Review: The role of atomic spectrometry in plant science†
Søren
Husted
*,
Daniel P.
Persson
,
Kristian H.
Laursen
,
Thomas H.
Hansen
,
Pai
Pedas
,
Michaela
Schiller
,
Josefine N.
Hegelund
and
Jan K.
Schjoerring
Plant and Soil Science Laboratory, Department of Agriculture and Ecology, Faculty of Life Sciences, University of Copenhagen, Thorvaldsensvej 40, DK-1871, Frederiksberg C, Denmark. E-mail: shu@life.ku.dk
First published on 1st December 2010
Abstract
Inductively coupled plasma-mass spectrometry (ICP-MS) is the state-of-the-art technique for multi-elemental analysis of plant tissue. It provides a powerful tool in functional genomics, linking altered elemental profiles of mutants with gene expression and function. In addition, with its unmatched sensitivity, ICP-MS enables characterization of the substrate specificity and regulation of membrane transport proteins. Digestion of plant tissue has traditionally represented a bottleneck due to the low capacity of commercially available equipment. However, recent developments in micro-scaled digestion, combined with semi-quantitative analysis and chemometrics, have enabled high-throughput multi-elemental profiling and multivariate classification of large sample sets, thereby supporting a range of new applications in molecular breeding, quality assessment and authenticity testing of plants. Novel hyphenated techniques based on liquid chromatography and ICP-MS (LC-ICP-MS) have significantly improved the understanding of elemental species and their importance for e.g. the bioactivity of metals in plants. Development of procedures for sample pre-treatment, extraction and multi-dimensional separation now allows characterization of important metallo-biomolecules in plants, such as the coordination complexes of phytochelatins, metallothioneins, nicotianamine and inositol phosphates. These are key ligands involved in ion homeostasis, translocation and long-term storage of elements. Much emphasis has also been given to studies of covalently bound Se and As species, primarily due to their impact on human health. LC-ICP-MS has extensively been complemented by molecular mass spectrometry for structural information of biologically relevant species. This review covers the most recent developments in multi-elemental analysis (Part A) and speciation analysis (Part B) in plant science. A number of relevant cases are presented in order to demonstrate how the analytical developments have unravelled the functional roles of elements in plants science. These cases show that ICP-MS is an essential technology in plant metallomic platforms.
 The ionomics and plant nutrition group at the University of Copenhagen, Faculty of Life Sciences. From the left: Professor Søren Husted, PhD student Michaela Schiller, Post Doc Pai Pedas, Professor Jan K. Schjoerring, PhD student Thomas H. Hansen, Post Doc Daniel Persson, PhD student Kristian Holst Laursen and Post Doc Josefine Nymark Hegelund | The research group is studying the functional roles of mineral elements in plants, including characterization of ion uptake, translocation and assimilation. Special attention is given to the essential plant micronutrients Fe, Zn and Mn in cereal species. Together with state-of-the-art plant molecular and biochemical techniques, the development of novel methods in atomic spectrometry plays an important role for these studies. Recently, we have developed semi-quantitative methods for multi-elemental fingerprinting of plant genotypes and transgenic lines, as well as a wide range of hyphenated techniques to study e.g. Fe and Zn speciation in plants. |
Introduction
The essential and beneficial plant nutrients
Plants are photoautotrophic organisms and are consequently able to use photochemical energy from the sun to synthesise all chemical components on the basis of CO2, H2O and 14 inorganic elements (Table 1). These inorganic elements are denoted essential plant nutrients and are by convention divided into two classes, comprising 6 macronutrients (N, P, K, Mg, Ca, S) and 8 micronutrients (Fe, Mn, Zn, Cu, Ni, B, Mo, Cl). The essential plant nutrients have one or more non-redundant functions in the primary metabolism of plants and as a consequence, plants are unable to complete a full life cycle from germination to maturity if just one of the essential plant nutrients is unavailable. In order to be classified as an essential plant nutrient, the elements must be required by all higher plants (tracheophytes: the vascular plants) without any exception. Some inorganic elements are classified as beneficial plant nutrients because they stimulate growth of some, but not all, higher plants species. The beneficial plant nutrients include Co for N2 fixing plants, Si for rice plants, Al for tea plants, Na for halophytes and Se for a number of plant species in very diverse families.1| Element | Barley (H. vulgare) | Rape leaves (B. napus) | Biochemical function |
|---|---|---|---|
| Macro (%) | |||
| N | 3.5–5.5 | 3.5–5.5 | Protein synthesis, nucleic acids, hormonal balance |
| P | 0.3–0.5 | 0.3–0.6 | Energy transport, biomembranes |
| K | 2.4–4.0 | 2.8–5.5 | Translocation, osmoregulation, pH homeostasis, enzyme activation |
| Ca | 0.2–0.5 | 1.4–3.0 | Cell wall synthesis and stabilisation, signal transduction |
| Mg | 0.13–0.3 | 0.2–0.65 | Photosynthesis (central atom of chlorophyll), enzyme activation |
| S | 0.15–0.4 | 0.2–0.9 | Protein synthesis and functionality |
| Micro (μg g−1) | |||
| Fe | 35–60 | 50–300 | Enzyme activation, electron transport, chlorophyll biosynthesis, oxidative stress protection |
| Mn | 25–100 | 30–250 | Enzyme activation, photosynthesis, electron transport, synthesis of cell wall components, oxidative stress protection |
| Zn | 15–70 | 20–80 | Gene expression, enzyme activation, hormone and nucleic acid synthesis, oxidative stress protection |
| Cu | 6–12 | 4–25 | Enzyme activation, synthesis of cell wall components, electron transport, oxidative stress protection |
| B | 5–10 | 35–60 | Stabilisation of cell walls, nucleotide biosynthesis, translocation, pollen germination |
| Mo | 0.1–0.5 | 0.3–1.0 | Enzyme activation, nitrogen fixation, nitrate reduction |
| Cl | — | — | Electroneutrality, osmoregulation, photosynthesis |
| Ni | — | — | Enzyme activation, urea metabolism |
Essential plant nutrients in metabolism
The essential nutrients serve a multitude of functions in plants as structural components, enzyme activators (cofactors) and regulators of electrochemical and osmotic potentials (Table 1). Some essential plant nutrients, e.g., Zn and Fe, have many different functions whereas others such as Cl, Ni and Mo are involved in just a few processes. Broadley et al. (2007)2 predicted that 2042 proteins in Arabidopsis thaliana contain one or more Zn binding domains. This contrasts with the number of Cl, Ni and Mo binding proteins of which less than a handful have been identified.3 Due to the multitude of metabolic processes and the links between them, changes in the activity of just one gene, protein or metabolite often affect the tissue concentration of several essential plant nutrients and not just the specific nutrient directly linked to a given process. This was recently illustrated in a yeast functional complementation assay, where Mn deficiency induced an elevated expression of the barley HvIRT gene, transporting Mn, Fe, Zn and Cd across plasmalemma.4 ICP-MS was used to screen the yeast cells for 31 elements and a marked change in multi-elemental composition was observed, including a doubling of the Cu concentration, which is not transported by the protein. Baxter et al. (2009)5 isolated a mutant of the model plant species Arabidopsis thaliana with increased deposition of suberin wax in the root endodermis, leading to reduced transpiration. This was accompanied by a decrease in Ca, Mn and Zn and an increase in Na, S, K, Mo, As and Se depending on whether the elements predominantly followed an apoplastic or symplastic loading pathway into the root stele.Plant nutrient homeostasis
Plants exhibit different physiological requirements for the essential plant nutrients. As a consequence, the elemental composition of plant tissue may vary significantly depending on the plant species considered (Table 1). The macronutrients usually constitute 0.1 to 5.5% of plant dry matter (DM), i.e. 1 to 55 mg g−1 DM. The corresponding range for the micronutrients is 0.1 to 100 μg g−1 DM (Table 1). In a plant species like wheat (Triticum aestivum) it is typical to find e.g. K leaf tissue concentrations close to 50![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 μg g−1 DM and Mo concentrations around 0.1 μg g−1 DM, implying a huge molar K/Mo ratio of 1.3 × 106. Despite their markedly different concentrations, both elements are essential in plant metabolism, and Mo deficiency would potentially be able to limit plant productivity as severely as K deficiency. Plants have developed a series of strategies to maintain the concentrations of individual essential plant nutrients within a narrow range (ion homeostasis, from greek: homoios “standing still”) under the fluctuating nutrient availabilities occurring in their root zone. This allows plants to balance tissue nutrient concentrations in a range ensuring essential metabolic functions and avoiding toxicity.
000 μg g−1 DM and Mo concentrations around 0.1 μg g−1 DM, implying a huge molar K/Mo ratio of 1.3 × 106. Despite their markedly different concentrations, both elements are essential in plant metabolism, and Mo deficiency would potentially be able to limit plant productivity as severely as K deficiency. Plants have developed a series of strategies to maintain the concentrations of individual essential plant nutrients within a narrow range (ion homeostasis, from greek: homoios “standing still”) under the fluctuating nutrient availabilities occurring in their root zone. This allows plants to balance tissue nutrient concentrations in a range ensuring essential metabolic functions and avoiding toxicity.
Atomic spectrometry has played a vital role in establishing the homeostatic concentration range for all the essential plant nutrients (Table 1). This information has been used to establish critical threshold values defining the concentration of individual plant nutrients at which plant productivity will be significantly impeded. Critical threshold values for all the essential nutrients in the most important crop and horticultural plants have been assembled in large databases.6 These values represent an invaluable resource of information with respect to optimising productivity and quality in commercial plant production. Most of these data have been collected continuously over several decades across different countries and soil types, using predominantly atomic absorption spectrometry (AAS), i.e. single element analysis. Typically, AAS based information on critical threshold values has been compiled from several independent sources, often employing different genotypes of the same plant species which creates uncertainties about the tabulated values.
The plant ionome and metallome
The single-element approach forming the basis for AAS has delayed the important discovery that ion homeostasis is regulated by a network of processes and that deficiency or surplus of one element leads to marked changes in the homeostatic levels of several others. However, during the last few decades, significant improvements in inductively coupled plasma spectrometry, either in the form of mass spectrometry (ICP-MS) or optical emission spectrometry (ICP-OES), have facilitated the simultaneous measurement of many essential and beneficial elements in plants. This multi-elemental approach has revealed the complex regulatory networks involved in plant nutrient homeostasis4,5,7,8 and has led to the introduction of the plant ionome as a concept. A derived term is ionomics, denoting the scientific discipline of analysing not only the essential and beneficial elements in plants, but in principle the complete elemental composition of a cell, tissue or organ. This novel approach allows dynamic studies of the plant ionome in response to changes in genetic constitution, nutrient availability and climate. Out of the 90 naturally occurring elements in the periodic table, Laursen et al. (2009)9 measured 47 elements in three different rice genotypes grown in soil. Watanabe et al. (2007)10 reported 42 elements in samples from 670 plant species. However, in most studies of the plant ionome significantly fewer elements have been analysed, typically less than 20 elements.11,12The introduction of high-throughput analytical platforms for plant sample digestion13 and multi-elemental analysis have paved the road for studying the complex networks between genes, proteins, metabolites and elements, thereby linking ionomics to genomics, proteomics and metabolomics in plant science. Williams (2001)14 suggested the term “metallome” which describes the distribution of metals between cell organelles, viz. cytoplasm, vacuole, chloroplast and mitochondria. This terminology has recently been refined by Mounicou et al. (2009),15 defining the metallome as the entirety and distribution of metal and metalloid chemical species in a cell or tissue type. Compared to the ionome, the metallome is a more focused term because it is confined to metal and metalloid ions, neglecting essential plant nutrients and non-metals such as P, N, S and Cl. Yet, at the same time, the metallome is a more comprehensive term, including information about the speciation of the metal and metalloid ions.
The speciation of an element characterises its distribution amongst defined chemical species in a system.16 This includes the isotopic composition, oxidation states and the molecular structure of an element. The speciation of elements in plants has major effects on uptake, translocation, assimilation and metabolic interactions between ions. Moreover, speciation regulates the bioavailability of elements in vegetable food and feed products. Technological developments over the last decade have revolutionised elemental speciation analysis by providing new and fundamental information showing the importance of focusing on elemental species rather than total elemental concentrations. The vast majority of speciation studies have so far focused on the differential toxicity of species of non-essential elements, e.g. As, Cr, Hg and Tl, and on how speciation affects the tolerance to heavy metals such as Cd, Ni and Pb.17–22 In addition, Se speciation in plants has received remarkable attention, primarily due to the hyper-accumulating ability of some plants23 and the putative anti-carcinogenic properties of specific Se species.24
Speciation studies focusing on essential plant nutrients are still relatively scarce. Fortunately, there is a growing interest to investigate how the speciation of essential elements influences ion uptake, translocation, assimilation and storage.25–27 The first papers linking the elemental speciation of Fe and Zn in the rice endosperm with bioavailability in human nutrition have recently appeared.28,29 In the following sections we review recent advances in multi-elemental analysis (Part A) and speciation analysis (Part B) in plant science.
Part A: Multi-elemental plant tissue analysis
Multi-elemental plant tissue analyses with ICP-MS and ICP-OES are gradually replacing most of the classical AAS-based single element analyses. Low detection limits combined with a high precision and accuracy have enabled high-throughput analysis of major parts of the periodic table in practically any plant sample matrix. This development has significantly improved our understanding of fundamental plant biology and plant nutrition.Sample pre-treatment
Due to the sensitivity of ICP based atomic spectrometry, decontamination of plant samples is often needed prior to multi-elemental analysis. Plant samples from field sites for example, always require efficient removal of dust-borne deposits on the surfaces. Usually this is accomplished by a simple rinse in several batches of de-ionised water with or without a detergent.13 Husted et al. (2004)30 found that cereal grains exposed to artificial dust containing radio-labelled 54Mn was completely cleaned by three rinses in Milli-Q-water. No changes in the ionic composition of the tissue were observed, except for K, which is known to be easily lost by leaching.After decontamination, a subsequent drying of plant samples is required before continuing with digestion and analysis. This is preferably conducted by lyophilisation to avoid sample contamination. Then follows homogenisation of the dried plant material, allowing representative sampling prior to digestion. Numerous milling techniques are available for this purpose. Laursen et al. (2009)9 homogenised rice grains with a grinding mill equipped with a titanium rotor to avoid trace element contamination.
Current principles and procedures for digestion of plant tissue
The analytical performances of ICP-OES and ICP-MS are highly dependent on efficient digestion of plant material prior to analysis. This is the case because organic residues can cause analytical biases and block the sample introduction system. Depending on the sample type, sample amount and elements of interest, different digestion methods are used. Plant leaves are generally easily digested, whereas grain, seed and fruit materials require more harsh digestion conditions.13 The most commonly used methods for digestion of organic matter are based on high temperature oxidation (dry ashing) or wet digestion using strong acids with or without the addition of oxidising agents.Dry ashing is based on incineration without ignition carried out at 550–600 °C in e.g. a muffle furnace. A large number of samples can be handled using this technique, but several problems exist. Significant quantities of volatile elements such as S, Hg, As and Se can be lost during the process and the ash have a tendency to adsorb to the surface of the porcelain or quartz cups used, leading to underestimation of all elements. Also the subsequent handling of the ash before analysis is challenging due to its fluffy structure and high hygroscopicity.31,32 However, dry ashing can be advantageous in the presence of high quantities of glycerol which is frequently used to stabilise chloroplasts obtained from tissue and cell fractionations. In combination with HNO3, glycerol can cause a safety risk in wet digestion of plant materials.33 In such case, advantage can be taken of the recent development of closed systems for dry ashing in which the risk of analyte loss is also reduced. Of these systems, microwave induced combustion (MIC) is the most promising technique as it employs inert quartz materials.34 This is advantageous for subsequent analysis of elements present in trace and ultra-trace concentrations. However, high-throughput dry ashing is not possible as only 8 samples can be processed simultaneously in the commercially available systems.32
Wet digestion, often referred to as wet ashing, relies on the use of one or more strong oxidising agent(s). The most commonly used oxidising agents are acids, e.g. HNO3, H2SO4 and HClO3, and H2O2. The latter is preferable as the reduction product is water. However, the oxidation power of H2O2 alone is not sufficient to digest biological material at ambient conditions. Consequently, it must be used at elevated temperatures, high pressure and/or in combination with other oxidation agents. The digestion power and boiling point of H2SO4 is much higher compared with other oxidising agents. This is advantageous as the oxidation power increases with temperature. H2SO4 is therefore often used in combination with other agents to increase the boiling point. However, as H2SO4 is not readily available in high purity it should be avoided for ultra-trace element analysis. In contrast, HNO3 and HClO3 are available in very high purity and create soluble salts (nitrates and chlorates) with most elements of interest. This means that mixtures of HNO3 and HClO3 are very effective for digestion and solubilisation of biological material, but the instability of HClO3 causes undesirable safety risks and it is therefore only used in open digestion systems. HNO3 is especially suitable for digestion of plant tissue as it enables oxidation at room temperature as well as at high temperature and high pressure. For these reasons HNO3 is by far the most frequently used oxidation agent in plant science, either alone or in combination with e.g., H2O2.35,36
Wet digestion may be carried out in open vessels with different heating devises such as hotplates, digestion blocks or microwave ovens, allowing high-throughput digestion.37,38 However, in open-vessel systems, degradation is limited by the boiling point of the oxidation agents. Therefore, only easily digestible plant tissues such as leaves can be fully degraded even when various oxidation agents are used (e.g. HNO3, H2SO4 and HClO3). Furthermore, the use of open-vessels may cause significant losses of volatile elements.37
Even a clear and transparent liquid after digestion may contain residual dissolved organic carbon. This can lead to a cascade of analytical problems, primarily related to instable nebulisation, plasma instability and carbon interferences caused by polyatomic ions (e.g.40Ar12C on 52Cr and 12C15N on 27Al). Tailing of 12C on 11B and carbon enhancement effects may also occur.39,40 In most cases the matrix effects of C can be overcome by implementation of a closed and more efficient digestion system. However, if dissolved organic carbon persist and analytes are present at trace levels, a common practise in ICP-MS analysis is the application of one or more internal standard(s) to correct for the matrix effect of C. The rule of thumb for selecting the most suitable internal standard is a close similarity in mass number and in the first ionisation energy between analyte and internal standard. However, there are plenty of examples in the literature, demonstrating that these guidelines are not always ideal. For example, 9Be is often used as internal standard when analysing 10B and/or 11B in biological samples, but it was found that C+ induces differential charge-transfer mechanisms on B and Be, making Be unsuitable as internal standard at more than 1500 μg μL−1 dissolved organic carbon.41 Likewise, Probst et al.42 found that 107Rh was a suitable internal standard for B despite their distant mass numbers. Thus, the choice of a suitable internal standard is not trivial and should always be carefully evaluated using a suite of relevant certified reference materials to evaluate accuracy after internal standard correction.41
A temperature of 300 °C facilitates a complete HNO3-based oxidation of plant tissue.39 Consequently, closed systems such as microwave ovens or autoclaves that safely combine the effects of high temperature and elevated pressure are required.37,43–46 Such systems have several advantages compared to open systems, including a significant lower risk of contamination and a reduced loss of volatiles.37,47 However, microwave oven digestion bombs often have maximum pressure and temperature limits at ∼40–80 bar and 220–250 °C, respectively. This is mainly due to the PTFE (polytetrafluoroethylene, TEFLON™) polymer used in liners and seals, which degrade and melt above this temperature. Addition of H2O2 improves the oxidation efficiency of HNO3 digestion and reduces the requirement for a high digestion temperature.39 Alternatively, microwave oven and autoclave techniques can be combined. This is for example the case in the Milestone UltraCLAVE® system which creates a high pressure by injection of an inert gas prior to heating samples with microwaves in a large digestion chamber. The boiling point of the acid is thereby increased, enabling digestion without boiling. As a consequence, risks of sample cross contamination are avoided compared to open vessels. However, due to safety issues, such systems are still costly thereby preventing them from being applied as a state-of-the-art technology.48 Further developments within sample digestion for multi-elemental analysis are thus required.
The labour consumption and costs per sample should be reduced while increasing sample throughput and digestion efficiency. Furthermore, plant samples containing high concentrations of elements that are not easily dissolved such as Si, Al and Cr may require special digestion methods. Rice straw for instance requires the addition of hydrogen fluoride (HF), which readily dissolves Si by formation of fluoride complexes.49–51 Unfortunately, HF can dissolve glassware in the sample introduction system of ICP-MS and ICP-OES instruments and cause Si, B and Al based interferences. Therefore, the HF concentration should not exceed 0.2% in the digested sample.13 If higher HF concentrations are required, a HF-inert sample introduction system must be used.
Most frequently, 200–500 mg of homogenised, dry plant material is used for multi-elemental plant tissue analysis.9,52 The large quantity of material significantly reduces the sample throughput as only low capacity rotors for microwave ovens (≤16 samples) can be used.53 Rotors with 24 and 48 vessel positions rotors have recently become commercially available, but they have not yet been implemented to any significant extent in plant science. In addition, the PTFE vessels in these rotors require time consuming steps associated with liquid transfer to autosampler vials prior to analysis. The PTFE vessels are expensive and need to be carefully cleaned between analytical cycles in order to avoid memory effects from the previous sample. For these reasons, sample digestion is usually a bottleneck in high-throughput analysis.
Sample throughput can be improved by a reduction in sample amount, allowing high capacity rotors to be used. A micro-scaling methodology was developed by Hansen et al. (2009)13 using a 64 position rotor to digest 1–20 mg of sample containing very different matrixes rich in proteins, lipids and carbohydrates. The rotor was equipped with standard auto-sampler vials, sealed with specially designed PTFE lip-seals and polyether ether ketone (PEEK) screw caps. Most of the essential plant nutrients could be measured with an accuracy exceeding 90% of certified reference values. This was even accomplished for important volatile elements in plants, such as Se and S. This digestion method is especially attractive for the application of multi-elemental analysis where large sample sets with limited quantity of material, e.g. grain tissue fractions or single seeds, have to be analysed. Digestion of small samples (0.5–3 mg) has previously been performed with several different vial-in-vial setups employing small sample vials fitted into larger digestion vials.54–56 However, when the number of samples increases, this approach is not ideal as only low capacity rotors (≤16 positions) can be used. Moreover, the duty cycle is long as both the outer and inner vial has to be heated and cooled. The high-throughput method developed by Hansen et al. (2009)13 thus represents an improvement compared to traditional digestion methods.
Full-quantitative multi-elemental analysis
For routine high-throughput applications ICP-OES is often considered superior compared to ICP-MS due to lower purchase and running cost, ease of operation, high matrix tolerance and less interference problems. However, as the demand for low detection limits, isotopic information and large linear dynamic concentration ranges is increasing, ICP-MS is becoming a routine analytical technique for commercial as well as scientific purposes. The elemental sensitivity of ICP-MS enables quantification of plant micronutrients present in ultra-trace concentrations. This ability has resulted in significant contributions to the history of plant nutrition such as the discovery and acceptance of Ni as an essential plant nutrient in 1987.57With a dynamic range of 6–7 orders of magnitude, ICP-MS enables multi-elemental quantification of most essential (Table 1) and beneficial plant nutrients. These nutrients are present in plant tissue in the mass range 1–96 m/z with 1H and 96Mo as the extremes. Studies across hundreds of terrestrial plant species have shown that the abundance of elements tend to decrease as a function of atomic mass.10 Since quantification of the elements H, C, N, O and Cl is not feasible with ICP-MS, due to a combination of high ionisation potentials and a high natural background, only 12 essential plant nutrients are included in most routine multi-elemental plant tissue analyses (B, Mg, P, S, K, Ca, Mn, Fe, Ni, Cu, Zn and Mo). These are normally present in concentrations ranging from 0.1 to 55 000 μg g−1 dry matter (Table 1) and are all required by plants in order to complete a full life-cycle.58 The simultaneous detection of these elements is a major advantage in e.g. agriculture and horticulture. Here, plant tissue analyses are frequently used during the growth season to adjust the fertilisation strategy and thereby optimise plant growth and quality.
ICP-MS quantification of several of the essential plant nutrients is challenging due to isobaric and polyatomic spectral interferences such as 40Ar on 40Ca, 38Ar1H on 39K, 40Ar16O on 56Fe, 44Ca16O on 60Ni and 40Ar23Na on 63Cu. Interferences are often found in the mass range between m/z 30–80 which covers the majority of the essential plant nutrients. Careful selection of “interference free” isotopes of each element is therefore essential. This often results in analysis of less abundant isotopes such as 34S to avoid 16O2 interference on 32S. The natural abundance of 34S is approximately 22 times lower compared to 32S thereby causing significant signal loss.28 State-of-the-art ICP-MS quadrupole instruments eliminate most spectral interferences using different variations of collision/reaction cell technologies. Ion-molecule collisions and reactions facilitate the removal of Ar and matrix based polyatomic interferences before entering the mass analyser and thereby increases the analytical accuracy.59 Other ways to eliminate interferences include the use of mathematical corrections/equations or the cool plasma technique which utilises the effect of a reduced radio frequency power on Ar+ formation.60 Furthermore, magnetic sector mass spectrometry has also been used for multi-elemental plant tissue analysis.61–63 This technique eliminates interferences by resolving spectral overlaps thereby avoiding isobaric overlay. However, a higher purchase price and more complicated instrumentation have prevented the introduction of magnetic sector ICP-MS for multi-elemental analysis of plant tissue on a routine basis.
After methods for interference elimination have been optimised, it is essential to validate the obtained multi-elemental results. However, standard additions or isotope dilutions, which are commonly used in single element analysis for verification of analytical results, are unsuitable for multi-elemental analysis. Furthermore, isotope dilutions are not applicable to all essential plant nutrients, because P and Mn are mono-isotopic. In multi-elemental analysis, a careful selection of suitable standards for external calibration is therefore of major importance. The calibration standards should include all elements of interest as well as ensure coverage of the wide concentration range between macro- and micronutrients. It is thus essential to use non-equimolar multi-elemental standards. Unfortunately, such standards are rarely commercially available and should be custom made to match different plant species.9
In full-quantitative ICP-MS or ICP-OES analysis, a 90% accuracy level is often used as data inclusion criteria.9,13,30 This is based on analysis of certified reference material (CRM). It is essential that the reference material matches the unknown sample because the elemental composition of different plant species and tissue types can vary significantly thus introducing different spectral interferences. Ca for example, is often present in 50 times higher concentration in leaves compared to seeds, while Fe is more uniformly distributed in plants.58 The high Ca/Fe ratio in leaves can thus cause 40Ca16O interferences on 56Fe.60 Another example is B which is often found in 5–10 times lower concentrations in monocots compared to dicots. The low B content of monocots combined with residual C from an incomplete digestion can cause tailing from 12C on 11B.64 Furthermore, different plant tissue types may induce varying non-spectral interferences (matrix-induced signal suppression or enhancement), which must be corrected for using internal standards.65 Thus, careful selection of suitable certified reference material is of major importance. However, the availability of plant based reference materials with certified multi-elemental profiles is still limited. To our knowledge it is for example not possible to purchase reference material of the most widely used model plant Arabidopsis with certified multi-elemental profiles. Moreover, for many reference materials, elements are certified in a broad concentration range, i.e. with a large uncertainty. Especially, certified values of trace elements such as Mo and Ni can have 10–40% relative standard deviations which complicate validation of analytical results.
Semi-quantitative multi-elemental analysis
Full-quantitative multi-elemental plant tissue analysis traditionally relies on a number of labour consuming analytical steps, such as the use of external calibration, which is a tedious prerequisite for acquisition of accurate data. As plant species and organs vary significantly regarding matrix composition and content of essential plant nutrients,58 several multi-elemental standards and certified reference materials must be available to enable validation of elemental results for e.g. leaves, fruits, vegetables and seeds. Moreover, accurate quantifications depend on the identification and elimination of spectral interferences which can be time-consuming if different collision and reaction cell gasses are required in the same sequence. Consequently, traditional full-quantitative multi-elemental analysis is often limited to 12–20 elements covering the essential plant nutrients, beneficial nutrients and occasionally elements relevant for e.g. human nutrition such as Co and Se. However, in many studies a more complete coverage of the periodic table is needed. This is currently unattainable with traditional full-quantitative ICP-MS and ICP-OES.Semi-quantitative ICP-MS is a strong alternative to traditional full-quantitative ICP-MS and ICP-OES analysis as it enables rapid assessment of the entire mass range from 7Li to 238U. This has revealed that plants contain traces of most elements of the periodic table, including the essential plant nutrients (Fig. 1). Several applications can benefit from this large amount of elemental information. Semi-quantitative multi-elemental profiles are for example often used for selection of suitable calibration standards and reference materials prior to traditional full-quantitative ICP-MS. The semi-quantitative technique is also suitable as a fast tool to screen and compare the elemental concentrations in unknown samples. This is enabled by recording the signals for most elements and isotopes which then serve as input to a series of mathematical algorithms. The algorithms are adjusted with a pre-calibrated internal standard containing only a few elements. As output, approximate concentrations of all elements in unknown samples are obtained. Semi-quantitative ICP-MS thus relies on a simple calibration procedure and generates quantifications of up to 80 elements in the periodic table.66,67 Analytical results obtained with semi-quantitative ICP-MS have been critically evaluated in several studies with different plant-based certified reference materials such as tomato leaves,68,69 citrus leaves,70 spinach,71 sea lettuce,72 hay70 and wheat flour.9,69 These studies have shown that the accuracy of semi-quantitative analysis is generally better than 70% of the true value for many elements. Furthermore, the data acquisition time is significantly reduced compared to full-quantitative analysis thereby increasing the sample throughput.
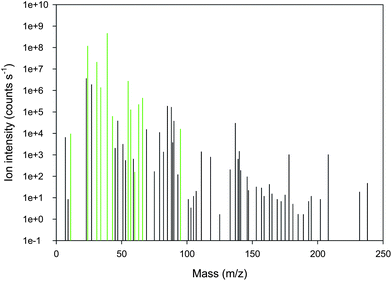 | ||
| Fig. 1 Background subtracted mass spectrum from semi-quantitative ICP-MS analysis of cereal grains showing traces of 62 masses. Essential plant nutrients are shown in green (from left to right: 11B, 24Mg, 31P, 34S, 39K, 43Ca, 55Mn, 57Fe, 60Ni, 63Cu, 66Zn, 95Mo). | ||
Several applications of ICP based atomic spectrometry in plant science require high-throughput and multi-elemental abilities. Despite fulfilling these needs the number of studies using semi-quantitative ICP-MS is still limited. This is partly due to the inherently lower accuracy compared to traditional full-quantitative analysis as well as an inevitable requirement for multivariate statistical methods for data analysis and interpretation. Laursen et al. (2009)9 recently compared full- versus semi-quantitative ICP-MS. In this study, multi-elemental fingerprints were analysed with multivariate statistics in order to discriminate three different rice genotypes. It was shown that the discrimination power of semi-quantitative ICP-MS based fingerprints was superior compared to the full-quantitative despite relying on a simplified calibration procedure. The inclusion of additional elements in semi-quantitative analysis thereby fully compensated for a lower accuracy. This demonstrates that semi-quantitative fingerprints constitute a promising alternative to the traditional full-quantitative procedures, thus representing a powerful tool for e.g. adulteration testing. Jiménez et al. (2007)73 analysed compost samples with semi-quantitative LA-ICP-MS and also concluded that semi-quantitative ICP-MS is a strong and practical alternative to the traditional full-quantitative approach. It is thus evident that semi-quantitative ICP-MS is a promising analytical technique with the potential to significantly reduce costs and labour requirements while providing additional elemental information.
Chemometric data analysis of multi-elemental plant fingerprints
Atomic spectrometry and multivariate data analysis (chemometrics) have developed in parallel during the past decades. Chemometrics enables fast data overviews and discovery of latent data structures even when the number of variables (elements) exceeds the number of samples. This is often the case when multi-elemental fingerprints are generated with ICP-OES or ICP-MS. Consequently, a combination of chemometrics and atomic spectrometry is beneficial for numerous plant based applications.Chemometrics efficiently extract the maximum useful information from large datasets regardless of the number of samples and variables. Some chemometric methods are unsupervised, such as principal component analysis (PCA) and cluster analysis (CA), which require no a priori assumptions regarding structure or likely groupings within the dataset. These methods search for the hidden data structure and are suitable for initial data analysis. This approach is often followed by supervised pattern recognition methods, where information about the class membership of samples to a certain group is utilised in order to classify new unknown samples.74 Some of the most popular methods for supervised pattern recognition in plant science are soft independent modelling of class analogy (SIMCA), linear discriminant analysis (LDA) and partial least squares regression discriminant analysis (PLS-DA).
The combination of multi-elemental plant profiles and chemometrics has been utilised in several studies. Especially, authenticity and adulteration testing of plant based food products have benefited from this strong partnership. The ability to trace the geographical origin of food or reveal adulteration has been shown for e.g., tea,75–78 rice,9,79 welsh onions,80,81 barley,30 hazelnuts,82 potatoes,83,84 pistachios,85 cannabis86 and different fruits and berries.87 These studies have shown that ICP-MS and ICP-OES based multi-elemental plant signatures contain valuable information reflecting the availability of elements in the soil. The geological imprint of the specific geographical location is thereby manifested in multi-elemental fingerprints of plants and is efficiently extracted with chemometrics (Fig. 2). This application is only possible by multi-elemental analysis covering essential as well as non-essential plant nutrients.
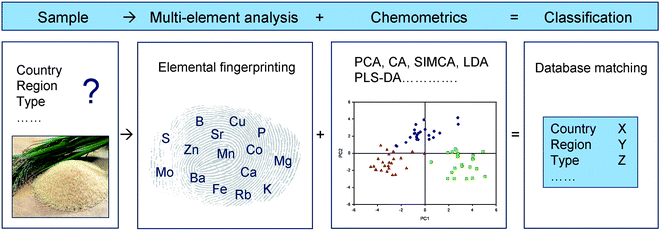 | ||
| Fig. 2 Principles for authenticity testing of plant products using multi-elemental fingerprints and chemometric data analysis. This approach is suitable for e.g., classification of genotypes or plants from different geographical locations (country, region). | ||
Multi-elemental analysis and chemometrics have also been combined in order to discriminate different agricultural production systems such as organic versus conventional. This was tested by Gundersen et al. (2000)61 using high resolution ICP-MS based plant fingerprints consisting of up to 63 elements. Of these, it was shown by principal component analysis that elements non-essential to plants such as Rb, Bi, Dy, Be, Lu, Gd, Y, Ti, Sr, U, Sc, Ba, La, Nd, Co and Ge were important for the discrimination of organic and conventional onions. However, a different elemental profile was necessary to enable discrimination of organic and conventional peas. This illustrates that further investigations across different plant species are required to be able to conclude that the elemental composition of organic and conventional plants is systematically different.
Baxter et al. (2008)88 utilised chemometrics to extract the information contained in ICP-MS based multi-elemental signatures to investigate plant responses to Fe or P nutrition. Multivariate fingerprints consisting of Mn, Co, Zn, Mo and Cd enabled discrimination of Fe deficient and Fe sufficient plants. This approach was more specific than analysis of the shoot Fe concentration. It was therefore suggested that ionomic signatures are suitable for detection of specific physiological responses to environmental or genetic perturbations.
Multi-elemental imaging of plant tissue
The distribution of inorganic elements in plants is characterised by high spatial and temporal variability (e.g. seed fractions, leaves and roots). Tight regulation of gradients in localisation of mineral elements is required for proper functions of plant cells and tissues. To study these regulatory processes, imaging techniques with high spatial resolution and low detection limits are required, enabling imaging of elements in tissue sections, single cells or sub-cellular regions. In situ imaging of trace metals started with histochemical methods more than 140 years ago. Highly sensitive mass spectrometry and synchrotron based techniques have now been developed and are increasingly used in plant science (see e.g. McRae et al. 2009,89 Becker et al. 2010,90 Lombi and Susini 200991).Elemental imaging in plant science have been dominated by synchrotron-based techniques such as proton-induced X-ray emission (PIXE)92 and micro X-ray fluorescence (μ-XRF) analysis.93–100 These studies have provided detailed information on the distribution of elements in specific tissue types, which play a fundamental role in molecular studies aiming at unravelling the mechanisms of loading, storage and unloading of elements in plants.
Imaging techniques based on atomic spectrometry include laser ablation ICP-MS (LA-ICP-MS) and secondary ion mass spectrometry (SIMS). These techniques differ greatly in their analytical performances. For sample vaporisation, atomisation and ionisation, SIMS and LA-ICP-MS utilise either a focused primary ion beam or a laser beam, respectively. Relative to SIMS, LA-ICP-MS has the advantage of a high sensitivity (sub ppm levels), high accuracy and precision. Quantification of analytical data in LA-ICP-MS is simple due to limited matrix effects. Different calibration strategies have been developed and matrix-matched laboratory standards can be prepared from standard reference material spiked with defined concentrations of analytes. SIMS has a better lateral resolution than LA-ICP-MS, but suffers from matrix effects and a high formation rate of polyatomic ions, which complicates quantification.89,90
Moore et al. (2010)101 recently provided a detailed investigation of Se distribution in wheat and As in rice using SIMS. The lateral resolution was <100 nm, which allowed visualisation of a sub-cellular As distribution. Another study revealed high concentrations of Ni in the peripheral regions of leaf hairs (trichomes) and in the epidermal cell layer of Alyssum leaves using high-resolution nano-SIMS.102 Recent technological improvements have enhanced the sensitivity (down to ppm) and resolution (down to 50 nm) of SIMS thus making it a very attractive tool for elemental imaging of plant tissues.89,90
LA-ICP-MS has so far only been used in a few plant based studies. Wirth and co-workers (2009)26 analysed the elemental content in cross sections of rice grains to investigate the effect of genetic engineering of Fe storage proteins (ferritin) and nicotianamine synthase. LA-ICP-MS was also used to study the accumulation and distribution of metals in different tissues of tobacco (Nicotiana tabaccum)103 and the Cu-tolerant hyper-accumulator species Elsholtzia splendens.104 These studies clearly showed that metals are heterogeneously distributed in plant tissues.
LA-ICP-MS and SIMS are more accessible than the synchrotron-based techniques and offer in addition superior sensitivity and potentially full-quantitative data. However, the higher lateral resolution of synchrotron techniques has prevented implementation of LA-ICP-MS as a standard tool for elemental imaging of plant tissue (Fig. 3).89,90 The resolution of LA-ICP-MS is limited by the diameter of the incident laser beam used for ablating the material.105 Resolution at nanometre scale is needed for quantitative analysis of elemental distribution in single cells (radius 10–100 μm) or cell organelles (radius 0.5–10 μm). Becker and co-workers (2006)106 developed a near-field LA-ICP-MS instrument operating at nanometre scale by using a thin silver needle to focus the laser beam on the sample surface. This reduced the crater size to 30–40 nm in diameter compared to 150 nm without the silver tip. The near-field ablation technique has not yet been applied for imaging of plant tissues but it represents a frontline methodology which is likely to mature into a powerful benchtop application for routine elemental imaging of tissue as it provides a resolution comparable to synchrotron-based techniques at much lower costs.
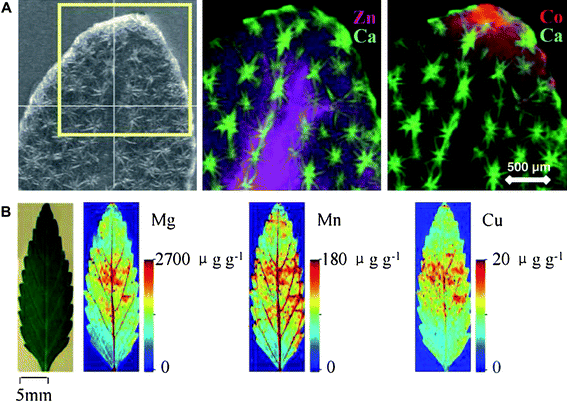 | ||
| Fig. 3 (A) Synchrotron X-ray micro-fluorescence (μ-XRF) images of the Zn and Co distribution in a hydrated Alyssum murale leaf pre-treated with a mixture of Ni, Co and Zn. Reproduced from Tappero et al. (2007),100 with permission. (B) Two-dimensional quantitative images of Mg, Mn and Cu measured by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) in the leaves of Elsholtzia splendens. Reproduced from Wu et al. (2009),104 with permission. μ-XRF is a non-destructive technique with high lateral resolution and moderate sensitivity, whereas LA-ICP-MS has high sensitivity but a relatively low lateral resolution. | ||
Applications of multi-elemental analysis in plant science
It is a continuous challenge within plant science to establish biological functions of elements, genes and proteins. Such knowledge enables engineering of crop plants with improved quality or tolerance to adverse soil conditions. Plant science often relies on well characterised model plants with sequenced genomes such as Arabidopsis, Lotus japonicus or Brachypodium distachyon. In addition, heterologous expression systems based on yeast (Saccharomyces cerevisiae), bacteria (Escherichia coli) or immature frog eggs (Xenopus oocytes) are increasingly used. ICP based atomic spectrometry has successfully complemented these model systems in numerous studies.Multi-elemental analysis of plant mutants
Mutant plants are often used for characterisation of the functional role(s) of a given gene by either reverse or forward genetics. Reverse genetics seeks to reveal what phenotypes arise as a result of particular genes by manipulation of their expression. The latter is achieved by establishing a population of knockout mutants or by gene silencing using RNAi or microRNA expression. Forward genetics attempts to find the genetic basis for a phenotype or trait occurring in a plant population. The functional role of genes of interest is examined by a variety of physiological and molecular techniques, including experiments with contrasting growth conditions verified by multi-elemental analysis of the resulting tissue composition.4,107–109 The elemental composition of plants is controlled by a network of gene products involved in uptake, chelation, sequestration and translocation.110 Many elements may be influenced directly or indirectly by one gene product or clusters of gene products implying that a defect in one specific gene may affect the accumulation of more than one element. This is illustrated by the Si transporter OsLsi1 which has an additional role in As transport.111 Similarly, Fe, Zn, Mn, Cd, Ni and Co share transport proteins.112–115 Down-stream mechanisms and biosynthetic pathways, not directly involved in ion homeostasis, may also affect the ion uptake and translocation. This is exemplified by the TaNAC gene involved in senescence, which also affect the remobilisation rate of Zn and Fe from leaves to seeds in wheat.116 In such cases, a multi-elemental profile is required to assess whether or not a modification in gene expression or protein phosphorylation causes changes in the ionome.The multi-elemental profile of a group of mutated lines within the same plant species can be used as a non-visual phenotypic screen. Based on this approach, an integrated functional genomics platform was recently developed (http://www.ionomicshub.org/home/PiiMS).12 Sixteen elements were analysed in more than 60 000 shoot samples of numerous inbred lines and synthetically mutated Arabidopsis plants. The dataset, consisting of more than 1 million full-quantitative elemental concentrations, was disseminated via an open-accessible web-platform. This platform is especially useful for studying gene networks involved in elemental acquisition and translocation in plants.11 As an example, a screening of Arabidopsis collections for elemental outliers resulted in isolation of lines with low Mo contents. The responsible genetic loci were thereafter characterised by use of association mapping revealing that the low Mo content was linked with a novel mitochondrial Mo transporter (AtMOT1).7 Using a similar approach, Co and Fe efflux transporters were identified.117 Unfortunately, the data in the current Arabidopsis platform is lacking information regarding elemental profiles obtained from plant growth under sub-optimal conditions. Such conditions will have a significant impact on the elemental composition10 and this information would therefore be valuable for identification of new genes that can be used in breeding of plant species with improved ability to grow under sub-optimal growth conditions.
Heterologous expression systems
Heterologous expression systems such as bacteria and yeast are often used to identify the substrate specificity and transport kinetics of transport proteins involved in ion uptake, efflux or intra-cellular translocation.4,113,118–120 As a eukaryote, yeast is often preferred as the expression system for plant membrane proteins. A number of yeast mutant strains are available for the study of elements, such as Zn, Mn, Fe, Cu, Cd, Co, Ca, Mg, K and Na.4,118,121–123 The yeast expression system in combination with atomic spectrometry can be used to screen the substrate specificity of transport proteins across the periodic table. This was done in a study of the multi-specific metal ion transporter AtIRT1 where yeast cells growing in liquid medium were supplied with non-toxic concentrations (0.01 μg mL−1) of 31 trace elements and metalloids (See method in Pedas et al. 20084). Following washing, drying and digestion of the yeast cells, ICP-MS analysis of cells transformed with AtIRT1 or the empty vector control (pFL6.1) revealed that AtIRT1 has the ability to transport Fe, Zn, Mn, Cd and Ni (Fig. 4), thereby confirming previous studies with AtIRT1.112–115 Subsequently, this method was applied for characterisation of the substrate specificity of the first identified Mn transporter, HvIRT1, from a monocot plant species.4 Thus, multi-elemental analysis of yeast complementation assays has proven to be a very valuable tool with respect to characterisation of the substrate specificity and transport kinetics of plant transport proteins.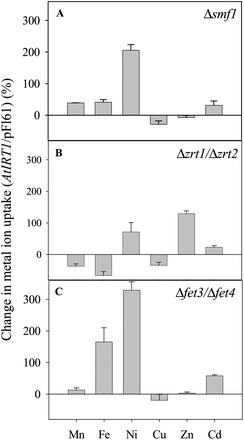 | ||
| Fig. 4 Uptake capacity of Mn, Fe, Ni, Cu, Zn, and Cd in yeast mutants transformed with the gene AtIRT1 which encodes a plasma-membrane localised protein transporting Mn, Fe, Zn and Cd in Arabidopsis. Three yeast mutants were analysed: (A) Δsmf1 (defective in Mn2+ uptake), (B) Δzrt1/Δzrt2 (defective in Zn2+ uptake), and (C) Δfet3/Δfet4 (defective in Fe2+ uptake). Uptake rates were measured over 5.5 h and normalised on the basis of the uptake rates in similar yeast mutants transformed with empty vector (pFL61), see protocol in Pedas et al. (2008).4 Negative values indicate a higher elemental accumulation in empty vector compared to AtIRT1 transformed yeast cells regulated by endogenous yeast genes.4 The experiment was replicated three times with different transformation lines with similar results, and representative data are shown. Data are the means +SE of three independent replicates. | ||
Yeast can be used for screening of plant cDNA libraries in order to identify proteins with a specific function, e.g. transport proteins or chelating ligands.4,124 Extraction of plasmids from viable yeast cells transformed with a cDNA library from Thlaspi caerulescens and grown at toxic Ni levels resulted in identification of a nicotianamine synthase homologue. Orthogonal chromatography, ICP-MS and Q-TOF-MS were used to verify the synthesis of a NA-Ni complex conferring Ni tolerance.125 As NA is not commercially available, this study demonstrates that yeast potentially may be used for production of precious metabolites and ligands.
Part A: Summary
It is evident that ICP based atomic spectrometry has contributed significantly to the scientific developments made in plant science. During the past decades various digestion methods, analytical techniques and data analysis tools have been combined in several ways in order to analyse different plant tissues (Fig. 5). This has resulted in novel scientific applications such as elemental analysis of specific seed compartments.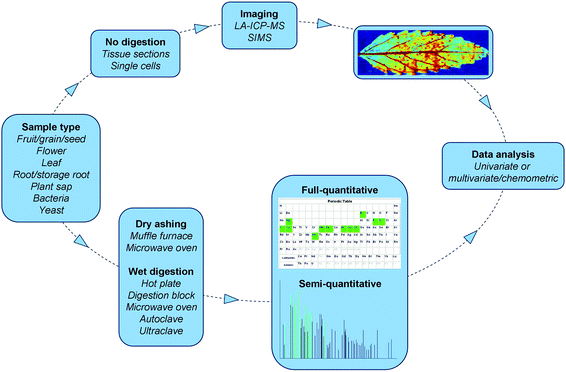 | ||
| Fig. 5 Workflow of atomic spectrometry based multi-elemental analysis in plant science. Imaging picture is from Wu et al. (2009),104 with permission. | ||
The superior sensitivity and multi-elemental capabilities of ICP-MS have also been used for analysis of heterologous expression systems, plant mutants, transformant lines and elite genotypes. This has contributed to a novel mechanistic understanding of the complex network of elements, genes, proteins and metabolites in plants. However, several challenges remain in multi-elemental analysis. Sample digestion is still a bottleneck for screening of large sample sets, such as breeding material and gene bank materials. Innovation in sample digestion procedures for biological materials is therefore urgently required. Data validation is another challenging area, which needs improvement regarding accessibility of certified multi-element reference materials for a much wider range of plant species and plant organs. Currently, too many plant journals publish non-validated elemental data, which seriously hampers interpretation and prevents comparison across studies. Finally, chemometric data analysis should be implemented, preferably as an integrated part of future software packages for ICP instruments. This would allow full utilisation of hidden information in inorganic mass spectra.
Part B. Speciation analysis of plant tissue
Traditionally, atomic spectrometry has been used to determine the identity and concentration of elements in a given sample. However, the determination of total concentrations does not provide information about the bioactive pool of elements. Such information can only be obtained by the analysis of chemical species. The modern speciation analysis aims to identify and quantify one or more chemical species in a sample and asses biologically relevant parameters related to biotoxicity,18 bioavailability28,29 and health promoting effects.126 In the following sections we review the analytical progress that has facilitated analysis of bioactive elemental species in plants.Ion homeostasis and speciation
The free ion activity in the cytosol or organelles of plant cells needs to be balanced at relatively narrow margins in order to support the essential key-functions of the elements. At the same time phytotoxic reactions should be avoided. The molecular speciation of elements, including both essential and non-essential plant nutrients, plays a fundamental role in the homeostatic balancing of the free ion activities in plant cells and in organelles. Finney and O'Hallohan (2003)127 highlighted the importance of speciation in controlling the bioactive pool of trace elements in cells. They showed that the response of several Zn and Cu sensing metalloproteins were in the femto- to zeptomolar range, i.e. concentrations 6–12 orders of magnitude lower than the cellular concentrations of these ions. These values correspond to less than one free Cu and/or Zn ion per cell, clearly illustrating the capacity of the plant cell to minimise the free ion activity. At the cellular level, ion homeostasis involves compartmentation in cell organelles,128 binding to the plasma membrane129 and cell walls,52,130 precipitation of biocrystallites131,132 and speciation with low molecular weight ligands133 as well as large biopolymers.134 Homeostasis at the whole plant level involves several processes, including ion mobilisation in the rhizosphere, ion uptake and exudation, ion storage in roots, translocation from roots to shoot and assimilation into the growing shoot tissue.135 Thus, investigating ion homeostasis in plants is a demanding task, causing significant analytical challenges.Extraction and stability of elemental species in plant tissues
The use of in situ vs. ex situ approaches to speciation analysis is a matter of much debate. Apart from LA-ICP-MS techniques, ICP-MS based speciation relies on ex situ analysis, where the elemental species have been extracted from the plant tissue into a liquid or gaseous phase. The most critical steps in such analyses are sample preparation and extraction of analyte species.136 Plant tissues constitute no exception to this rule.137–139 The plant cell contains species of highly variable kinetic and thermodynamic stabilities. Some species are chemically stable and can be recovered from the tissue without significant changes in composition. However, it must be realised that a significant proportion of the elemental species may degrade, dissociate or be exposed to ligand exchange already during the first seconds of tissue homogenisation, generating artefacts or loss of important species.140,141Many metallobiomolecules exhibit chemical polymorphism142 which denotes the ability of a ligand to form coordination complexes with different ions when exposed to the multi-ion environment in the fluid component of plant cells.143 This makes the analysis of e.g. Cys and His rich peptides in biological tissues a major challenge because these amino acids have a high affinity for cationic transition elements.144,145 Furthermore, during extraction a wide range of enzymes such as proteases, amylases, pectinases, cellulases, lipases and phosphatases are being released from the disintegrated cellular compartments and vesicles of the cell. As a result, the original species composition may change due to the activity of the enzymes present in a given extraction mixture. Some species are highly unstable when exposed to O2, making extraction in an inert atmosphere consisting of N2 or Ar mandatory.145 Furthermore, several species such as Cd and Zn may be occluded in bio-crystallites in vacuoles or co-precipitated with polysaccharides and/or storage proteins in specific tissues.26 Thus, plant cells contain species of highly variable solubility for which reason the choice of extractant(s) is important.28 The vacuole is significantly more acidic (pH ∼5.5) than the cytosol (pH ∼7.5), making pH of the solvents a principal parameter to optimise in the extraction procedure. In addition, the following key parameters must be carefully considered: solvent, buffer composition, ionic strength, temperature, atmospheric composition during extraction, addition of reducing agents and enzyme inhibitors, homogenisation procedure and the extraction time needed to recover the intact species from the extract. A reproducible and biologically meaningful speciation analysis of plant tissue can only be undertaken if the extraction procedure is critically optimised for the species of interest. Validation of the extraction is often impossible due to the lack of suitable certified reference materials. However, certified reference materials for Se species are now available for selenised yeast146 but unfortunately this is not the case for plant tissues. Consequently, the lack of reference materials represents a considerable shortcoming in validation of the extraction procedure for most elements of interest.
Removal of lipids from plant tissue prior to speciation analysis is usually only performed for oil-rich tissues from seeds or nuts. Typically, the lipid rich tissue is extracted with organic solvents such as a chloroform–ethanol mixture150 or petroleum ether.151 Sussulini et al. (2007)152 evaluated two de-lipidation procedures and observed major differences in the ability to preserve metal binding proteins extracted from soybean using hexane or petroleum ether. The largest differences were found for K, Mg and Ca binding, whereas Cu and Mn were only marginally affected.
State-of-the-art extraction procedures
The most frequently used procedure for homogenisation and extraction of species from plant material involves an initial step in which fresh or freeze-dried material is pulverised in liquid N2 at 77 K using an acid-rinsed mortar and a pestle.20,133,143,153–155 This approach has been applied to species of highly variable chemical identities, such as: glutathione (GSH), phytochelatins (PCs) and metallothioneins (MTs),155,156 nicotianamine (NA),125 pectin,157 selenocysteine (SeCys)-derivatives158 and various As149 and Tl-species.22 Other approaches have been based on a hydraulic press to collect root and leaf sap159 or grinding of e.g. apples and carrots in a blender.160The solvents used for extraction of species from plant tissue span from de-ionised water20,161 to highly complex solvents containing several buffers, electrolytes, reducing agents, various enzyme inhibitors and antibacterial agents.162,163 Amino acid and peptide based coordination complexes involving NA, GSH, PC and MT ligands, are among the most frequently studied in plant tissues and are usually extracted in dilute aqueous (mM) buffers such as PBS, TRIS-HCl or ammonium acetate adjusted to a pH between 5.5 and 7.5. A small amount of acid rinsed quartz sand is often added to the mortar in order to facilitate cell disruption and increase the extractability.143,164 The extraction efficiency may also be improved by continuous stirring, shaking or rotation for minutes143 or several hours133 at temperatures ranging from ice-cold to heated conditions at 50–90 °C.161 Subsequently, samples are pelleted by centrifugation, the supernatant filtered and very often lyophilized before the speciation analysis is initiated.125,155
Ultrasound probes or baths are often applied in order to shorten the time needed for efficient extraction.19,149 Fang et al. (2009)165 showed that a reduction in extraction time from 20 to 5 h using ultrasound, prevented the oxidation of selenomehionine (SeMet) to SeOMet in rice grains. This agrees with Larsen et al. (2003)166 concluding that sonication generally reduces the risk of oxidisation. Therefore, ultrasound assisted extraction is frequently used in As and Se speciation studies as it decreases extraction time without changing species composition. This appears to be the case for Se-amino acids167 and methylated As species168 even under conditions where the solvent is slightly heated due to the sonication procedure, implying that these species are relatively stable during extraction. The same is definitely not the case for most peptide based coordination complexes such as GSH, PC and MT where great care must be taken to minimise disintegration at all steps from homogenisation to final analysis. This typically involves sample preparation under an inert atmosphere (N2 or Ar) in combination with ice-cold and N2-sparged extractants with or without reducing agents such as β-mercaptoethanol, dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP).155,160,169
Often different species of the same element can not be quantitatively extracted by use of only one extractant, primarily due to differences in solubility and surface affinities towards cell organelles and biopolymers (primarily proteins and carbohydrates). To solve this problem, sequential170,164 as well as parallel extraction techniques140,165,168 have been developed. In a sequential extraction, a series of different extractants are applied to the same sample in order to sub-divide the total element content and/or the content of individual species in predefined solvent fractions. The sequential extractions typically starts with mild extractants such as water or dilute salt solutions followed by targeted steps involving enzymes or organic solvents, and ending with very harsh conditions using strong acids or bases. Sequential extractions have been implemented on Se species from garlic170 and Cd species from Arabidopsis.164 The Se species from garlic were first extracted in water and the remaining pellet exposed to 5 sequential steps, (i) lysis of cell-walls with a mixture of cellulose, protease, chitinase and glucanase; (ii) proteolysis with protease XIV; (iii) 0.1 M HCl; (iv) 1 M Na2SO3 (pH 7) and (v) 25% CS2 solution. This procedure enabled extraction of 90% of the total Se species which were assigned to water soluble, cell-wall bound, protein bound, elemental and selenide Se fractions. The dominating species were elemental Se°, selenate and selenite, accounting together for nearly 95% of the extracted Se. This finding markedly contrasts with previous reports on Se speciation in lyophilised garlic by Larsen et al. (2006).140 Using a parallel extraction procedure with aqueous hydroxylamine (aliinase enzyme inhibitor) or proteolysis (protease XIV), these authors found that proteolysis enabled an extraction efficiency of nearly 100%. In the following speciation analysis it was found that inorganic Se species accounted for less than 10% and that 67% was γ-glutamyl Se-methyl-selenocysteine (γ-Se-MeSeCys).
It must be noted over the last decades there has been remarkably little development in extraction procedures for speciation analysis of plant tissues. The main reason for this is probably that speciation analysis is still a relatively new application which has yet to be developed to a stage where fast and efficient extraction procedures are prioritised in plant science. Examples of automated extraction techniques are accelerated solvent extraction (ASE) and microwave-assisted extraction, which use a combination of pressure and heat to increase the extractability. ASE has been used to extract As species from carrots171 and microwave extraction in combination with degrading enzymes have been used for Se and As species in rice,172 oilseed rape and Arabidopsis.173
Size exclusion chromatography (SEC) is frequently used as the first analytical step following extraction. It is used to fractionate and isolate the species of interest, using ICP-MS as a sensitive elemental detector, either off- or on-line with the chromatographic procedure. Size-exclusion chromatography is a gentle fractionation technique, based primarily on a molecular sieve effect separating molecules according to size and to some extent also on the shape of molecules. One of the most frequently used stationary phases in SEC is cross-linked agarose-dextran (for example the Superdex Peptide 10/300 GL, Amersham Biosciences) with an optimum separation range between 100 and 7000 Da. However, SEC columns for fractionation of biomolecules with molecular sizes up to 500 kDa are available on the market from several commercial suppliers. Even agarose-dextran based SEC columns may induce some dissociation of labile coordination complexes during separation steps, which typically last from 20 to 40 min. This may lead to poor column recovery for labile species and also to a marked risk of ligand exchange with free ions trapped on the residual charges of the agarose-dextran stationary phase.143,153 Consequently, residual elements need to be rigorously removed in order to obtain reproducible results. Persson et al. (2006)143 did for example use repetitive injections of EDTA between every sample to obtain highly reproducible fractionation of Cd-PC complexes.
Following the SEC-ICP-MS species fractionation, second dimension or orthogonal chromatography is often performed on the collected fractions in order to identify the ligands and/or intact elemental species. Reverse phase separation and subsequent MS identification of different PC ligands are usually performed using an acidic mobile phase, e.g. a gradient of formic acid/methanol or formic acid/acetonitrile. This frequently leads to a marked dissociation of the metal-PC complexes, increasing the risk of ligand exchange and artefact formation and caution in the interpretation of data should be taken at this step.
Xuan et al. (2006)175 investigated the stability of phytosiderophore (PS) species during zwitterionic hydrophilic chromatography (ZIC-HILIC) coupled to ESI-MS. Small metal-PS complexes, especially Fe containing ones, may dissociate or oxidise during chromatography, leading to ligand exchange and irreproducible retention times in the subsequent analyses. An EDTA injection was used to check the stability of Fe-PS species. In case of no dissociation, a peak of the pure EDTA ligand (m/z 290.9) was observed, whereas a Fe-EDTA peak (m/z 344.0) appeared as an indication of residual Fe trapped in the column due to species destabilisation. The study also showed that a fast, isocratic chromatographic separation could decrease the amount of dissociated Fe-PS species.175
State-of-the-art hyphenated techniques
Hyphenated ICP-MS techniques have gained increasing importance in plant science during the last decade due to their superior elemental sensitivity, high matrix tolerance and easy coupling to various chromatographic techniques. Under ideal conditions, ICP-MS detection limits are at the part per quadrillion level,178 which makes hyphenated ICP-MS an ideal tool for the initial screening of fractions rich in one or more elemental ions (multi-elemental species fractionation). Combined with molecular mass spectrometry, it becomes possible to take full advantage of the unmatched elemental sensitivity of ICP-MS, while at the same time using the power of molecular mass spectrometry to obtain structural information on metallo-biomolecules in plants (Fig. 6).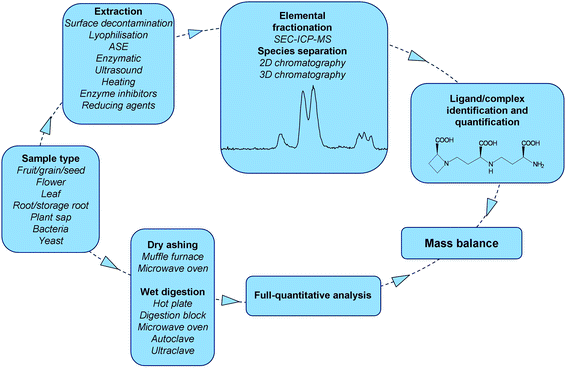 | ||
| Fig. 6 Workflow of parallel speciation and full-quantitative analysis. The extraction is followed by elemental fractionation and complex/ligand identification and quantification. The quantified species should eventually be presented in a mass balance to determine their accumulated amount relative to the total. | ||
The resolution of SEC is usually not sufficient to enable separation of individual species varying in sizes with less than a few hundred Da. This leads to co-elution of species which prevents a reliable identification and quantification. Hence, due to the poor resolution of SEC (typically H−1 < 35.000 m−1), one or more orthogonal dimensions using liquid chromatography columns with higher resolution such as ion exchange (IE), reverse phase (RP) (including ion pairing chromatography), hydrophilic interaction liquid chromatography (HILIC) or capillary zone electrophoresis (CZE) have been applied (see methodological reviews by: Szpunar and Lobinski 2002;180 Sanz-Medel et al. 2003;134 Meija et al. 2005;181 Lobinski et al. 2006;182 Mounicou et al. 2009).15 Among the orthogonal chromatographic procedures, RP-HPLC or CZE, using ESI-MS for structural identification, has been extensively used for the analysis of metallo-PC complexes, especially focusing on Cd143,153–155,164,174 and As.137,139 In addition, the orthogonal SEC/RP principle has been used on quite diverse phyto- metallo-biomolecules such as Ni-NA,20,125 Se-peptides,158,183 rare earth elements (REE) binding proteins184 and in multi-elemental speciation analysis of e.g. Fe, Cu, Zn binding peptides from cereals.28,176 Dernovics et al. (2007)185 used multi-dimensional SEC-/IE-/RP-nanoHPLC with ICP-MS detection to isolate three Se species from Lecythis minor (monkey pot nuts) which subsequently were identified as two isoforms of γ-glutamyl-selenocystathione and selenocystathione using standard-less ESI-Q-TOF-MS/MS.
Bioactivity and bioavailability
The total concentration of a given element provides limited information on the biological activity that is to be expected in planta or upon human consumption of the plant parts. ICP-MS based speciation analyses combined with molecular mass spectrometry have significantly improved the understanding of nutritional and health promoting effects of different Se species, the bioavailability of essential micronutrients like Fe and Zn as well as the toxicity of As and Cd. Selected cases to illustrate this will be presented below.NA is ubiquitously found in all higher plants and plays a non-redundant role in both ion transport and homeostasis.112,194,196–198 The NA deficient tomato mutant chloronerva has been intensively used as a model plant in this context, since it is unable to translocate Fe and Zn to the growing tissue which leads to chlorosis.199 By over-expression of the NA aminotransferase gene (NAAT) in tobacco plants, NA was quantitatively converted to phytosiderophores (PS) which eventually were excreted by the roots. Hence, an artificial NA deficiency was induced, which resulted in lowered contents of Fe, Zn, Cu and Mn in the developing leaves.200 Moreover, these plants were unable to flower, which illustrates the importance of NA for translocation and efficient assimilation of cationic transition elements. By over-expressing NA synthase (NAS), one of the genes controlling NA synthesis, Zn and Fe levels in tobacco plants were 2.5 and 1.9 times higher, respectively, compared to the wild type plants. Similar results were obtained in rice, where over-expression of NAS has been used to improve the content and bioavailability of Fe and Zn in the endosperm.26,29,201–203
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric complexes is only 0.9841 amu, and as the complexes are detected as [M-3H + Fe(II)] and [M-4H + Fe(III)], this means that the difference between Fe(II)-NA and Fe(III)2′DMA is only 0.0239 amu, representing an analytical challenge which was resolved using the mass accuracy of ESI-FTICR-MS.204
:1 stoichiometric complexes is only 0.9841 amu, and as the complexes are detected as [M-3H + Fe(II)] and [M-4H + Fe(III)], this means that the difference between Fe(II)-NA and Fe(III)2′DMA is only 0.0239 amu, representing an analytical challenge which was resolved using the mass accuracy of ESI-FTICR-MS.204
NA coordination complexes have only been identified in a very few cases, the limitations mainly caused by analytical challenges related to stability during extraction and separation rather than by limits of detection. A Zn-NA complex in yeast strains over-expressing the NAS gene was recently identified using EXAFS and XANES.192 Pioneering analytical work on native, intact NA complexes in plants has mainly been performed with ICP-MS based techniques, applied on hyper-accumulating species. Sebertia acuminata is a rare endemic tree found in New Caledonia, locally known as Sevé bleue (blue sap). It can accumulate as much as 20% Ni in the latex DM fraction which is the highest Ni concentration ever measured in a living organism.205 Nickel coordination complexes from this tree were investigated by Schaumlöffel et al. (2003)20 who used SEC-ICP-MS for screening and preparative collection of Ni species in the latex, followed by orthogonal separation by CZE-ICP-MS and structure elucidation by ESI-MS/MS. It was found that Ni-citrate was the dominating Ni species and that only 0.3% of the Ni was complexed to NA. Thlaspi caerulescens is a hyperaccumulator able to accumulate large amounts (>0.1% DM) of Cd, Zn and Ni in its leaves without displaying toxicity symptoms. Vacchina et al. (2003)125 exposed plants of this species to elevated Ni concentrations in hydroponics and isolated the Ni rich fractions from the root tissue by SEC-ICP-MS. The fractions were pooled, lyophilised, dissolved in water and further analysed by direct injection ESI-MS/MS, showing a Ni-complex at m/z 360 to be the dominating complex. Extraction of Ni-NA from a functional yeast complementation assay, using a Ni resistant strain, produced a mass spectrum identical to the one obtained from the plants, confirming the identity of Ni-NA and its role in hypertolerance. This approach was subsequently refined using orthogonal separation of several other Ni coordination complexes from T. caerulescens by SEC-/HILIC-ICP-MS.133 The Ni-rich HILIC fractions were analysed by direct injection using ESI-Q-TOF-MS. Apart from the anticipated Ni-NA complex, this approach also allowed identification of malate, citrate and His Ni-complexes as well as a Ni-EDTA complex originating from the hydroponic cultivation. The transport routes of Ni-NA complexes in plants were further analysed and described by Mari et al. (2006).206 Using a ZIC-HILIC separation method coupled to ESI-MS, Xuan et al. (2006)175 and Weber et al. (2008)159 detected Ni-NA in the xylem sap of Arabidopsis and Cu-2′DMA and Zn-2′DMA in press sap from Fe deficient wheat roots. Unlike conventional HILIC columns, the stationary phase of the ZIC-HILIC column has a sulfobetaine-type zwitterionic functionality, covalently attached to porous silica, which facilitates hydrophilic partitioning as well as weak electrostatic interactions with either positive or negative charges.175 The same group also used a special zwitterionic capillary CE-capillary for the separation of native NA and 2′DMA-metal complexes in wheat and Arabidopsis.207
Bioavailability of plant derived Fe and Zn
Analysing the bioavailability of micronutrients in plant-based foods is challenging due to the large number of factors potentially influencing human uptake. The degree of bioavailability involves not only the native chemical binding forms of the element, but also depends on diet composition, food processing and ligand exchange reactions between metals and ligands in the stomach and intestines. Human physiology factors like pregnancy, age, nutritional status and co-ingestion of pharmaceutical drugs may also have an impact on the net uptake of certain micronutrients.208,209Based on average values from legume and cereal seeds, only 5% Fe and 25% Zn of the total concentrations are present in a bioavailable form in the edible plant parts.210,211 Hence, the analytical focus for essential micronutrients is changing from quantification of total element concentrations to analysis of their speciation, i.e. the amount of bioavailable species of a nutrient in a given sample. Whereas ICP-MS have frequently been used to analyse stable isotopes in absorption trials,212 speciation analyses involving elemental and molecular speciation techniques have only recently been applied to essential plant micronutrients.
Ferritin is a Fe storage protein in the endosperm and has been a potential target for biotechnological biofortification. Plant ferritin is mainly composed of 28 kDa subunits which aggregate in a 540 kDa protein complex, with the ability to store as many as 4500 Fe atoms.220 Wirth et al. (2009)26 increased the Fe concentration in the rice endosperm 6-fold by co-expression of a NA synthase gene from Arabidopsis (AtNAS1) and the ferritine gene from bean. Seeds were analysed by LA-ICP-MS and μ-XRF, showing a heterogeneous Fe distribution, including some hotspots, supposedly dominated by precipitated Fe-ferritin species. Using a comparable transgenic approach, Lee et al. (2009)29 constitutively over-expressed the NA synthase gene OsNAS3 in rice, which resulted in a 2.6-fold increase of Fe and a 2.2-fold increase of Zn in the endosperm. Interestingly, it was found that these transformed rice seeds could ameliorate Fe deficiency in mice within 2 weeks. The polished rice seeds were extracted in 50 mM TRIS-HCl (pH 7.5) and the subsequent SEC-ICP-MS analysis showed that the transformant and the wild type contained equal amounts of Fe bound to IP6, whereas Fe bound in a low molecular weight Fe-NA complex was 7-fold higher in the transformed rice. This difference in speciation was most likely linked to the superior Fe bioavailability in these seeds.29 In a following study, Lee at al. (2011)195 selected a rice line over-expressing OsNAS2 and showed that these plants accumulated 2.5-fold more Zn and 20-fold more NA in the endosperm. When analysed by SEC-ICP-MS and orthogonal HILIC-ESI-TOF-MS it was found that the transformant synthesised Zn-NA and Zn-2′DMA, which was not present in the wild type (Fig. 8). The total amount of speciated Zn was increased 16-fold which indicates that the marked increase in NA levels may induce ligand exchange mobilizing Zn from otherwise insoluble Zn species. Zn deficient mice fed with transgenic seeds for two weeks recovered to normal Zn levels in blood plasma, whereas mice fed with the wild type rice failed to recover, indicating a higher bioavailable pool of Zn in the transgenic rice lines.195 However, it is worth noting that the mere presence of NA may be responsible for an increase in mineral bioavailability from plants with increased NA synthesis, as was recently shown in an in vitro Caco-2 cell model simulating the human digestive system.203
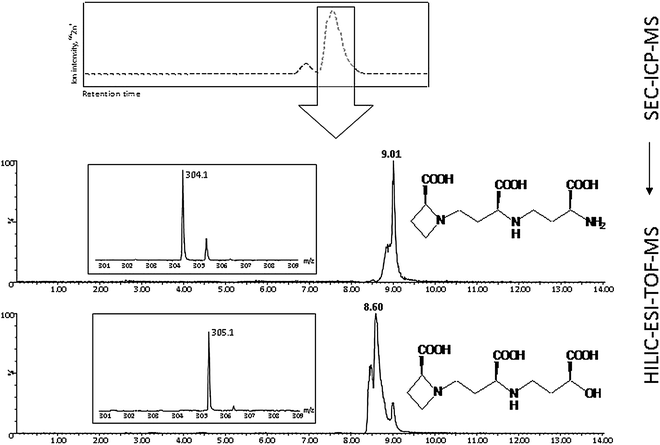 | ||
| Fig. 8 Elemental Zn fractionation by SEC-ICP-MS, followed by nicotianamine (NA: m/z 304.1566) and 2′deoxy mugineic acid (2′DMA: m/z 305.1363) ligand identification by second dimension HILIC-chromatography coupled to ESI-TOF-MS. Modified from Lee et al. (2011).195 | ||
Se speciation in plants and human health
Selenium (Se) is characterised as a non-essential plant nutrient, but it is evident that Se has a beneficial role in the metabolism of many higher plants (see part A). Being an essential element to humans and animals, the ability of vascular plants to accumulate and transform Se into bioactive species has important implications for human health.23,229,230For the identification of Se species, four simultaneous approaches have been taken in order to improve the quality of the hyphenated ICP-MS techniques: i) development of efficient extraction procedures; ii) improvement of the chromatographic resolution for accurate peak identification, for example by RPC, IEC, IPC or HILIC iii) development of complementary molecular mass spectrometry techniques for standard-less peak identification; iv) incorporation of classical proteomic approaches, including analysis of gels and tryptic digests of seleno proteins.
Gas chromatography has frequently been applied in order to analyse volatile Se compounds in e.g. garlic.231 However, since most Se species in plants are non-volatile,161 LC has generally been preferred.126 However, peaks in both first and second dimensional chromatography (typically SEC followed by RPC) often contain more than one Se species and proper Se standards matching retention times for many Se analytes occurring in the chromatograms are rarely available. To date, RPC,161 IPC using TFA or HFBA as ion-pairing agents,232 and IEC233 have been the most frequently used techniques for second dimension chromatography of Se species.
The multi-dimensional chromatographic approach to species separation is necessary not only to purify the Se compounds but also to remove matrix components that may interfere with the molecular identification. The growing interest in identification of both low molecular weight Se species and seleno proteins has led to the implementation of proteomic approaches, including separation by 1D and 2D gel electrophoresis and trypsin digests followed by MALDI-TOF-MS, LA-ICP-MS and/or LC-ESI-MS/MS.234 However, as most seleno proteins present in plant tissue remain to be identified, these techniques still face substantial analytical challenges before being integrated as a proteomic tools in plant science.
The chemical similarity between sulfate and selenate raises the question of whether selenate may be reduced by the same enzymes as sulfate. In vitro studies have shown that the major enzyme in this pathway, ATP sulfurylase, indeed can accept selenate as its substrate, forming adenosine 5′-phosphoselenate (APSe). After reduction, APSe may be non-enzymatically transformed into selenide or selenopersulfide (GSSeH) by GSH, and may subsequently enter into the metabolic pathway.126 Here, SeCys is a key metabolite for the synthesis of other seleno amino acids and volatile compounds. In animals, seleno phosphate acts as a Se donor for SeCys synthesis. Whether or not seleno phosphate also exists in plants remain to be resolved. Interestingly, more Se species have been identified in plants than in animals,126 even though Se is a non-essential plant nutrient. However, there is some evidence that plants may have evolved specific Se enzymes which serve non-redundant functions in plants,1 implying that the classification of Se as a non-essential plant nutrient might need re-evaluation in the future.
In general, the major Se species in plants are inorganic selenate (Se(VI)) and selenite (Se(IV)), organic forms like SeMet, SeCys, Se-methyl-selenocysteine (Se-MeSeCys) and γ-glutamyl Se-methyl-selenocysteine (γ-Se-MeSeCys) or seleno proteins where SeMet and SeCys have been incorporated in place of Met and Cys. Plants may also volatilise Se as dimethyl-selenide or dimethyl-diselenide.23,24,238 Many other identified Se-species exist in plants, see for example Casiot et al. (1999)233 and Dernovics et al. (2007),239 Larsen et al. (2006)140 and the continuous progress in the development of sophisticated analytical methods implies that yet many others remain to be identified.
Hence, the composition of plant species in the human diet has a major impact on food Se concentrations. The Se concentration range between toxicity and deficiency is narrow in plant based foods. In general, consumption of foodstuffs with a concentration above 1 mg kg−1 DW is considered toxic, whereas a concentration below 0.1 mg kg−1 DW may induce deficiency.231,237 On a global scale, the Se concentrations differ widely among different soils, having a decisive influence on local plant food Se concentrations as well as those in the urine, serum and breast milk of people living on these soils.237 In the UK it was recently estimated that wheat contributes to 22% of the dietary Se intake.240
Organic, plant derived Se species seem to have different efficacy with respect to their anti-carcinogenic properties.245 In mice trials, for example, it was shown that the presence of some low molecular weight seleno compounds in garlic could reduce colon cancer and mammary tumours in rats.242 These anti-carcinogenic effects are believed to be related to the presence of the CH3SeH metabolite, which can be formed directly from Se-MeSeCys or γ-Se-MeSeCys by the action of β-lyase.161,245 The identification of these compounds was pioneered by Casiot et al. (1999)233 and Kotrebai et al. (1999),232 using SEC and orthogonal IEC or IPC facilitated by perflourinated carboxylic acid ion pairing agents, in combination with HPLC-ESI-MS. Following two-dimensional HPLC, including SEC and RPC, McSheehy et al. (2000)161 introduced the use of ESI tandem mass detection which enabled identification of γ-Se-MeSeCys without the need for an authentic standard. Along with the analytical progress, identification and quantification of beneficial Se species in plants have gained enormous interest. The progress of these efforts was compiled by Rayman et al. (2008).24 From this review it can be stated that Se-MeSeCys and γ-Se-MeSeCys, as well as a range of other Se compounds, are found in various proportions in a number of edible plants including cereals, nuts, garlic, onion, broccoli and many other species.24 The concentration range of low molecular weight Se compounds reported in plants is still incomplete. In garlic, for example, the reported contents of Se-MeSeCys and γ-Se-MeSeCys ranged from 5 to more than 90% of the total Se in two different investigations.161,170
Foliar Se application may be used to increase the Se concentration in a range of food plants, for example in carrots where SeMet and γ-Se-MeSeCys were shown to be the dominating species.246 Se uptake is also influenced by the symbiotic fungi mychorriza, which upon addition to the soil was shown to increase Se uptake in garlic tenfold. However, the addition of mychorriza did not change the distribution of Se species.140
The total Se content of the plant growth media may affect not only the total uptake, but also the distribution of Se species, as was shown by Kotrebai et al. (2000).247 In this study, the absolute content of organic Se compounds in garlic increased along with increasing Se supplementation and at tissue concentrations above 300 μg g−1 DW, Se-MeSeCys was found to be the dominant species. Furthermore, the distribution of Se species is remarkably different depending on the inorganic Se species used for plant cultivation, as was reviewed by Pyrzynska (2009)230 and Zhu et al. (2009).235
Of major concern to the bioactivity of Se is also whether the beneficial compounds remain active and accessible after consumption. Hence, in vitro gastrointestinal digestion studies of onions, chives, garlic, Brazil nuts (Bertholletia excelsa) and Se-supplements have been investigated, using HPLC-ICP-MS and HPLC-ESI-MS/MS.237,248 Dumont et al. (2006)237 concluded that the SeMet, SeCys2 and Se-MeSeCys remained stable under these conditions, and that γ-Se-MeSeCys lost its glutamine part.
Brazil nuts are known for their high Se concentration. These nuts were found to prevent mammary cancer in rats, possibly by the action of increased Se retention in the liver, kidney, mammary gland and plasma,249 but these characteristics could not be attributed to γ-Se-MeSeCys or Se-MeSeCys, since the low molecular weight fraction was dominated by SeMet, selenoethionine and selenocystine-selenomethionine.250 Furthermore, 15 novel seleno proteins were characterised by ICP-MS assisted nanoHPLC-ESI-Q-TOF-MS/MS in these nuts, again illustrating the complexity and variety of Se speciation in plant derived foods.185
Developments in speciation analysis methodology have made major scientific contributions, enabling sensitive and accurate quantification as well as structural characterisation of bioactive Se species. However, the majority of the accumulated data is still to be regarded as tentative as it relies on retention time matching with standards or inconclusive MS identification.24 Speciation analyses have mapped the major biosynthetic pathways of Se compounds in plants, but many gaps in knowledge persist regarding the complexity of seleno protein synthesis and degradation. Hence, the absolute contents of cancer protective and health promoting Se compounds and the factors determining the dominating Se species under certain circumstances still require intensive research.
Speciation and elemental toxicity
Terrestrial plants have developed defence systems in order to cope with exposure to toxic elements like As, Hg and Cd.251–253 Hyperaccumulating plants have the unique ability to store elevated concentrations of elements in their tissues. These plants may accumulate toxic metals/metalloids many orders of magnitude higher than their non-accumulating relatives, as was reviewed by Verbruggen et al. (2009).254 However, hyper-accumulation is not the common defence mechanism that plants use against toxic element concentrations. Instead, plants as well as algae, bacteria, yeast and some fungi, generally defend themselves by inducing the synthesis of detoxifying peptides known as phytochelatins (PCs), first identified and characterised by Grill et al. (1989).255 PCs are polymeric units of reduced GSH, which is the precursor of all PCs. PCs have the general structure (γ-Glu-Cys)n-X, where n = 2–11 and X may be glycine, β-alanine, serine, glutamate, glutamine or missing. Another variation is lack of γ-Glu linked N-terminal (des-γ-Glu-PCs). To date seven families of PCs which vary among plant species have been described.143,164 All PCs chelate metals and function similarly by forming oligopeptide based coordination complexes. PCs have the highest affinity for the thiophilic Cd ion, which becomes approximately 1000 times less toxic to most plant enzymes when bound in a PC complex.256 Besides PCs, plants synthesise metallothioneins (MTs), which are proteins that may contribute to metal detoxification (see section below), but their exact role(-s) in planta remains unclear.The first native plant MT to be isolated was an early cysteine labeled protein (Ec), purified from mature wheat embryos.271–274 The Ec protein, belonging to the MT4 subgroup, was found to be Zn binding. Despite being discovered more than 25 years ago, only a few MT proteins have been purified from plant tissues since then. In Arabidopsis, partial protein sequences from the MT1, MT2 and MT3 subgroups have been obtained using sequential SEC, Cu and thiol affinity chromatography.275 Recently, a proteomic approach directed towards translational responses to Cu stress in rice embryos enabled identification of an MT protein (MT2c) in a pool of Cu inducible proteins.310
Multi-elemental characterisation of plant lines with genetically altered MT expression has been used to investigate the homeostatic functions of MTs in planta. A study of transgenic Arabidopsis over-expressing the seed-specific MT4, a homolog to the wheat Ec, found that roots and leaves of mature plants accumulated Cu, but not Zn when compared to the wild type plants. However, the ectopic expression of MT4 in leaves and roots rendered the plants tolerant to both Cu and Zn when applied in excess.277 Conversely, Arabidopsis RNAi plants with MT1 expression reduced to 5–10% of the wild type level were Cd sensitive. Multi-elemental ICP-MS analysis revealed that the MT1 RNAi plants growing on a Cd media accumulated less Cd and Zn compared to the wild type whereas the Cu levels were unaffected.278 In rice, expression of the OsMT1a gene was induced by Zn but not by Mn, Fe and Cu. Upon over-expression of the same gene, Zn accumulation in both vegetative parts and grains increased by 44–54% compared to the wild type.276 The above mentioned research illustrates that some plant MTs are involved in Zn and Cu homeostasis as well as in Cd detoxification. In planta there appears to be a remarkable discrepancy between mRNA and protein levels, since MT transcripts are generally highly abundant in all tissues whereas protein levels tend to be very low. The reasons for this low protein abundance are not resolved but may be related to a high protein turn-over, post-translational modifications or a confined expression of MT proteins in highly specialised cells or plant structures.275 In contrast to vertebrate MTs, plant MTs are characterised by having so-called linker regions devoid of Cys residues. These regions appear to be highly susceptible to proteolytic attack from endogenous plant proteases, making them unstable during extraction and purification processes.279 Thus, due to the difficulties in extracting sufficient amounts of intact MT proteins from plant tissue, most information on their functional roles have been obtained from gene expression studies. Recombinant E. coli systems have been the dominating source of plant MTs in bioanalytical studies on metal binding which raises some concern regarding the equivalency of the recombinant proteins to the native ones, because the multi-ion environment in bacteria differs considerably from the plant cytosol. Moreover, it is worth noting that most MT fusion proteins are purified using various tags which may change conformation and metal speciation relative to the native MTs.
Commercially available MTs have served as model proteins in numerous analytical studies focusing on identification and quantification of individual MTs and their metal binding stoichiometry. During the last decade, ESI-MS has been the most frequently used analytical tool to study these fundamental aspects as reviewed by Blindauer and Leszczyszyn (2010).260 ESI provides a soft ionisation source where optimisation of the ionisation voltage allows preservation of the coordination between ligand and metal/metalloid. Such optimisation is important in order not to dissociate the labile coordination bonds and misinterpret the metal binding stoichiometry of the MTs. Leszczyszyn et al. (2007)144 used ESI-MS to characterise the speciation of the wheat Ec protein (obtained from the MT batch purified by Lane et al. (1987)272) and observed that Zn6Ec was the dominating MT complex. The Zn/Ec complex was subsequently purified by SEC, followed by off-line ICP-OES analysis for the determination of the Zn:S ratio.
Even though ESI-MS has the potential to identify metal-MT species at low concentrations, the matrix composition of most plant tissue extracts suppresses ionisation and leads to a poor and often inadequate sensitivity, unless a gentle and efficient matrix removal procedure is applied. The sensitivity of ESI-MS under these conditions represents a critical shortcoming for the quantitative determination of MTs. Hence, analytical chemists have taken advantage of the matrix tolerance and sensitivity of ICP-MS in order to develop hyphenated techniques with HPLC or CE for an efficient separation and quantification of MT metal complexes. For the determination of the metal:S stoichiometry, methods with pressurised reaction cells have been developed in order to increase the S sensitivity.28,280 MT speciation analyses were pioneered by Lobinski et al. (1998)281 and reviewed by Prange and Schaumloffel (2002).282 This has subsequently promoted several attractive HPLC- or CE-ICP-MS based techniques for the separation, identification and quantification of various MTs. These techniques have been applied in human,283,284 fish285,286 and animal studies.287 However, to date these techniques have not been used to quantify or characterise any plant MTs. Considering the wealth of bioanalytical papers being published on this subject, it is remarkable that ICP-MS based analytical achievements on vertebrate MTs have not yet been widely implemented in studies focusing on the biological chemistry and functional roles of plant MT proteins.
The behaviour of As in the environment has received a lot of interest due to widespread groundwater contamination in South-East Asia. Millions of people have been exposed to high levels of As in drinking water, with reported values between 1000–3000 μg As L−1. Furthermore, large agricultural areas in these countries are continuously irrigated with contaminated ground water, implying that As also enters the food chain via crops and live-stock feeding on As contaminated vegetation.290 Hence, understanding the biological and metabolic pathways of As in plants is essential for evaluating the health risks posed to humans.
Both inorganic and organic forms of As are potential human carcinogens.288,291 It is now generally accepted that the toxicity of As critically depends on the chemical form in which it is found in the environment17,292 Recent progress on As speciation in plants also shows that As biochemistry in planta has a huge impact on its translocation and storage.149,293,294 Thus, As speciation determines not only its toxicity, but also its translocation to edible plant parts and entrance into the food chain.
HPLC techniques have developed along with the progress made in As speciation analyses, IPC and IEC being the most successful techniques. Kohlmeyer et al. (2002)295 used IEC with a HNO3 mobile phase together with an ion pairing agent (0.05 mM benzene 1, 2-disulfonate) which allowed separation of anions and cations in the same chromatographic run. This approach enabled separation of 17 As species from oyster tissue extracts. Sloth et al. (2003)296 used cation exchange chromatography with pyridine formate, which enabled separation of 23 As species in a single chromatographic run. Nischwitz and Pergantis (2006)297 employed a combination of cation exchange pre-columns and an anion exchange column for the separation and identification of 50 arsenic species in clams by selected reaction monitoring (SRM) using ESI-MS/MS. However, most of these analyses were performed on samples in which the majority of the As was either inorganic or covalently bound. This contrasts with most non-hyperaccumulating plants, in which a substantial part of As forms thiol based coordination complexes with PCs.298–300 As-PC complexes tend to decompose during anion exchange chromatography.139,179 Instead, gentle SEC followed by second dimension chromatography has frequently been used. Recently, successful separation of several As-PC complexes was obtained by conventional RPC using a C18 column and an acidic gradient of 0.1% formic acid and MeOH.139,179 An overview on speciation methods used for plant samples was given by Quaghebeur and Rengel (2005).293
Being element specific, and not species specific, quantitative HPLC-ICP-MS analysis needs to be combined with retention time matching, using As-standards, or molecular mass spectrometry for standard-less identification, as has been reviewed elsewhere.17 In general As species identification and structural elucidation face the similar limitations and possibilities as described previously for Se speciation.
The distribution of As species is a matter of controversy and the reported proportion of thiol-bound As shows large deviations among studies. Using X-ray adsorption spectroscopy (XAS) it was found that 96–100% of total As was bound to thiol groups in the roots of Brassica juncae and Arabidopsis,298,299 whereas less than 60% was thiol bound in sunflower.300 Obviously, plant species are different, but deviations may also stem from bias introduced by the analytical method of choice. The issues of species stability during extraction, storage and chromatographic separation are generally considered one of the major challenges with LC-ICP-MS, which relies on extracted and disintegrated tissues (see previous sections). X-ray adsorption spectroscopy (XAS) and X-ray adsorption near edge spectroscopy (XANES) are non-disruptive techniques, but have a much higher limit of detection, and require model compounds for species identification.18 Bluemlein et al. (2009)137 obtained similar results for As speciation in Thunbergia alata using one-dimensional LC-ICP-MS/ESI-MS analysis and XAS analysis. Both methods showed that 53–64% of total As was thiol bound.137 In another study, Lombi et al. (2009)97 found 36% DMA in the rice endosperm using HPLC-ICP-MS, but only 26% by XANES analysis.97
The bioactivity of As species has been increasingly elucidated by speciation analyses combining HPLC-ICP-MS, molecular mass spectrometry and X-ray adsorption spectroscopy techniques. Such progress is crucial if As toxicity and mobility is to be fully understood. Method validation with respect to species stability and extractability has moved towards standardised analytical procedures and improved data quality. In spite of this progress, major questions and details remain unresolved regarding As uptake, reduction and transport in plants.
Part B. Summary
As evident from the cases highlighted above, hyphenated ICP-MS techniques have successfully been used for speciation analysis in plant science in order to unravel the bioactivity and functional role of elements. Improvements have been accomplished in all important steps and procedures in the speciation analysis, viz. sample pre-treatment, extraction, separation, species identification and quantification. Especially the procedures for analysis of Se and As species in plants have progressed significantly over the last decade and the analytical achievements seem well implemented in plant, soil and environmental sciences. In contrast, speciation analysis of various coordination complexes with essential plant nutrients still suffers from a substantial lack of analytical method development. Studies of the chemical binding forms of Fe and Zn in plants should be prioritised due to the importance of Fe/Zn species in relation to bioavailability. New insight into Fe and Zn speciation constitutes an ideal case to demonstrate the strength of integrating cutting-edge analytical speciation analysis with plant science and human nutrition in order to solve the major global problem of Fe/Zn malnutrition.Perspectives
ICP-MS has become the method of choice for multi-elemental analysis of plant tissue, thanks to the development of novel high-throughput procedures for sample digestion, semi- and full-quantitative analysis as well as chemometric data treatment. However, the lack of suitable and commercially available certified reference materials remains a problem in quality assurance of plant tissue analysis. Currently, no reference materials exist for the most frequently used model plant Arabidopsis, which seriously limits interpretation of multi-elemental analyses on this species.Speciation analysis has developed tremendously during the last decade, thanks to a very active scientific community working in this area. The future challenge will be to further integrate the analytical developments into the biological sciences, including plant science. ICP-MS based analytical procedures for covalently bound Se and As species seem well-integrated, whereas procedures for analysis of coordination complexes remain an emerging discipline with a rather poor interaction with the biological sciences. This is especially true for MTs, where hyphenated ICP-MS has proven to be a valuable tool for quantifying vertebrate MTs and characterising the metal binding stoichiometry of these proteins. However, hyphenated ICP-MS techniques to study MT speciation have not yet managed to cross the interdisciplinary barrier between the analytical chemistry communities and those involved in plant science.
Undoubtedly, novel hyphenated ICP-MS techniques will continuously develop and contribute to the advances made in plant science. This includes the emerging role of ICP-MS in quantitative proteomics, either using naturally occurring heteroatoms in proteins (S, P, metals and metalloids) or by lanthanide tagging of conjugated proteins.309 This principle allows sensitive multiplexed protein quantification and constitutes one of the most promising analytical frontiers in atomic spectrometry currently.
List of abbreviations
Acknowledgements
The authors acknowledge financial support from the EU-FP6 projects META-PHOR (FOOD-CT-2006-03622), PHIME (FOOD-CT-2006-016253), the Danish Ministry of Food, Agriculture and Fisheries (3304-FVFP-09-B-004) and Ministry of Sciences, Technology and Innovation (grant no. 274-06-0325).References
- E. A. H. Pilon-Smits, C. F. Quinn, W. Tapken, M. Malagoli and M. Schiavon, Current Opinion in Plant Biology, 2009, 12, 267–274 CrossRef CAS.
- M. R. Broadley, P. J. White, J. P. Hammond, I. Zelko and A. Lux, New Phytologist, 2007, 173, 677–702 Search PubMed.
- R. Hansch and R. R. Mendel, Current Opinion in Plant Biology, 2009, 12, 259–266 CrossRef CAS.
- P. Pedas, C. K. Ytting, A. T. Fuglsang, T. P. Jahn, J. K. Schjoerring and S. Husted, Plant Physiology, 2008, 148, 455–466 CrossRef CAS.
- I. Baxter, P. S. Hosmani, A. Rus, B. Lahner, J. O. Borevitz, B. Muthukumar, M. V. Mickelbart, L. Schreiber, R. B. Franke and D. E. Salt, PLOS Genetics, 2009, 5 Search PubMed , Article e1000492.
- D. J. Reuter and J. B. Robinson, ed., Plant Analysis - an Interpretation Manual, Second edn, CSIRO Publishing, Collingwood, Australia, 1997 Search PubMed.
- I. Baxter, B. Muthukumar, H. C. Park, P. Buchner, B. Lahner, J. Danku, K. Zhao, J. Lee, M. J. Hawkesford, M. L. Guerinot and D. E. Salt, PLOS Genetics, 2008, 4 Search PubMed , Article e1000004.
- Z. Chen, T. Watanabe, T. Shinano, K. Okazaki and M. Osaki, New Phytologist, 2009, 181, 795–801 Search PubMed.
- K. H. Laursen, T. H. Hansen, D. P. Persson, J. K. Schjoerring and S. Husted, J. Anal. At. Spectrom., 2009, 24, 1198–1207 RSC.
- T. Watanabe, M. R. Broadley, S. Jansen, P. J. White, J. Takada, K. Satake, T. Takamatsu, S. J. Tuah and M. Osaki, New Phytologist, 2007, 174, 516–523 Search PubMed.
- B. Lahner, J. Gong, M. Mahmoudian, E. L. Smith, K. B. Abid, E. E. Rogers, M. L. Guerinot, J. F. Harper, J. M. Ward, M. McIntyre, J. I. Schroeder and D. E. Salt, Nature Biotechnology, 2003, 12, 1215–1221 CrossRef.
- I. Baxter, M. Ouzzani, S. Orcun, B. Kennedy, S. S. Jandhyala and D. E. Salt, Plant Physiology, 2007, 143, 600–611 CAS.
- T. H. Hansen, K. H. Laursen, D. P. Persson, P. Pedas, S. Husted and J. K. Schjoerring, Plant Methods, 2009, 5 Search PubMed.
- R. J. P. Williams, Coordination Chemistry Reviews, 2001, 216, 583–595 CrossRef.
- S. Mounicou, J. Szpunar and R. Lobinski, Chemical Society Reviews, 2009, 38, 1119–1138 RSC.
- D. M. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure and Applied Chemistry, 2000, 72, 1453–1470 CrossRef CAS.
- K. A. Francesconi and D. Kuehnelt, Analyst, 2004, 129, 373–395 RSC.
- F. J. Zhao, J. F. Ma, A. A. Meharg and S. P. McGrath, New Phytologist, 2009, 181, 777–794 Search PubMed.
- A. P. Navaza, M. Montes-Bayon, D. L. LeDuc, N. Terry and A. Sanz-Medel, Journal of Mass Spectrometry, 2006, 41, 323–331 CrossRef CAS.
- D. Schaumloffel, L. Ouerdane, B. Bouyssiere and R. Lobinski, J. Anal. At. Spectrom., 2003, 18, 120–127 RSC.
- E. M. Krupp, B. F. Milne, A. Mestrot, A. A. Meharg and J. Feldmann, Analytical and Bioanalytical Chemistry, 2008, 390, 1753–1764 CrossRef CAS.
- A. Nolan, D. Schaumloffel, E. Lombi, L. Ouerdane, R. Lobinski and M. McLaughlin, J. Anal. At. Spectrom., 2004, 19, 757–761 RSC.
- D. R. Ellis and D. E. Salt, Current Opinion in Plant Biology, 2003, 6, 273–279 CrossRef CAS.
- M. P. Rayman, H. G. Infante and M. Sargent, British Journal of Nutrition, 2008, 100, 238–253 CAS.
- N. von Wiren, S. Klair, S. Bansal, J. F. Briat, H. Khodr, T. Shioiri, R. A. Leigh and R. C. Hider, Plant Physiology, 1999, 119, 1107–1114 CrossRef CAS.
- J. Wirth, S. Poletti, B. Aeschlimann, N. Yakandawala, B. Drosse, S. Osorio, T. Tohge, A. R. Fernie, D. Gunther, W. Gruissem and C. Sautter, Plant Biotechnology Journal, 2009, 7, 631–644 Search PubMed.
- P. J. White and M. R. Broadley, New Phytologist, 2009, 182, 49–84 Search PubMed.
- D. P. Persson, T. H. Hansen, K. H. Laursen, J. K. Schjoerring and S. Husted, Metallomics, 2009, 1, 418–426 RSC.
- S. Lee, U. S. Jeon, S. J. Lee, Y. K. Kim, D. P. Persson, S. Husted, J. K. Schjorring, Y. Kakei, H. Masuda, N. K. Nishizawa and G. An, Proceedings of the National Academy of Sciences of the United States of America, 2009, 106, 22014–22019 CrossRef CAS.
- S. Husted, B. F. Mikkelsen, J. Jensen and N. E. Nielsen, Analytical and Bioanalytical Chemistry, 2004, 378, 171–182 CrossRef CAS.
- J. Dombovari and L. Papp, Microchemical Journal, 1998, 59, 187–193 CrossRef CAS.
- E. M. M. Flores, J. S. Barin, M. F. Mesko and G. Knapp, Spectrochimica Acta Part B-Atomic Spectroscopy, 2007, 62, 1051–1064 Search PubMed.
- S. Husted, K. H. Laursen, C. A. Hebbern, s. B. Scmidt, P. Pedas, A. Haldrup and P. E. Jensen, Plant Physiology, 2009, 150, 825–833 CrossRef CAS.
- E. M. D. Flores, J. S. Barin, J. N. G. Paniz, J. A. Medeiros and G. Knapp, Analytical Chemistry, 2004, 76, 3525–3529 CrossRef.
- Z. B. Alfassi and C. M. Wai, Preconcentration techniques for trace elements, CRC Press, Boca Raton, 1992 Search PubMed.
- E. Engstrom, A. Stenberg, S. Senioukh, R. Edelbro, D. C. Baxter and I. Rodushkin, Analytica Chimica Acta, 2004, 521, 123–135 CrossRef CAS.
- B. Bocca, A. Alimonti, G. Forte, F. Petrucci, C. Pirola, O. Senofonte and N. Violante, Analytical and Bioanalytical Chemistry, 2003, 377, 65–70 CrossRef CAS.
- J. M. C. Danku, L. Gumaelius, I. Baxter and D. E. Salt, J. Anal. At. Spectrom., 2009, 24, 103–107 RSC.
- H. J. Reid, S. Greenfield and T. E. Edmonds, Analyst, 1995, 120, 1543–1548 RSC.
- M. Wasilewska, W. Goessler, M. Zischka, B. Maichin and G. Knapp, J. Anal. At. Spectrom., 2002, 17, 1121–1125 RSC.
- A. S. Al-Ammar, E. Reitznerova and R. M. Barnes, Spectrochimica Acta Part B-Atomic Spectroscopy, 1999, 54, 1813–1820 Search PubMed.
- T. U. Probst, N. G. Berryman, P. Lemmen, L. Weissfloch, T. Auberger, D. Gabel, J. Carlsson and B. Larsson, J. Anal. At. Spectrom., 1997, 12, 1115–1122 RSC.
- J. K. Friel, C. S. Skinner, S. E. Jackson and H. P. Longerich, Analyst, 1990, 115, 269–273 RSC.
- S. L. Wu, X. B. Feng and A. Wittmeier, J. Anal. At. Spectrom., 1997, 12, 797–806 RSC.
- J. Dombovari, Z. Varga, J. S. Becker, J. Matyus, G. Kakuk and L. Papp, Atomic Spectroscopy, 2001, 22, 331–335 CAS.
- M. Azizi, C. Emanueli, S. Peyrard, P. Madeddu, F. Alheno-Gelas and D. J. Campbell, Journal of Hypertension, 2008, 26, 1890–1890 Search PubMed.
- M. Dapiaggi, F. Leva, D. Rabuffetti, S. Ayrault, A. Gaudry and R. M. Cenci, Environmental Technology, 2001, 22, 1183–1192 Search PubMed.
- M. Krachler, C. Mohl, H. Emons and W. Shotyk, J. Anal. At. Spectrom., 2002, 17, 844–851 RSC.
- P. Quevauviller, E. A. Maier, K. Vercoutere, H. Muntau and B. Griepink, Fresenius Journal of Analytical Chemistry, 1992, 343, 335–338 Search PubMed.
- E. Bulska, W. Kandler, P. Paslawski and A. Hulanicki, Mikrochimica Acta, 1995, 119, 137–146 CAS.
- R. Koplik, E. Curdova and M. Suchanek, Fresenius Journal of Analytical Chemistry, 1998, 360, 449–451 Search PubMed.
- P. Pedas, C. A. Hebbern, J. K. Schjoerring, P. E. Holm and S. Husted, Plant Physiology, 2005, 139, 1411–1420 CrossRef CAS.
- B. Bocca, A. Lamazza, A. Pino, E. De Masi, M. Iacomino, D. Mattei, S. Rahimi, E. Fiori, A. Schillaci, A. Alimonti and G. Forte, Rapid Communications in Mass Spectrometry, 2007, 21, 1776–1782 CrossRef CAS.
- J. Millos, M. Costas-Rodriguez, I. Lavilla and C. Bendicho, Talanta, 2009, 77, 1490–1496 CrossRef CAS.
- J. Dombovari, J. S. Becker and H. J. Dietze, Fresenius Journal of Analytical Chemistry, 2000, 367, 407–413 Search PubMed.
- M. K. Jakobsen, L. R. Poulsen, A. Schulz, P. Fleurat-Lessard, A. Moller, S. Husted, M. Schiott, A. Amtmann and M. G. Palmgren, Genes & Development, 2005, 19, 2757–2769 Search PubMed.
- P. H. Brown, R. M. Welch and E. E. Cary, Plant Physiol., 1987, 85, 801–803 CrossRef CAS.
- H. Marschner, Mineral Nutrition of Higher Plants, Second edition edn, Academic Press, London, 1995 Search PubMed.
- Y. C. Yip and W. C. Sham, Trac-Trends in Analytical Chemistry, 2007, 26, 727–743 CrossRef CAS.
- R. Thomas, Practical guide to ICP-MS, Marcel Dekker, Inc, New York, 2004 Search PubMed.
- V. Gundersen, I. E. Bechmann, A. Behrens and S. Stürup, Journal of Agricultural and Food Chemistry, 2000, 48, 6094–6102 CrossRef CAS.
- I. Rodushkin, Fresenius Journal of Analytical Chemistry, 1998, 362, 541–546 Search PubMed.
- I. Rodushkin, E. Engstrom, D. Sorlin and D. Baxter, Science of the Total Environment, 2008, 392, 290–304 CrossRef CAS.
- A. M. S. Nyomora, R. N. Sah, P. H. Brown and R. O. Miller, Fresenius Journal of Analytical Chemistry, 1997, 357, 1185–1191 Search PubMed.
- P. Masson, T. Dalix and S. Bussiere, Communications in Soil Science and Plant Analysis, 2010, 41, 231–243 CrossRef CAS.
- M. Broadhead, R. Broadhead and J. W. Hager, Atomic Spectroscopy, 1990, 11, 205–209 CAS.
- E. R. Denoyer, J. Anal. At. Spectrom., 1992, 7, 1187–1193 RSC.
- C. J. Amarasiriwardena, B. Gercken, M. D. Argentine and R. M. Barnes, J. Anal. At. Spectrom., 1990, 5, 457–462 RSC.
- D. Amarasiriwardena, S. F. Durrant, A. Lásztity, A. Krushevska, M. D. Argentine and R. M. Barnes, Microchemical Journal, 1997, 56, 352–372 CrossRef CAS.
- J. I. García-Alonso, M. G. Camblor, M. M. Bayón, J. M. Marchante-Gayón and A. Sanz-Medel, Journal of Mass Spectrometry, 1997, 32, 556–564 CrossRef CAS.
- K. Günther, A. von Bohlen, G. Paprott and R. Klockenkämper, Fresenius' Journal of Analytical Chemistry, 1992, 342, 444–448 Search PubMed.
- Y. Hu, F. Vanhaecke, L. Moens and R. Dams, Analytica Chimica Acta, 1997, 355, 105–111 CrossRef CAS.
- M. S. Jimenez, M. T. Gomez and J. R. Castillo, Talanta, 2007, 72, 1141–1148 CrossRef CAS.
- K. H. Esbensen, Multivariate Data Analysis-in practice, 5th edn, Camo Process AS, Oslo, Norway, 2002 Search PubMed.
- A. Marcos, A. Fisher, G. Rea and S. J. Hill, J. Anal. At. Spectrom., 1998, 13, 521–525 RSC.
- A. Moreda-Piñeiro, A. Fisher and S. J. Hill, Journal of Food Composition and Analysis, 2003, 16, 195–211 CrossRef CAS.
- Y. X. Chen, M. G. Yu, J. Xu, X. C. Chen and J. Y. Shi, Journal of the Science of Food and Agriculture, 2009, 89, 2350–2355 Search PubMed.
- T. S. Pilgrim, R. J. Watling and K. Grice, Food Chemistry, 2010, 118, 921–926 CrossRef CAS.
- A. Yasui and K. Shindoh, Bunseki Kaguru, 2000, 49, 405–410 Search PubMed.
- K. Ariyama, H. Horita and A. Yasui, Analytical Sciences, 2004, 20, 871–877 CAS.
- K. Ariyama, H. Horita and A. Yasui, Journal of Agricultural and Food Chemistry, 2004, 52, 5803–5809 CrossRef CAS.
- M. Oddone, M. Aceto, M. Baldizzone, D. Musso and D. Osella, Journal of Agricultural and Food Chemistry, 2009, 57, 3404–3408 CrossRef CAS.
- K. A. Anderson, B. A. Magnuson, M. L. Tschirgi and B. Smith, Journal of Agricultural and Food Chemistry, 1999, 47, 1568–1575 CrossRef CAS.
- F. Di Giacomo, A. Del Signore and M. Giaccio, Journal of Agricultural and Food Chemistry, 2007, 55, 860–866 CrossRef CAS.
- K. A. Anderson and B. W. Smith, Journal of Agricultural and Food Chemistry, 2005, 53, 410–418 CrossRef CAS.
- E. K. Shibuya, J. E. S. Sarkis, O. Negrini-Neto and J. P. H. B. Ometto, Journal of the Brazilian Chemical Society, 2007, 18, 205–214 CAS.
- A. L. Perez, B. W. Smith and K. A. Anderson, Journal of Agricultural and Food Chemistry, 2006, 54, 4506–4516 CrossRef CAS.
- I. R. Baxter, O. Vitek, B. Lahner, B. Muthukumar, M. Borghi, J. Morrissey, M. L. Guerinot and D. E. Salt, Proceedings of the National Academy of Sciences, 2008, 105, 12081–12086 CrossRef CAS.
- R. McRae, P. Bagchi, S. Sumalekshmy and C. J. Fahrni, Chemical Reviews, 2009, 109, 4780–4827 CrossRef CAS.
- J. S. Becker, M. Zoriy, A. Matusch, B. Wu, D. Salber and C. Palm, Mass Spectrometry Reviews, 2010, 29, 156–175 CAS.
- E. Lombi and J. Susini, Plant and Soil, 2009, 320, 1–35 CrossRef CAS.
- A. P. Mazzolini, C. K. Pallaghy and G. J. F. Legge, New Phytologist, 1985, 100, 483–509 CrossRef CAS.
- G. Sarret, G. Willems, M.-P. Isaure, M. A. Marcus, S. C. Fakra, H. Frérot, S. Pairis, N. Geoffroy, A. Manceau and P. Saumitou-Laprade, New Phytologist, 2009, 184, 581–595 Search PubMed.
- M.-P. Isaure, B. Fayard, G. Sarret, S. Pairis and J. Bourguignon, Spectrochimica Acta Part B: Atomic Spectroscopy, 2006, 61, 1242–1252 Search PubMed.
- S. A. Kim, T. Punshon, A. Lanzirotti, L. Li, J. M. Alonso, J. R. Ecker, J. Kaplan and M. L. Guerinot, Science, 2006, 314, 1295–1298 CrossRef CAS.
- A. A. Meharg, E. Lombi, P. N. Williams, K. G. Scheckel, J. Feldmann, A. Raab, Y. G. Zhu and R. Islam, Environmental Science & Technology, 2008, 42, 1051–1057 Search PubMed.
- E. Lombi, K. G. Scheckel, J. Pallon, A. M. Carey, Y. G. Zhu and A. A. Meharg, New Phytologist, 2009, 184, 193–201 Search PubMed.
- M. Takahashi, T. Nozoye, N. Kitajima, N. Fukuda, A. Hokura, Y. Terada, I. Nakai, Y. Ishimaru, T. Kobayashi, H. Nakanishi and N. K. Nishizawa, Plant and Soil, 2009, 325, 39–51 CrossRef CAS.
- P. N. Williams, E. Lombi, G. X. Sun, K. Scheckel, Y. G. Zhu, X. B. Feng, J. M. Zhu, A. M. Carey, E. Adomako, Y. Lawgali, C. Deacon and A. A. Meharg, Environmental Science & Technology, 2009, 43, 6024–6030 Search PubMed.
- R. Tappero, E. Peltier, M. Gräfe, K. Heidel, M. Ginder-Vogel, K. J. T. Livi, M. L. Rivers, M. A. Marcus, R. L. Chaney and D. L. Sparks, New Phytologist, 2007, 175, 641–654 Search PubMed.
- K. L. Moore, M. Schroder, E. Lombi, F. J. Zhao, S. P. McGrath, M. J. Hawkesford, P. R. Shewry and C. R. M. Grovenor, New Phytologist, 2010, 185, 434–445 Search PubMed.
- K. E. Smart, M. R. Kilburn, C. J. Salter, J. A. C. Smith and C. R. M. Grovenor, International Journal of Mass Spectrometry, 2007, 260, 107–114 CrossRef CAS.
- J. S. Becker, R. C. Dietrich, A. Matusch, D. Pozebon and V. L. Dressler, Spectrochimica Acta Part B: Atomic Spectroscopy, 2008, 63, 1248–1252 Search PubMed.
- B. Wu, M. Zoriy, Y. Chen and J. S. Becker, Talanta, 2009, 78, 132–137 CrossRef CAS.
- D. Cleveland and R. G. Michel, Applied Spectroscopy Reviews, 2008, 43, 93–110 CrossRef CAS.
- J. S. Becker, A. Gorbunoff, M. Zoriy, A. Izmer and M. Kayser, J. Anal. At. Spectrom., 2006, 21, 19–25 RSC.
- P. Pedas, J. K. Schjoerring and S. Husted, Plant Physiology and Biochemistry, 2009, 47, 377–383 Search PubMed.
- Y. Ishimaru, M. Suzuki, T. Kobayashi, M. Takahashi, H. Nakanishi, S. Mori and N. K. Nishizawa, Journal of Experimental Botany, 2005, 56, 3207–3214 Search PubMed.
- Y. Ishimaru, M. Suzuki, T. Tsukamoto, K. Suzuki, M. Nakazono, T. Kobayashi, Y. Wada, S. Watanabe, S. Matsuhashi, M. Takahashi, H. Nakanishi, S. Mori and N. K. Nishizawa, Plant Journal, 2006, 45, 335–346 CrossRef CAS.
- S. Clemens, M. G. Palmgren and U. Krämer, TRENDS in Plant Science, 2002, 7, 309–315 CrossRef CAS.
- J. F. Ma, N. Yamaji, N. Mitani, X.-Y. Xu, Y.-H. Su, S. P. McGrath and F.-J. Zhao, Proceedings of the National Academy of Sciences, 2008, 105, 9931–9935 CrossRef CAS.
- G. Vert, N. Grotz, F. Dedaldechamp, F. Gaymard, M. L. Guerinot, J.-F. Briat and C. Curie, Plant Cell, 2002, 14, 1223–1233 CrossRef CAS.
- Y. O. Korshunova, D. Eide, W. G. Clark, M. L. Guerinot and H. B. Pakrasi, Plant Molecular Biology, 1999, 40, 37–44 CrossRef CAS.
- G. Schaaf, A. Honsbein, A. R. Meda, S. Kirchner, D. Wipf and N. von Wiren, Journal of Biological Chemistry, 2006, 281, 25532–25540 CrossRef CAS.
- E. L. Connolly, J. P. Fett and M. L. Guerinot, The Plant Cell, 2002, 14, 1347–1357 Search PubMed.
- C. Uauy, A. Distelfeld, T. Fahima, A. Blechl and J. Dubcovsky, Science, 2006, 314, 1298–1301 CrossRef CAS.
- J. Morrissey, I. R. Baxter, J. Lee, L. Li, B. Lehner, N. Grotz, J. Kaplan, D. E. Salt and M. L. Guerinot, The Plant Cell, 2009, 21, 3326–3338 Search PubMed.
- E. Peiter, B. Montanini, A. Gobert, P. Pedas, S.r. Husted, F. J. M. Maathuis, D. Blaudez, M. Chalot and D. Sanders, Proceedings of the National Academy of Sciences, 2007, 104, 8532–8537 CrossRef CAS.
- S. Frietsch, Y.-F. Wang, C. Sladek, L. R. Poulsen, S. M. Romanowsky, J. I. Schroeder and J. F. Harper, Proceedings of the National Academy of Sciences of the USA, 2007, 104, 14531–14536 CrossRef CAS.
- P. Pedas and S. Husted, Plant Signal Behav, 2009, 4, 842–845 Search PubMed.
- F.t. Hoopen, T. A. Cuin, P. Pedas, J. N. Hegelund, S. Shabala, J. K. Schjoerring and T. P. Jahn, Journal of Expermental Botany, 2010 Search PubMed.
- L. Li, A. F. Tutone, R. S. M. Drummond, R. C. Gardner and S. Luan, The Plant Cell, 2001, 13, 2761–2775 Search PubMed.
- A. Ruiz and J. Ariño, Eukaryotic Cell, 2007, 6, 2175–2183 CrossRef CAS.
- N. H. Roosens, C. Bernard, R. Leplae and N. Verbruggen, FEBS Letters, 2004, 577, 9–16 CrossRef CAS.
- V. Vacchina, S. Mari, P. Czernic, L. Marques, K. Pianelli, D. Schaumloffel, M. Lebrun and R. Lobinski, Analytical Chemistry, 2003, 75, 2740–2745 CrossRef CAS.
- Y. Ogra and Y. Anan, J. Anal. At. Spectrom., 2009, 24, 1477–1488 RSC.
- L. A. Finney and T. V. O'Halloran, Science, 2003, 300, 931–936 CrossRef CAS.
- J. K. Pittman, New Phytologist, 2005, 167, 733–742 Search PubMed.
- W. Y. Song, E. Martinoia, J. Lee, D. Kim, D. Y. Kim, E. Vogt, D. Shim, K. S. Choi, I. Hwang and Y. Lee, Plant Physiology, 2004, 135, 1027–1039 CrossRef CAS.
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- R. N. Reese, C. A. White and D. R. Winge, Plant Physiology, 1992, 98, 225–229 CAS.
- F. Van Belleghem, A. Cuypers, B. Semane, K. Smeets, J. Vangronsveld, J. d'Haen and R. Valcke, New Phytologist, 2007, 173, 495–508 Search PubMed.
- L. Ouerdane, S. Mari, P. Czernic, M. Lebrun and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 676–683 RSC.
- A. Sanz-Medel, M. Montes-Bayon and M. L. F. Saanchez, Analytical and Bioanalytical Chemistry, 2003, 377, 236–247 CrossRef CAS.
- J. Jeong and M. L. Guerinot, Trends in Plant Science, 2009, 14, 280–285 CrossRef CAS.
- M. A. Z. Arruda and R. A. Azevedo, Annals of Applied Biology, 2009, 155, 301–307 Search PubMed.
- K. Bluemlein, A. Raab and J. Feldmann, Analytical and Bioanalytical Chemistry, 2009, 393, 357–366 CrossRef CAS.
- E. M. Krupp, A. Mestrot, J. Wielgus, A. A. Meharg and J. Feldmann, Chemical Communications, 2009, 4257–4259 RSC.
- A. Raab, A. A. Meharg, M. Jaspars, D. R. Genney and J. Feldmann, J. Anal. At. Spectrom., 2004, 19, 183–190 RSC.
- E. H. Larsen, R. Lobinski, K. Burger-Meyer, M. Hansen, R. Ruzik, L. Mazurowska, P. H. Rasmussen, J. J. Sloth, O. Scholten and C. Kik, Analytical and Bioanalytical Chemistry, 2006, 385, 1098–1108 CrossRef CAS.
- R. Rellan-Alvarez, J. Abadia and A. Alvarez-Fernandez, Rapid Communications in Mass Spectrometry, 2008, 22, 1553–1562 CrossRef CAS.
- H. Chassaigne and R. Lobinski, Analytical Chemistry, 1998, 70, 2536–2543 CrossRef CAS.
- D. P. Persson, T. H. Hansen, P. E. Holm, J. K. Schjoerring, H. C. B. Hansen, J. Nielsen, I. Cakmak and S. Husted, J. Anal. At. Spectrom., 2006, 21, 996–1005 RSC.
- O. I. Leszczyszyn, R. Schmid and C. A. Blindauer, Proteins-Structure Function and Bioinformatics, 2007, 68, 922–935 Search PubMed.
- X. Wan and E. Freisinger, Metallomics, 2009, 1, 489–500 RSC.
- Z. Mester, S. Willie, L. Yang, R. Sturgeon, J. A. Caruso, M. L. Fernandez, P. Fodor, R. J. Goldschmidt, H. Goenaga-Infante, R. Lobinski, P. Maxwell, S. McSheehy, A. Polatajko, B. B. M. Sadi, A. Sanz-Medel, C. Scriver, J. Szpunar, R. Wahlen and W. Wolf, Analytical and Bioanalytical Chemistry, 2006, 385, 168–180 CrossRef CAS.
- S. R. Troelstra and F. Berendse, Plant and Soil, 1982, 64, 277–281 CrossRef.
- A. Raab, P. N. Williams, A. Meharg and J. Feldmann, Environmental Chemistry, 2007, 4, 197–203 CrossRef CAS.
- W. J. Liu, B. A. Wood, A. Raab, S. P. McGrath, F. J. Zhao and J. Feldmann, Plant Physiology, 2010, 152, 2211–2221 CrossRef CAS.
- J. L. Gomez-Ariza, T. Garcia-Barrera, F. Lorenzo, V. Bernal, M. J. Villegas and V. Oliveira, Analytica Chimica Acta, 2004, 524, 15–22 CrossRef CAS.
- S. Mounicou, J. Szpunar, R. Lobinski, D. Andrey and C. J. Blake, J. Anal. At. Spectrom., 2002, 17, 880–886 RSC.
- A. Sussulini, J. S. Garcia, M. F. Mesko, D. P. Moraes, E. M. M. Flores, C. A. Perez and M. A. Z. Arruda, Microchimica Acta, 2007, 158, 173–180 CrossRef CAS.
- V. Vacchina, K. Polec and J. Szpunar, J. Anal. At. Spectrom., 1999, 14, 1557–1566 RSC.
- D. Baralkiewicz, M. Kozka, A. Piechalak, B. Tomaszewska and P. Sobczak, Talanta, 2009, 79, 493–498 CrossRef CAS.
- L. Q. Chen, Y. F. Guo, L. M. Yang and Q. Q. Wang, J. Anal. At. Spectrom., 2007, 22, 1403–1408 RSC.
- B. B. M. Sadi, A. P. Vonderheide, J. M. Gong, J. I. Schroeder, J. R. Shann and J. A. Caruso, Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 2008, 861, 123–129 Search PubMed.
- K. Polec-Pawlak, R. Ruzik, E. Lipiec, M. Ciurzynska and H. Gawronska, J. Anal. At. Spectrom., 2007, 22, 968–972 RSC.
- M. Shah, S. S. Kannamkumarath, J. C. A. Wuilloud, R. G. Wuilloud and J. A. Caruso, J. Anal. At. Spectrom., 2004, 19, 381–386 RSC.
- G. Weber, N. von Wiren and H. Hayen, Journal of Separation Science, 2008, 31, 1615–1622 CrossRef CAS.
- J. Szpunar, P. Pellerin, A. Makarov, T. Doco, P. Williams and R. Lobinski, J. Anal. At. Spectrom., 1999, 14, 639–644 RSC.
- S. McSheehy, W. J. Yang, F. Pannier, J. Szpunar, R. Lobinski, J. Auger and M. Potin-Gautier, Analytica Chimica Acta, 2000, 421, 147–153 CrossRef CAS.
- O. I. Leszczyszyn, C. D. Evans, S. E. Keiper, G. Z. L. Warren and C. A. Blindauer, Inorganica Chimica Acta, 2007, 360, 3–13 CrossRef CAS.
- E. Eren, D. C. Kennedy, M. J. Maroney and J. M. Arguello, Journal of Biological Chemistry, 2006, 281, 33881–33891 CrossRef CAS.
- K. Polec-Pawlak, R. Ruzik, K. Abramski, M. Ciurzynska and H. Gawronska, Analytica Chimica Acta, 2005, 540, 61–70 CrossRef CAS.
- Y. Fang, Y. F. Zhang, B. Catron, Q. L. Chan, Q. H. Hu and J. A. Caruso, J. Anal. At. Spectrom., 2009, 24, 1657–1664 RSC.
- E. H. Larsen, J. J. Sloth, M. Hansen and S. Moesgaard, J. Anal. At. Spectrom., 2003, 18, 310–316 RSC.
- M. Montes-Bayon, M. J. D. Molet, E. B. Gonzalez and A. Sanz-Medel, Talanta, 2006, 68, 1287–1293 CrossRef CAS.
- J. A. Caruso, D. T. Heitkemper and C. B'Hymer, Analyst, 2001, 126, 136–140 RSC.
- I. Leopold and D. Gunther, Fresenius Journal of Analytical Chemistry, 1997, 359, 364–370 Search PubMed.
- S. Mounicou, M. Dernovics, K. Bierla and J. Szpunar, Talanta, 2009, 77, 1877–1882 CrossRef CAS.
- N. P. Vela, D. T. Heitkemper and K. R. Stewart, Analyst, 2001, 126, 1011–1017 RSC.
- J. L. G. Mar, L. H. Reyes, G. A. M. Rahman and H. M. S. Kingston, Journal of Agricultural and Food Chemistry, 2009, 57, 3005–3013 CrossRef.
- M. Quaghebeur, Z. Rengel and M. Smirk, J. Anal. At. Spectrom., 2003, 18, 128–134 RSC.
- V. Vacchina, R. Lobinski, M. Oven and M. H. Zenk, J. Anal. At. Spectrom., 2000, 15, 529–534 RSC.
- Y. Xuan, E. B. Scheuermann, A. R. Meda, H. Hayen, N. von Wiren and G. Weber, Journal of Chromatography A, 2006, 1136, 73–81 CrossRef CAS.
- R. Koplik, M. Borkova, B. Bicanova, J. Polak, O. Mestek and J. Kominkova, Food Chemistry, 2006, 99, 158–167 CrossRef CAS.
- K. A. Francesconi and M. Sperling, Analyst, 2005, 130, 998–1001 RSC.
- G. Hieftje, J. Anal. At. Spectrom., 2010, 25, 611–612 RSC.
- A. Raab, J. Feldmann and A. A. Meharg, Plant Physiology, 2004, 134, 1113–1122 CrossRef CAS.
- J. Szpunar and R. Lobinski, Analytical and Bioanalytical Chemistry, 2002, 373, 404–411 CrossRef CAS.
- J. Meija, M. Montes-Bayon, J. A. Caruso and A. Sanz-Medel, Trac-Trends in Analytical Chemistry, 2006, 25, 44–51 CrossRef CAS.
- R. Lobinski, D. Schaumloffel and J. Szpunar, Mass Spectrometry Reviews, 2006, 25, 255–289 CrossRef CAS.
- V. Gergely, M. Montes-Bayon, P. Fodor and A. Sanz-Medel, Journal of Agricultural and Food Chemistry, 2006, 54, 4524–4530 CrossRef CAS.
- Y. Lai, Q. Q. Wang, W. W. Yan, L. M. Yang and B. L. Huang, J. Anal. At. Spectrom., 2005, 20, 751–753 RSC.
- M. Dernovics, P. Giusti and R. Lobinski, J. Anal. At. Spectrom., 2007, 22, 41–50 RSC.
- T. Y. Yen, J. A. Villa and J. G. DeWitt, Journal of Mass Spectrometry, 1999, 34, 930–941 CrossRef CAS.
- M. E. V. Schmoger, M. Oven and E. Grill, Plant Physiology, 2000, 122, 793–801 CrossRef CAS.
- D. G. Mendoza-Cozatl, E. Butko, F. Springer, J. W. Torpey, E. A. Komives, J. Kehr and J. I. Schroeder, Plant Journal, 2008, 54, 249–259 CrossRef CAS.
- U. P. Rodrigues-Filho, S. Vaz, M. P. Felicissimo, M. Scarpellini, D. R. Cardoso, R. C. J. Vinhas, R. Landers, J. F. Schneider, B. R. McGarvey, M. L. Andersen and L. H. Skibsted, Journal of Inorganic Biochemistry, 2005, 99, 1973–1982 CrossRef CAS.
- H. Kupper, A. Mijovilovich, W. Meyer-Klaucke and P. M. H. Kroneck, Plant Physiology, 2004, 134, 748–757 CrossRef.
- D. E. Salt, R. C. Prince, A. J. M. Baker, I. Raskin and I. J. Pickering, Environmental Science & Technology, 1999, 33, 713–717 Search PubMed.
- A. Trampczynska, H. Kupper, W. Meyer-Klaucke, H. Schmidt and S. Clemens, Metallomics, 2010, 2, 57–66 RSC.
- C. Curie and J. F. Briat, Annual Review of Plant Biology, 2003, 54, 183–206 Search PubMed.
- C. Curie, G. Cassin, D. Couch, F. Divol, K. Higuchi, M. Jean, J. Misson, A. Schikora, P. Czernic and S. Mari, Annals of Botany, 2009, 103, 1–11 Search PubMed.
- S. Lee, Y.-S. Kim, U. S. Jeon, Y.-K. Kim, D. P. Persson, T. Hansen, S. Husted, J. K. Schjoerring, Y. Kakei, H. Masuda, N. K. Nishizawa and G. An, Plant Biotech. J., 2010 (in press) Search PubMed.
- G. Scholz, R. Becker, A. Pich and U. W. Stephan, Journal of Plant Nutrition, 1992, 15, 1647–1665 CrossRef CAS.
- A. Pich, S. Hillmer, R. Manteuffel and G. Scholz, Journal of Experimental Botany, 1997, 48, 759–767 Search PubMed.
- A. Pich, R. Manteuffel, S. Hillmer, G. Scholz and W. Schmidt, Planta, 2001, 213, 967–976 CrossRef CAS.
- K. Higuchi, N. Nishizawa, V. Romheld, H. Marschner and S. Mori, Journal of Plant Nutrition, 1996, 19, 1235–1239 CrossRef CAS.
- M. Takahashi, Y. Tereda, I. Nakai, H. Nakanishi, E. Yoshimura, S. Mori and N. K. Nishizawa, The Plant Cell, 2003, 15, 1263–1280 Search PubMed.
- H. Inoue, K. Higuchi, M. Takahashi, H. Nakanishi, S. Mori and N. K. Nishizawa, Plant Journal, 2003, 36, 366–381 CrossRef CAS.
- D. Douchkov, C. Gryczka, U. W. Stephan, R. Hell and H. Baumlein, Plant Cell and Environment, 2005, 28, 365–374 CrossRef CAS.
- L. Q. Zheng, Z. Q. Cheng, C. X. Ai, X. H. Jiang, X. S. Bei, Y. Zheng, R. P. Glahn, R. M. Welch, D. D. Miller, X. G. Lei and H. X. Shou, Plos One, 2010, 5 Search PubMed.
- G. Weber, N. von Wiren and H. Hayen, Rapid Communications in Mass Spectrometry, 2006, 20, 973–980 CrossRef CAS.
- T. Jaffre, R. R. Brooks, J. Lee and R. D. Reeves, Science, 1976, 193, 579–580 CAS.
- S. Mari, D. Gendre, K. Pianelli, L. Ouerdane, R. Lobinski, J. F. Briat, M. Lebrun and P. Czernic, Journal of Experimental Botany, 2006, 57, 4111–4122 Search PubMed.
- Y. Xuan, E. B. Scheuermann, A. R. Meda, P. Jacob, N. von Wiren and G. Weber, Electrophoresis, 2007, 28, 3507–3519 CrossRef CAS.
- M. Y. Chu and D. Beauchemin, J. Anal. At. Spectrom., 2004, 19, 1213–1216 RSC.
- K. M. Hambidge and N. F. Krebs, Journal of Nutrition, 2007, 137, 1101–1105 Search PubMed.
- W. H. Pfeiffer and B. McClafferty, Crop Science, 2007, 47, S88–S105 Search PubMed.
- W. Maret and H. H. Sandstead, Journal of Trace Elements in Medicine and Biology, 2006, 20, 3–18 Search PubMed.
- J. R. Turnlund, Critical Reviews in Food Science and Nutrition, 1991, 30, 387–396 CAS.
- H. E. Bouis, C. Hotz, B. McClafferty, J. V. Meenakshi, W. H. Pfeiffer, P. Eozenou and A. Rahman, Annals of Nutrition and Metabolism, 2009, 55, 57–58 Search PubMed.
- J. E. Mayer, W. H. Pfeiffer and P. Beyer, Current Opinion in Plant Biology, 2008, 11, 166–170 CrossRef CAS.
- L. Ozturk, M. A. Yazici, C. Yucel, A. Torun, C. Cekic, A. Bagci, H. Ozkan, H. J. Braun, Z. Sayers and I. Cakmak, Physiologia Plantarum, 2006, 128, 144–152 CrossRef CAS.
- V. Raboy, K. A. Young, J. A. Dorsch and A. Cook, Journal of Plant Physiology, 2001, 158, 489–497 Search PubMed.
- J. N. A. Lott, I. Ockenden, V. Raboy and G. D. Batten, Seed Science Research, 2000, 10, 11–33 Search PubMed.
- I. Cakmak, Plant and Soil, 2008, 302, 1–17 CrossRef CAS.
- M. Vasconcelos, K. Datta, N. Oliva, M. Khalekuzzaman, L. Torrizo, S. Krishnan, M. Oliveira, F. Goto and S. K. Datta, Plant Science, 2003, 164, 371–378 CrossRef CAS.
- F. Goto, T. Yoshihara, N. Shigemoto, S. Toki and F. Takaiwa, Nature Biotechnology, 1999, 17, 282–286 CrossRef CAS.
- B. Lonnerdal, Journal of Nutrition, 2000, 130, 1378S–1383S Search PubMed.
- R. P. Glahn and D. R. VanCampen, Journal of Nutrition, 1997, 127, 642–647 Search PubMed.
- B. Lonnerdal, Journal of Nutrition, 2003, 133, 1490S–1493S Search PubMed.
- U. B. Kutman, B. Yildiz, L. Ozturk and I. Cakmak, Cereal Chemistry, 2010, 87, 1–9 Search PubMed.
- A. Scalbert, C. Manach, C. Morand, C. Remesy and L. Jimenez, Critical Reviews in Food Science and Nutrition, 2005, 45, 287–306 CrossRef CAS.
- E. N. C. Mills, M. L. Parker, N. Wellner, G. Toole, K. Feeney and P. R. Shewry, Journal of Cereal Science, 2005, 41, 193–201 Search PubMed.
- A. Helfrich and J. Bettmer, J. Anal. At. Spectrom., 2004, 19, 1330–1334 RSC.
- A. W. Peck, G. K. McDonald and R. D. Graham, Journal of Cereal Science, 2008, 47, 266–274 Search PubMed.
- T. G. Sors, D. R. Ellis and D. E. Salt, Photosynthesis Research, 2005, 86, 373–389 CrossRef CAS.
- K. Pyrzynska, Food Chemistry, 2009, 114, 1183–1191 CrossRef CAS.
- Z. Pedrero and Y. Madrid, Analytica Chimica Acta, 2009, 634, 135–152 CrossRef CAS.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Analytical Communications, 1999, 36, 249–252 Search PubMed.
- C. Casiot, J. Szpunar, R. Lobinski and M. Potin-Gautier, J. Anal. At. Spectrom., 1999, 14, 645–650 RSC.
- L. Tastet, D. Schaumloffel and R. Lobinski, J. Anal. At. Spectrom., 2008, 23, 309–317 RSC.
- Y. G. Zhu, E. A. H. Pilon-Smits, F. J. Zhao, P. N. Williams and A. A. Meharg, Trends in Plant Science, 2009, 14, 436–442 CrossRef CAS.
- H. F. Li, S. P. McGrath and F. J. Zhao, New Phytologist, 2008, 178, 92–102 Search PubMed.
- E. Dumont, F. Vanhaecke and R. Cornelis, Analytical and Bioanalytical Chemistry, 2006, 385, 1304–1323 CrossRef CAS.
- J. Meija, M. Montes-Bayon, D. L. Le Duc, N. Terry and J. A. Caruso, Analytical Chemistry, 2002, 74, 5837–5844 CrossRef.
- M. Dernovics, T. Garcia-Barrera, K. Bierla, H. Preud'homme and R. Lobinski, Analyst, 2007, 132, 439–449 RSC.
- M. P. Rayman, Lancet, 2000, 356, 233–241 CrossRef CAS.
- L. C. Clark, G. F. Combs, B. W. Turnbull, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glover, G. F. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith and J. R. Taylor, Jama-Journal of the American Medical Association, 1996, 276, 1957–1963 CrossRef.
- C. Ip, M. Birringer, E. Block, M. Kotrebai, J. F. Tyson, P. C. Uden and D. J. Lisk, Journal of Agricultural and Food Chemistry, 2000, 48, 2062–2070 CrossRef CAS.
- C. Ip and H. E. Ganther, Cancer Research, 1990, 50, 1206–1211 CAS.
- C. Ip, D. J. Lisk and G. S. Stoewsand, Nutrition and Cancer-an International Journal, 1992, 17, 279–286 Search PubMed.
- M. P. Rayman, Proceedings of the Nutrition Society, 2005, 64, 527–542 CAS.
- E. Kapolna, P. R. Hillestrom, K. H. Laursen, S. Husted and E. H. Larsen, Food Chemistry, 2009, 115, 1357–1363 CrossRef.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Analyst, 2000, 125, 71–78 RSC.
- E. Kapolna and P. Fodor, International Journal of Food Sciences and Nutrition, 2007, 58, 282–296 CrossRef CAS.
- C. Ip and D. J. Lisk, Nutrition and Cancer-an International Journal, 1994, 21, 203–212 Search PubMed.
- A. P. Vonderheide, K. Wrobel, S. S. Kannamkumarath, C. B'Hymer, M. Montes-Bayon, C. P. De Leon and J. A. Caruso, Journal of Agricultural and Food Chemistry, 2002, 50, 5722–5728 CrossRef CAS.
- T. Vamerali, M. Bandiera and G. Mosca, Environmental Chemistry Letters, 2010, 8, 1–17 Search PubMed.
- A. R. Memon and P. Schroder, Environmental Science and Pollution Research, 2009, 16, 162–175 CrossRef CAS.
- E. Pilon-Smits, Annual Review of Plant Biology, 2005, 56, 15–39 Search PubMed.
- N. Verbruggen, C. Hermans and H. Schat, New Phytologist, 2009, 182, 781–781 Search PubMed.
- E. Grill, S. Loffler, E. L. Winnacker and M. H. Zenk, Proceedings of the National Academy of Sciences of the United States of America, 1989, 86, 6838–6842 CAS.
- M. H. Zenk, Gene, 1996, 179, 21–30 CrossRef CAS.
- V. Loreti, D. Toncelli, E. Morelli, G. Scarano and J. Bettmer, Analytical and Bioanalytical Chemistry, 2005, 383, 398–403 CrossRef CAS.
- J. M. Gong, D. A. Lee and J. I. Schroeder, Proceedings of the National Academy of Sciences of the United States of America, 2003, 100, 10118–10123 CrossRef CAS.
- M. Margoshes and B. L. Vallee, Journal of the American Chemical Society, 1957, 79, 4813–4814 CrossRef CAS.
- C. A. Blindauer and O. I. Leszczyszyn, Nat. Prod. Rep., 2010, 27, 720–741 RSC.
- E. Freisinger, Dalton Trans, 2008, 6663–6675 RSC.
- C. Cobbett and P. Goldsbrough, Annu. Rev. Plant Biol., 2002, 53, 159–182 CrossRef CAS.
- J. Heise, S. Krejci, J. Miersch, G.-J. Krauss and K. Humbeck, Crop Sci, 2007, 47, 1111–1118 CrossRef CAS.
- K. Akashi, N. Nishimura, Y. Ishida and A. Yokota, Biochem Bioph Res Co, 2004, 323, 72–78 Search PubMed.
- J. Yuan, D. Chen, Y. J. Ren, X. L. Zhang and J. Zhao, Plant Physiol, 2008, 146, 1637–1650 CrossRef CAS.
- G. K. Zhou, Y. F. Xu and J. Y. Liu, J Plant Physiol, 2005, 162, 686–696 CrossRef CAS.
- W. Zhu, D. X. Zhao, Q. Miao, T. T. Xue, X. Z. Li and C. C. Zheng, J Plant Biol, 2009, 52, 585–592 Search PubMed.
- T. Xue, X. Li, W. Zhu, C. Wu, G. Yang and C. Zheng, J Exp Bot, 2009, 60, 339–349 CAS.
- H. L. Wong, T. Sakamoto, T. Kawasaki, K. Umemura and K. Shimamoto, Plant Physiol, 2004, 135, 1447–1456 CrossRef CAS.
- S. H. Kim, H. S. Lee, W. Y. Song, K. S. Choi and Y. Hur, J Plant Biol, 2007, 50, 1–7 Search PubMed.
- L. Hanley-Bowdoin and B. G. Lane, Eur J Biochem, 1983, 135, 9–15 CrossRef CAS.
- B. Lane, R. Kajioka and T. Kennedy, Biochem Cell Biol, 1987, 65, 1001–1005 Search PubMed.
- E. A. Peroza, R. Schmucki, P. Guntert, E. Freisinger and O. Zerbe, J Mol Biol, 2009, 387, 207–218 CrossRef CAS.
- O. I. Leszczyszyn, C. R. J. White and C. A. Blindauer, Mol Biosyst, 2010 Search PubMed.
- A. Murphy, J. Zhou, P. B. Goldsbrough and L. Taiz, Plant Physiol., 1997, 113, 1293–1301 CrossRef CAS.
- Z. Yang, Y. R. Wu, Y. Li, H. Q. Ling and C. C. Chu, Plant Molecular Biology, 2009, 70, 219–229 CrossRef CAS.
- I. D. Rodríguez-Llorente, P. Pérez-Palacios, B. Doukkali, M. A. Caviedes and E. Pajuelo, Plant Sci, 2010, 178, 327–332 CrossRef CAS.
- A. M. Zimeri, O. P. Dhankher, B. McCaig and R. B. Meagher, Plant Mol Biol, 2005, 58, 839–855 CrossRef CAS.
- P. Kille, D. R. Winge, J. L. Harwood and J. Kay, FEBS Lett, 1991, 295, 171–175 CrossRef CAS.
- D. Profrock, P. Leonhard and A. Prange, Analytical and Bioanalytical Chemistry, 2003, 377, 132–139 CrossRef CAS.
- R. Lobinski, H. Chassaigne and J. Szpunar, Talanta, 1998, 46, 271–289 CrossRef CAS.
- A. Prange and D. Schaumloffel, Analytical and Bioanalytical Chemistry, 2002, 373, 441–453 CrossRef CAS.
- K. Gellein, P. M. Roos, L. Evje, O. Vesterberge, T. P. Flaten, M. Nordberg and T. Syversen, Brain Res., 2007, 1174, 136–142 CrossRef CAS.
- M. Malavolta, F. Piacenza, L. Costarelli, R. Giacconi, E. Muti, C. Cipriano, S. Tesei, S. Spezia and E. Mocchegiani, J. Anal. At. Spectrom., 2007, 22, 1193–1198 RSC.
- A. Rodriguez-Cea, A. R. L. Arias, M. R. de la Campa, J. C. Moreira and A. Sanz-Medel, Talanta, 2006, 69, 963–969 CrossRef CAS.
- H. G. Infante, F. Cuyckens, K. Van Campenhout, R. Blust, M. Claeys, L. Van Vaeck and F. C. Adams, J. Anal. At. Spectrom., 2004, 19, 159–166 RSC.
- C. N. Ferrarello, J. R. Encinar, G. Centineo, J. I. G. Alonso, M. R. F. de la Campa and A. Sanz-Medel, J. Anal. At. Spectrom., 2002, 17, 1024–1029 RSC.
- C. B'Hymer and J. A. Caruso, Journal of Chromatography A, 2004, 1045, 1–13 CrossRef CAS.
- M. Bissen and F. H. Frimmel, Acta Hydrochimica Et Hydrobiologica, 2003, 31, 9–18 CrossRef CAS.
- A. A. Meharg and J. Hartley-Whitaker, New Phytologist, 2002, 154, 29–43 Search PubMed.
- M. Vahter, Toxicology, 2002, 181, 211–217 CrossRef.
- S. McSheehy, J. Szpunar, R. Morabito and P. Quevauviller, Trac-Trends in Analytical Chemistry, 2003, 22, 191–209 CrossRef CAS.
- M. Quaghebeur and Z. Rengel, Microchimica Acta, 2005, 151, 141–152 CrossRef CAS.
- A. M. Carey, K. G. Scheckel, E. Lombi, M. Newville, Y. Choi, G. J. Norton, J. M. Charnock, J. Feldmann, A. H. Price and A. A. Meharg, Plant Physiology, 2010, 152, 309–319 CrossRef CAS.
- U. Kohlmeyer, J. Kuballa and E. Jantzen, Rapid Communications in Mass Spectrometry, 2002, 16, 965–974 CrossRef CAS.
- J. J. Sloth, E. H. Larsen and K. Julshamn, J. Anal. At. Spectrom., 2003, 18, 452–459 RSC.
- V. Nischwitz and S. A. Pergantis, J. Anal. At. Spectrom., 2006, 21, 1277–1286 RSC.
- I. J. Pickering, R. C. Prince, M. J. George, R. D. Smith, G. N. George and D. E. Salt, Plant Physiology, 2000, 122, 1171–1177 CrossRef CAS.
- O. P. Dhankher, Y. J. Li, B. P. Rosen, J. Shi, D. Salt, J. F. Senecoff, N. A. Sashti and R. B. Meagher, Nature Biotechnology, 2002, 20, 1140–1145 CrossRef.
- A. Raab, H. Schat, A. A. Meharg and J. Feldmann, New Phytologist, 2005, 168, 551–558 Search PubMed.
- G. P. Bienert, M. Thorsen, M. D. Schussler, H. R. Nilsson, A. Wagner, M. J. Tamas and T. P. Jahn, Bmc Biology, 2008, 6 Search PubMed.
- S. B. Ha, A. P. Smith, R. Howden, W. M. Dietrich, S. Bugg, M. J. O'Connell, P. B. Goldsbrough and C. S. Cobbett, Plant Cell, 1999, 11, 1153–1163 CrossRef CAS.
- F. E. C. Sneller, L. M. Van Heerwaarden, F. J. L. Kraaijeveld-Smit, W. M. Ten Bookum, P. L. M. Koevoets, H. Schat and J. A. C. Verkleij, New Phytologist, 1999, 144, 223–232 Search PubMed.
- K. Francesconi, P. Visoottiviseth, W. Sridokchan and W. Goessler, Science of the Total Environment, 2002, 284, 27–35 CrossRef CAS.
- E. Lombi, F. J. Zhao, M. Fuhrmann, L. Q. Ma and S. P. McGrath, New Phytologist, 2002, 156, 195–203 Search PubMed.
- W. J. Liu, Y. G. Zhu, Y. Hu, P. N. Williams, A. G. Gault, A. A. Meharg, J. M. Charnock and F. A. Smith, Environmental Science & Technology, 2006, 40, 5730–5736 Search PubMed.
- P. N. Williams, A. H. Price, A. Raab, S. A. Hossain, J. Feldmann and A. A. Meharg, Environmental Science & Technology, 2005, 39, 5531–5540 Search PubMed.
- A. L. Juhasz, E. Smith, J. Weber, M. Rees, A. Rofe, T. Kuchel, L. Sansom and R. Naidu, Environmental Health Perspectives, 2006, 114, 1826–1831 CAS.
- J. Bettmer, M. M. Bayon, J. R. Encinar, M. L. F. Sanchez, M. D. F. de la Campa and A. S. Medel, J. Proteomics, 2009, 72, 989–1005 CrossRef CAS.
- H. Zhang, C. Lian and Z. Shen, Ann. Bot., 2009, 923–930 CrossRef CAS.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |