromium, apo-enzyme has presented a more intense peak than the enzyme in the presence of metallic ions, which can be observed in Fig. 4a, 4b and 4c. This fact suggests partial binding between enzyme and chromium, while apo-enzyme still maintains itself in its apo form. The binding of Se4+ and Se6+ can be demonstrated in Fig. 4d and 4e. Considering the intensity of the apo-anhydrase peak, especially for Se6+, the anhydrase has a higher affinity for selenium than chromium, since both apo-anhydrase consumption and selenium peak were more intense. The presence of potential interferents, such as sodium from selenium salts and chloride and potassium in chromium salts, was also observed, but did not interfere in the identification of selenium or chromium-bound species, since these additions greatly differ for the selenium and chromium salts.
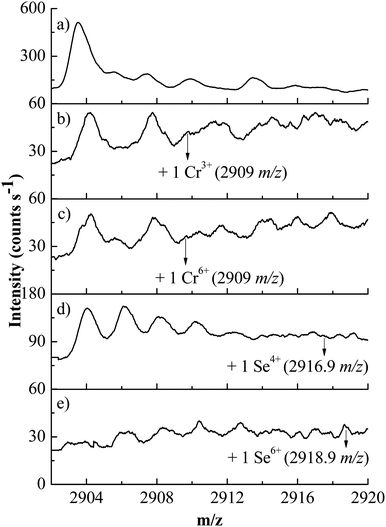 | ||
| Fig. 4 BCA mass spectra at 8.5 μmol L−1, (a) in the apo-form, and in the presence of metals (b) Cr3+, (c) Cr6+, (d) Se4+ and (e) Se6+, at 85.0 μmol L−1. | ||
Ion mobility as tool for speciation analysis in biomolecules
As previously observed in MS experiments, zinc and copper can bind up to three atoms in BCA structure. The binding of first metal per protein molecule was already enough to alter the ion mobility of the metallo-enzyme, in comparison with apo-enzyme, while the second and third cations bound in the BCA structure did not affect the mobility of the species. This fact can suggest that the first metal bound to BCA can be present in the metal binding site belonging to the structure of the enzyme, while other additions may refer to metals bound to amino acids side chain in the protein surface.36 Thus, the first addition would be responsible for the most significant structural change; while others bound atoms would not be enough to cause a significant variation on enzyme mobility.For all metals studied in this work, the ions of BCA that have been bound to one or more metals had a lower mobility than ions present in the apo-enzyme itself, as shown in Fig. 5. This suggests that the addition of metals increases the collision cross section (CCS) of such protein ions, resulting in longer arrival times for metal-bound species, and, consequently, in higher separation efficiency by TWIMS-MS. The metal affinity also had an effect over the mobility of holo-species. The highest mobility was observed for lead (Fig. 5b), while the lowest mobility was for zinc and copper (Fig. 5d and 5e, respectively). It suggests that the enzyme affinity could be related to the ability of the enzyme to bind metal in its structure.37,38 The smallest mobility was obtained with metals with higher affinity to the BCA, since metals with greater affinity for the enzyme have been bound in the metal binding site, causing a more significant structural change.
 | ||
| Fig. 5 Arrival time distributions for (a) apo-BCA at 8.5 µmol L−1, and in the presence of metals: (b) Pb2+, (c) Ba2+, (d) Zn2+ and (e) Cu2+, at 85.0 µmol L−1. | ||
Separation strategies must allow the identification of oxidation states of a metal or metalloid, that interact with the structure of a biomolecule in a non aggressive ambient or under physiological conditions.28 These characteristics were obtained in the TWIMS-MS speciation analysis of Se4+/Se6+, Cr3+/Cr6+, bound to the enzyme. None of the species showed the same apo-carbonic anhydrase mobility.
In the chromium speciation analysis, anhydrase bound to Cr3+ had a lower mobility than the anhydrase bound to Cr6+, as shown in Fig. 6b and 6c. Considering selenium speciation, the anhydrase bound to Se4+ presented a higher mobility than the anhydrase bound to Se6+, as shown in Fig. 6d and 6e. The oxidation state seems to influence the protein mobility, even when comparing the same charge states of the enzyme at different conditions. This may suggest that the enzyme allows a slight opening in its three-dimensional structure to accommodate more charged species, in contrast to a form free-of-metal.39,40
 | ||
| Fig. 6 Arrival time distributions for (a) apo-BCA at 8.5 µmol L−1, and in the presence of metals: (b) Cr3+, (c) Cr6+, (d) Se4+ and (e) Se6+, at 85.0 µmol L−1. | ||
Although there is no pKD established for chromium and selenium species bound to BCA, some inferences can be established. As noted in the case of divalent metals, the lower mobility reflects an increased interaction between BCA and ions. Thus, the chromium and selenium mobilities suggest that the enzyme affinity follows the order: Se4+ < Cr6+ < Se6+ < Cr3+. It is an intriguing fact to note that more toxic species, such as Se4+ and Cr6+, have a lower affinity for the enzyme than the others. At this moment, the ionic radius has not been shown to be an important factor in the mobilities of these species. Therefore, this study opens up new possibilities for research, regarding the interaction between metals in different oxidation states and biomolecules.
Conclusions
Travelling wave ion mobility mass spectrometry technique was successfully applied as an alternative technique, when focusing on speciation without chromatographic separation in bovine carbonic anhydrase (BCA). In this way, those divalent species, such as Ba, Pb, Zn and Cu bound to BCA, were differentially identified through their ionic mobility, making possible individual species identification in this enzyme. Additionally, Se4+/Se6+ and Cr3+/Cr6+ speciation in BCA was also possible, putting in evidence the TWIMS-MS as an important and complementary technique when focusing on speciation studies.As the modus operandi of the ion mobility technique and all the used conditions in the ionization source applied to this technique are generally soft ones (i.e., low ionization energy, absence of hard organic solvents and high pressures for separations, among others), all the measurements can be carried out at atmospheric pressure, producing perfect conditions for working with molecules that present stable (inorganic) and label (organic) fractions, simultaneously, in their structures. Then, inorganic speciation from those elemental species present in biomolecules, opens new possibilities in terms of investigations regarding conformation studies of the biomolecules when considering their interactions with metals at different oxidation states and biomolecules. This aspect is useful in metalloproteomics studies.
Notes and references
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- J. L. Gomez-Ariza, T. García-Barrera, F. Lorenzo, V. Bernal, M. J. Villegas and V. Oliveira, Anal. Chim. Acta, 2004, 524, 15–22 CrossRef CAS.
- A. Polatajko, N. Jakubowski and J. Szpunar, J. Anal. At. Spectrom., 2006, 21, 639–654 RSC.
- A. Z. Mason, R. Moeller, K. A. Thrippleton and D. Loyd, Anal. Biochem., 2007, 369, 87–104 CrossRef CAS.
- J. Giner, J. G. Martínez-Sierra, F. M. Sanz, P. H. Espílez, R. Santamaria-Fernandez, J. M. M. Gay and J. I. G. Alonso, J. Anal. At. Spectrom., 2010, 25, 989–997 RSC.
- M. Benassi, Y. E. Corilo, D. Uria, R. Augusti and M. N. Eberlin, J. Am. Soc. Mass Spectrom., 2009, 20, 269–277 CrossRef CAS.
- H. H. Hill, C. H. Hill, G. R. Asbury, C. Wu, L. M. Matz and T. Ichiye, Int. J. Mass Spectrom., 2002, 219, 23–37 Search PubMed.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS.
- E. V. Duijn, A. Barendregt, S. Synowsky, C. Verluis and A. J. R. Heck, J. Am. Chem. Soc., 2009, 131, 1452–1459 CrossRef CAS.
- B. Michalke, S. Halbach and V. Nischwitz, J. Environ. Monit., 2009, 11, 939–954 RSC.
- A. Gojova, B. Guo, R. S. Kota, J. C. Rutledge, I. M. Kennedy and I. A. Barakat, Environ. Health Perspect., 2007, 115, 403–409 CAS.
- A. Sanz-Medel, M. Montes-Bayon and M. L. F. Sanchez, Anal. Bioanal. Chem., 2003, 377, 236–247 CrossRef CAS.
- R. Lobinski, D. Schaumloffel and J. Szpunar, Mass Spectrom. Rev., 2006, 25, 255–289 CrossRef CAS.
- R. Jirasko, M. Holcapek and E. Rosenberg, Int. J. Mass Spectrom., 2009, 280, 198–203 Search PubMed.
- R. Jirasko, M. Holcapek, L. Kolarova and T. S. B. Baul, J. Mass Spectrom., 2007, 42, 918–928 CrossRef CAS.
- B. Ells, D. A. Barnett, R. W. Purves and R. Guevremont, Anal. Chem., 2000, 72, 4555–4559 CrossRef CAS.
- R. Handy, D. A. Barnett, R. W. Purves, G. Horlick and R. Guevremont, J. Anal. At. Spectrom., 2000, 15, 907–911 RSC.
- Y. Guo, Y. Ling, B. A. Thomson and K. W. M. Siu, J. Am. Soc. Mass Spectrom., 2005, 16, 1787–1794 CrossRef CAS.
- P. A. Faull, K. E. Korkeila, J. M. Kalapothakis, A. Gray and P. E. Barran, Int. J. Mass Spectrom., 2009, 283, 140–146 Search PubMed.
- S. L. Shirran and P. E. Barran, J. Am. Soc. Mass Spectrom., 2009, 20, 1159–1171 CrossRef CAS.
- B. J. McCullough, J. Kalapothakis, H. Eastwood, P. Kemper, D. MacMillan, K. Taylor, J. Dorin and P. E. Barran, Anal. Chem., 2008, 80, 6336–6344 CrossRef CAS.
- S. J. Ray, F. Andrade, G. Gamez, D. McClenathan, D. Rogers, G. Schilling, W. Wetzel and G. M. Hieftje, J Chrom. A., 2004, 1050, 3–34 CrossRef CAS.
- B. T. Ruotolo, K. J. Gillig, A. S. Woods, T. F. Egan, M. V. Ugarov, J. A. Schultz and D. H. Russel, Anal. Chem., 2004, 76, 6727–6733 CrossRef CAS.
- K. Thalassinos, M. Grabenauer, S. E. Slade, G. R. Hilton, M. T. Bowers and J. H. Scrivens, Anal. Chem., 2009, 81, 248–254 CrossRef CAS.
- L. F. A. Santos, A. H. Iglesias, E. J. Pilau, A. F. Gomes and F. C. Gozzo, J. Am. Soc. Mass Spectrom., 2010, 21, 2062–2069 CrossRef CAS.
- R. Saito, T. Sato, A. Ikai and N. Tanaka, Acta Cryst., 2004, 60, 792–795.
- S. Lindskog and P. O. Nyman, Biochim. Biophys. Acta., 1964, 85, 462–474 CAS.
- S. H. Lomeli, S. R. R. Yin, R. R. O. Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2000, 20, 593–596.
- S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers and J. H. Scrivens, Int. J. Mass Spectrom., 2007, 261, 1–12 Search PubMed.
- S. McSheehy and Z. Mester, J. Anal. At. Spectrom., 2004, 19, 373–380 RSC.
- D. A. Barnett, R. W. Purves and R. Guevremont, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 450, 179–185 CrossRef CAS.
- J. Lim and R. W. Vachet, Anal. Chem., 2004, 76, 3498–3504 CrossRef CAS.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS.
- T. K. Hurst, D. Wang, R. B. Thompson and C. A. Fierke, Biochim. Biophys. Acta., 2010, 1804, 393–403 CAS.
- L. A. Calhoun and D. L. Livesey, J. Inorg. Biochem., 1985, 25, 261–275 CrossRef CAS.
- L. A. Finney and T. V. O'Halloran, Science, 2003, 300, 931–936 CrossRef CAS.
- J. A. Hunt, M. Ahmed and C. A. Fierke, Biochemistry, 1999, 38, 9054–9062 CrossRef CAS.
- K. A. McCall and C. A. Fierke, Biochemistry, 2004, 43, 3979–3986 CrossRef CAS.
- M. Bohm, S. Sturzebecher and G. Klebe, J. Med. Chem., 1999, 42, 458–477 CrossRef CAS.
- D. W. Christianson and C. A. Fierke, Acc. Chem. Res., 1996, 29, 331–339 CrossRef CAS.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |