Loss of di- and trimethylarsine on Nafion membrane dryers following hydride generation†
Petra
Taurková
ab,
Milan
Svoboda
ab,
Stanislav
Musil
ab and
Tomáš
Matoušek
*a
aInstitute of Analytical Chemistry of the ASCR, v.v.i., Veveří 97, 602 00, Brno, Czech Republic. E-mail: matousek@biomed.cas.cz
bCharles University in Prague, Faculty of Science, Department of Analytical Chemistry, Hlavova 2030, 128 43, Prague 2, Czech Republic
First published on 18th November 2010
Abstract
Complete loss of trimethylarsine upon passage through a Nafion membrane dryer was identified. Pronounced loss of dimethylarsine—19 and 66%, depending on dryer dimensions—was also observed; arsine and methylarsine did not exhibit significant losses. Arsines were generated from corresponding precursors at the 2 ng ml−1 level by tetrahydroborate reduction, after passing through the dryer separated by cryotrapping and thermal desorption and detected by atomic absorption spectrometry with a quartz multiatomizer. Since Nafion membrane dryers have been commonly used in analytical atomic spectroscopy, implications for trace and speciation analysis of arsenic are discussed. A dryer based on sodium hydroxide pellets is proposed as an alternative, safe for all arsines.
Introduction
In the well known sample introduction technique for elemental analytical spectroscopy-hydride generation, HG, (or more generally volatile compound generation), the analyte is chemically volatilized and separated from the matrix and introduced into the detector in the gaseous phase. As an undesirable by-product of the process, the gaseous phase contains a fair amount of water, both as aerosol droplets and vapor. The most critical demands for water removal from the gaseous phase containing volatile analyte species is prior to cryofocusing and separation,1–5 where water can possibly block the cryotrap, and prior to atomic fluorescence detection, where aerosol droplets can cause light scattering and water molecules fluorescence quenching resulting in sensitivity decrease and baseline drift.6Varied approaches for water removal were applied throughout the years, typically based either on condensation at temperatures below 0 °C, or on various desiccants (e.g.sulfuric acid, calcium chloride, silicagel, calcium sulfate, magnesium perchlorate, sodium hydroxide1,7 or soda lime8). Nafion membrane dryers were introduced to analytical spectroscopy in the early 1990's.6,9–11 It is a simple, elegant and inexpensive solution, commercially available (from Perma Pure), very simple to use and allowing continuous and maintenance free operation. Signals of chromatographic separations are not adversely influenced thanks to the low volume of the dryers. The dryer consists of a tubular membrane, encased in a plastic outer tube. Gaseous phase containing volatilized analyte passes inside the membrane tube, while its outside surface is dried by a counter-flow of drying gas.
Nafion is a copolymer of tetrafluoroethylene and perfluoro-3,6-dioxa-4-methyl-7-octene-sulfonic acid. Similarly to Teflon, Nafion has a very good chemical resistance, but at the same time it is able to absorb up to 13 molecules of water per sulfonic group. Nafion removes water by absorption (hydration). This absorption process is a first order reaction and equilibrium is achieved very quickly (typically in the order of milliseconds), in contrast to the very slow diffusion process of water transfer through the microporous permeation membrane. Nafion membranes are fairly selective, although other compounds, such as ammonia, alcohols, amines and some polar compounds are also removed from the gaseous phase according to manufacturer's web pages.
Nafion membrane dryers found their way not only to prototype instruments,4,5 but became a standard part of a commercial atomic fluorescence spectrometer.9 Generally, no problems were reported with the membrane dryers over the years. Good efficiency of water removal and negligible losses of mercury vapor, arsine and selenium hydride were reported in flow injection HG setups with quartz tube atomic absorption detection.11 No loss of arsine, mono- and dimethylarsine on a membrane dryer was found in a HG-cryotrapping-AAS system.10 The only analyte species with reported losses we are aware of was methylmercury hydride.8,12
With the ever increasing importance of the speciation analysis, we tested the membrane dryers as well as a sodium hydroxide cartridge for use in HG based analysis of As species, with AAS as the detection method. In particular, losses of individual methylsubstituted hydrides were investigated.
Experimental
Instrumentation
Standards and reagents
A 1000 mg l−1 As AAS standard solution (Merck, Darmstadt, Germany) was used as an iAs(V) stock standard solution. A stock solution of 1000 mg As l−1 was prepared for each of the other arsenic species in water using the following compounds: Na2(CH3)AsO3·6H2O, Chem. Service, West Chester, PA, USA (MA); (CH3)2As(O)OH, Strem Chemicals, Inc., Newburyport, MA, USA (DMA). (CH3)3AsO (TMAO) was obtained courtesy of Dr William Cullen (University of British Columbia, Vancouver, Canada). Working standards were prepared for individual species by serial dilution of the stock solutions in water. Mixed standards were used only in the last dilution, i.e. at the ng ml−1 level.The reducing solution containing 1% (m/v) NaBH4 (Fluka, Buchs, Switzerland) in 0.1% (m/v) KOH (p.a., Lachema, Brno, Czech Rep.) was prepared daily. TRIS buffer—a 0.75 M Tris(hydroxymethyl)aminomethane-hydrochloride buffer (pH 6)—was prepared from a reagent grade Trizma® hydrochloride (Sigma), pH adjusted to 6 according to pH meter readings (Mettler, Toledo) by addition of approx. 1.5 ml of 10% (m/v) KOH per 250 ml of buffer solution.
Dryers
Results and discussion
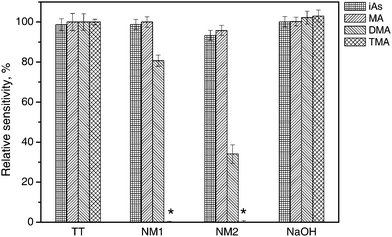
Relative peak area absorbances for individual arsines after passing through the dryer tubes are shown in Fig. 1. The areas are related to the setup without any dryer tube present, i.e. phase separator output connected directly to the U-tube. As evident, there are absolutely no losses of any species in the TT tube, which rules out adsorption or condensation losses. There are also no losses of arsine and methylarsine in the shorter NM1 tube and a small loss in the longer NM2 one. There is a different situation for dimethylarsine and especially for trimethylarsine, though. Pronounced losses of approx. 19% and 66% of dimethylarsine were observed in the Nafion dryers NM1 and NM2, respectively (see Fig. 1). Trimethylarsine was lost completely in both dryers, despite a very short residence time in the dryer (0.53 s and 0.50 s, respectively). A contradicting report of no observed losses of dimethylarsine (trimethylarsine was not tested) in ref. 10 might be related to quite different experimental parameters: a dryer of different size (membrane 6.3 mm o.d., 1828 mm long), a 40 times higher amount of As taken for the tests as well as purge gas of different composition at much higher flow rate (90 ml min−1 N2 + 300 ml min−1 H2, i.e. about 4 times higher than the flow rate used in this study).The peak shapes, retention times and resolutions of all species (except the completely disappearing peak of trimethylarsine) were not changed by Nafion tubes, as illustrated by the chromatogram in Fig. 2. Please note that TMAO must be determined without pre-reduction, since upon pre-reduction by L-cys prior to analysis, volatile trimethylarsine is partially produced and lost;14,15 on the other hand, dimethylarsine is generated from DMA at pH 6 to a small extent even without pre-reduction.14
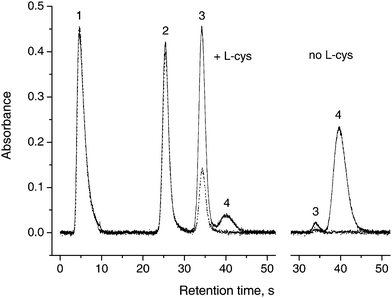 | ||
| Fig. 2 Chromatograms without dryer (solid line) and with long Nafion dryer (NM2, dotted line), with and without L-cys pre-reduction. 1: iAs; 2: MA; 3: DMA; 4: TMAO, 1 ng (as As) each form. | ||
Implications of the fact of dimethylarsine and trimethylarsine losses in Nafion tubes are obvious. The complete loss of trimethylarsine on Nafion membrane dryers remained overlooked for a long time in the literature (it led even the authors of a recent review on speciation analysis by AFS to the erroneous statement that trimethylarsenic oxide does not readily form volatile hydride16). Due to poor transport efficiency, DMA and TMAO sensitivity is much lower compared to iAs, and in the samples containing As in the forms of di- and trimethylated species, the use of a Nafion dryer can lead to underestimation of total (or total toxicologically relevant) As content and impaired accuracy of analysis. Methylated As species are present in surface and sea water, and they constitute the major As species in biological samples. A sample pre-treatment (mineralization) converting all species to the iAs form is then essential.
Concerning other important As species, arsenosugars were reported to produce a mixture of methyl- and dimethylarsine, with efficiencies below 10%.17 There might be a small decrease in dimethylarsine signal caused by the Nafion dryer, but due to the low efficiency of generation, a decomposition prior to HG of arsenosugars is advisable. Arsenobetaine (AB) and arsenocholine do not produce volatile hydrides, and therefore mineralization is required for those species.
Complete conversion of all As species into iAs may not be easily achieved, especially if resistant compounds such as AB are targeted. For instance, alkaline digestion converts AB into trimethylated species.18Ref. 19 can be another good example of this issue, showing that several methods of decomposition convert AB to a mixture of species of various levels of methylation. Incidentally, only 22% and 5% sensitivity was reported for DMA and TMAO, respectively, compared to iAs and MA. This was attributed to incomplete generation from these species,19 while in fact the Nafion dryer included in that system might have been the true cause.
In speciation studies employing HPLC with HG and a Nafion dryer containing an AFS detector, which is a viable alternative to a more expensive HPLC-ICP MS setup, an on-line mineralization step is typically involved.16 This is most often performed by UV photooxidation in the presence of peroxosulfate, which converts even AB and arsenocholine to hydride forming forms;20,21 it is assumed that this procedure breaks organoarsenicals all the way to iAs(V)22 (which is a reasonable assumption as long as a similar peak area sensitivity is achieved for all forms, but it cannot be taken for granted) and therefore analysis should not be influenced by losses on Nafion dryers. Sensitivities of 45% and 0% of iAs for DMA and TMAO, respectively, were reported in an AFS system featuring a dryer analogous to Tube 1, if zero concentration of K2S2O8 was tested in the course of on-line oxidation step optimization study.22
Although an increase in the purge gas flow rate might decrease the loss of dimethylarsine, this solution would also mean a worse drying efficiency and may not be compatible with the experimental setup (e.g. pressure build-up in a cryotrap) or with optimum conditions for the detection technique.
As an alternative, sodium hydroxide pellets as water absorbent were tested. This approach was already popular in the early days of speciation by cryoseparation.23 As evident from Fig. 1, NaOH did not significantly absorb any As species. An additional advantage is that a background absorption peak partially coinciding with an iAs peak, appearing as if L-cys pre-reduction was applied and attributed to hydrogen sulfide,14 completely disappeared. On the other hand, NaOH has to be replaced regularly, depending on the design of the hydride generator. In our case, the period was approximately two days/200 measurement cycles. Also, an influence of the NaOH cartridge on transient signals need to be considered in cases of on-line coupling of chromatographic methods to HG and the detector.
Conclusions
Dryer tubes based on tubular Nafion membranes cause significant losses of dimethylarsine and complete loss of trimethylarsine. Although acceptable when complete mineralization of As species to iAs is performed, other strategies for water removal from the gaseous phase need to be adopted if volatile di- and trimethylarsine are produced by HG procedure. This would be common in the case of incomplete or non mineralization of biological and environmental samples as well as in speciation analysis. A sodium hydroxide absorber is a possible safe alternative for all As volatile species. The losses of methylsubstituted hydrides of other elements on the Nafion membrane are yet to be tested.Acknowledgements
We gratefully acknowledge financial support from the Czech Science Foundation (grant No. 203/09/1783) and Academy of Sciences of the Czech Republic (Institutional Research Plan no. AV0Z40310501), Grant Agency of the Charles University (projects Nos. 133008 and SVV 261204), the Czech Ministry of Education, Youths and Sports (project MSM 0021620857) and from the Analytical Laboratory for Development of Biomarkers of Environmental Exposures to Arsenic supported by a Gillings Innovation Laboratory award from the UNC Gillings School of Global Public HealthReferences
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley & Sons, Inc., Chichester, 1995 Search PubMed.
- C. Pecheyran, D. Amouroux and O. F. X. Donard, J. Anal. At. Spectrom., 1998, 13, 615–621 RSC.
- E. A. Crecelius, Anal. Chem., 1978, 50, 826–827 CrossRef CAS.
- A. Wasik, R. Lobinski and J. Namiesnik, Instrum. Sci. Technol., 2001, 29, 393–405 CrossRef CAS.
- A. Wasik, I. R. Pereiro, C. Dietz, J. Szpunar and R. Lobinski, Anal. Commun., 1998, 35, 331–335 RSC.
- W. T. Corns, L. Ebdon, S. J. Hill and P. B. Stockwell, Analyst, 1992, 117, 717–719 RSC.
- A. de Diego, C. Pecheyran, C. M. Tseng and O. F. X. Donard, in A. Sanz-Medel, (Ed.), Flow Analysis with Atomic Spectrometric Detection; Elsevier: Amsterdam, 1999, Chap. 12, pp. 375–406 Search PubMed.
- T. Stoichev, R. C. R. Martin-Doimeadios, E. Tessier, D. Amouroux and O. F. X. Donard, Talanta, 2004, 62, 433–438 CrossRef CAS.
- W. T. Corns, P. B. Stockwell, L. Ebdon and S. J. Hill, J. Anal. At. Spectrom., 1993, 8, 71–77 RSC.
- J. T. van Elteren, H. A. Das, C. L. DeLigny, J. Agterdenbos and D. Bax, J. Radioanal. Nucl. Chem., 1994, 179, 211–219 CAS.
- N. G. Sundin, J. F. Tyson, C. P. Hanna and S. A. McIntosh, Spectrochim. Acta, Part B, 1995, 50, 369–375 CrossRef.
- J. Qvarnström, Q. Tu, W. Frech and C. Lüdke, Analyst, 2000, 125, 1193–1197 RSC.
- T. Matoušek, J. Dědina and A. Selecká, Spectrochim. Acta, Part B, 2002, 57, 451–462 CrossRef.
- T. Matoušek, A. Hernández-Zavala, M. Svoboda, L. Langerová, B. M. Adair, Z. Drobná, D. J. Thomas, M. Stýblo and J. Dědina, Spectrochim. Acta, Part B, 2008, 63, 396–406 CrossRef.
- S. Musil and T. Matoušek, Spectrochim. Acta, Part B, 2008, 63, 685–691 CrossRef.
- D. Sanchez-Rodas, W. T. Corns, B. Chen and P. B. Stockwell, J. Anal. At. Spectrom., 2010, 25, 933–946 RSC.
- R. Regmi, B. F. Milne and J. Feldmann, Anal. Bioanal. Chem., 2007, 388, 775–782 CrossRef CAS.
- W. H. Geng, R. Komine, T. Ohta, T. Nakajima, H. Takanashi and A. Ohki, Talanta, 2009, 79, 369–375 CrossRef CAS.
- Z. Šlejkovec, J. T. van Elteren and U. D. Woroniecka, Anal. Chim. Acta, 2001, 443, 277–282 CrossRef CAS.
- D. L. Tsalev, M. Sperling and B. Welz, Spectrochim. Acta, Part B, 2000, 55, 339–353 CrossRef.
- A. G. Howard and L. E. Hunt, Anal. Chem., 1993, 65, 2995–2998 CrossRef CAS.
- K. Ito, C. D. Palmer, W. T. Corns and P. J. Parsons, J. Anal. At. Spectrom., 2010, 25, 822–830 RSC.
- R. S. Braman, D. L. Johnson, C. C. Foreback, J. M. Ammons and J. L. Bricker, Anal. Chem., 1977, 49, 621–625 CrossRef CAS.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |