L-5F, an apolipoprotein A-I mimetic, inhibits tumor angiogenesis by suppressing VEGF/basic FGF signaling pathways†‡
Feng
Gao
a,
Sergio X.
Vasquez
b,
Feng
Su
a,
Svetlana
Roberts
a,
Neha
Shah
b,
Victor
Grijalva
a,
Satoshi
Imaizumi
a,
Arnab
Chattopadhyay
a,
Ekambaram
Ganapathy
a,
David
Meriwether
a,
Brad
Johnston
b,
G. M.
Anantharamaiah
c,
Mohamad
Navab
a,
Alan M.
Fogelman
a,
Srinivasa T.
Reddy§
*a and
Robin
Farias-Eisner¶
*a
aDavid Geffen School of Medicine, University of California Los Angeles, 10833 Le Conte Avenue, Los Angeles, CA 90095-5347, USA. E-mail: rfeisner@mednet.ucla.edu; sreddy@mednet.ucla.edu
bNumira Biosciences, 560 Arapeen Drive, Suite 250, Salt Lake City, UT 84108, USA
cDepartment of Medicine University of Alabama at Birmingham, Birmingham, AL 35294, USA
First published on 1st February 2011
Abstract
We recently reported that apolipoprotein A-I (apoA-I) and apoA-I mimetic peptides inhibit tumor growth and improve survival in a mouse model of ovarian cancer. The current study was designed to examine whether inhibition of angiogenesis is one of the mechanisms for the observed anti-tumorigenic effects. The apoA-I mimetic peptide L-5F had no affect on proliferation and cell viability of human umbilical vascular endothelial cells (HUVECs) in the basal state; however, treatment with L-5F at 1, 3, and 10 μg ml−1, dose-dependently inhibited both vascular endothelial growth factor (VEGF)- and basic fibroblast growth factor (bFGF)-induced proliferation, cell viability, migration, invasion and tube formation in HUVECs. L-5F inhibited VEGF- and bFGF-induced activation of their corresponding receptors, VEGFR2 and FGFR1, as well as downstream signaling pathways, including Akt and ERK1/2. MicroCT scanning and immunohistochemistry staining demonstrated that daily injection of L-5F (10 mg kg−1) decreased both the quantity and size of tumor vessels in mice. L-5F treated mice showed significantly reduced levels of VEGF in both tumor tissue and the circulation, which is consistent with in vitro data showing that L-5F inhibited production and secretion of VEGF from mouse and human ovarian cell lines in the absence and presence of exogenously added lysophosphatidic acid, a potent tumor promoter. In conclusion, our data that L-5F inhibits angiogenesis suggests that apoA-I mimetic peptides may serve as novel anti-angiogenesis agents for the treatment of angiogenesis-associated diseases, including cancer.
Insight, innovation, integrationExtracellular input is an essential aspect of cell integrity. Unlike the immediate milieu, the cues from vascular flow are important mediators of information for maintaining cell/tissue/organ homeostasis. We report here a very innovative paradigm, which for the first time, suggests that apolipoprotien A-I mimetic peptides may modulate tumorigenesis by inhibiting both vascular endothelial growth factor and basic fibroblast growth factor signaling pathways. We postulate that apoA-I mimetic peptides will become a novel class of anti-angiogenesis agents used for therapies in a number of diseases whose etiology is governed by angiogenesis, including cancer. |
Introduction
Ovarian cancer is the leading cause of death from gynecologic malignancies in the Western world. The 5-year survival for ovarian cancer is only 50% in part because of (i) the lack of tests to diagnose ovarian cancer at an early stage and (ii) the absence of effective therapeutic strategies. Our previous studies suggested that apolipoprotein A-I (apoA-I), the main protein constituent of high density lipoprotein (HDL), is not only a biomarker for detection of early stage ovarian cancer, but is also a promising therapeutic target for the treatment of ovarian cancer.1–4Peptides that mimic the properties of apoA-I, termed apoA-I mimetic peptides, hold promise for the treatment of many inflammatory diseases.5,6 Recently, we have reported that L-5F, one of the apoA-I mimetic peptides, greatly inhibits tumor growth in a mouse model.4 However, the mechanism behind the anti-tumorigenic effects of apoA-I and apoA-I mimetic peptides remains unknown.Angiogenesis plays a critical role in growth and metastasis of solid tumors, which account for more than 85% of cancer mortality7 including ovarian cancer.8,9Angiogenesis is a complicated process, requiring the coordinated action of a variety of growth factors and cell-adhesion molecules in endothelial and mural cells. Vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) are critical pro-angiogenic factors, involved in all phases of angiogenesis including growth, migration, and differentiation of endothelial cells; and they have been implicated in etiology of ovarian cancer.10–14 In the present study, we examined whether the anti-tumorigenic effects of L-5F are due, in part, to the inhibition of VEGF/bFGF-mediated angiogenesis.
Both microCT scanning and immunohistochemistry (IHC) staining demonstrated that the quantity and size of vessels in tumor tissues were decreased in mice receiving L-5F peptide, compared to mice that did not receive L-5F peptide. Our results demonstrate that L-5F dose-dependently inhibits VEGF/bFGF-induced proliferation, migration, invasion, and tube formation of human umbilical vascular endothelial cells (HUVECs). These effects are mediated by the inhibitory effect of L-5F on the activation of VEGF receptor-2 (VEGFR2) and FGF receptor-1 (FGFR1), and the phosphorylation of downstream signaling molecules. We further show that VEGF levels are reduced in tumor tissue and the circulation in mice treated with L-5F. Moreover, L-5F inhibits VEGF production from ovarian cancer cell lines consistent with our in vivo findings. Our results demonstrate that L-5F, an apoA-I mimetic peptide, inhibits angiogenesis and may serve as a therapeutic agent for the treatment of angiogenesis-associated diseases, especially ovarian cancer.
Results
L-5F suppresses tumor angiogenesisin vivo
Our previous data showed that apoA-I mimetic peptides, L-4F and L-5F, inhibit tumor growth in an immunocompetent mouse model of ovarian cancer that utilizes the epithelial cancer cell line ID8.4 Given that growth of solid tumors is angiogenesis-dependent and suppression of angiogenesis can inhibit tumor growth, we further examined the inhibitory function of tumor angiogenesis by L-4F and L-5F using the same model. L-4F treatment inhibited angiogenesis in tumors as measured by CD31 staining (Supplemental Fig. 1‡). Both microCT scanning and IHC staining showed that L-5F treatment decreased the quantity and size of perfused vessels and total vessels, respectively, compared to the vehicle-treated group (Fig. 1A and B).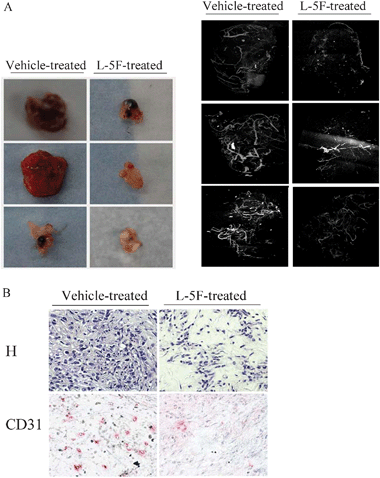 | ||
| Fig. 1 L-5F inhibits tumor angiogenesis in vivo. Flank tumors were established in wild-type C57BL6/J mice as described under Materials and Methods. After treatment with ABCT buffer or L-5F (10 mg kg−1, s.c., daily injection) for 5 weeks, mice were perfused with PBS to remove blood, and then the vasculatures were filled with MICROFIL. Dissected tumors were subjected to microCT scanning or IHC staining. A, microCT imaging analysis shows that L-5F decreases the quantity and size of vessels, compared to vehicle-treated group. Left, Dissected tumors from 3 mice that were vehicle treated and 3 mice that were L-5F treated. Right, the vessel images from the tumors shown on the left. B, IHC staining of tumor tissues. H, tissues were stained with hematoxylin to define tissue morphology. CD31, Immunostaining was performed with anti-CD31 for detection of endothelial cells in microvessels. | ||
L-5F inhibits VEGF/bFGF-induced proliferation and viability of HUVECs, but has no effect on HUVECs in the basal state
To determine whether apoA-I mimetic peptides inhibit angiogenesis, we first examined the effect of L-5F on the growth of endothelial cells. Cell proliferation and viability assays showed that L-5F did not inhibit the proliferation and viability of HUVECs in the basal state, but pretreatment with L-5F significantly inhibited VEGF/bFGF-induced cell proliferation and viability at 24 h and 48 h as shown in Fig. 2. Moreover, treatment with a closely related, scrambled control peptide (SP)4 had no effect on VEGF/bFGF-induced proliferation and viability of HUVECs, confirming the specific effects of L-5F (Fig. 2). | ||
| Fig. 2 L-5F inhibits VEGF- and bFGF-stimulated proliferation and viability of HUVECs. HUVECs were treated with vehicle or different concentrations of L-5F (1, 3, and 10 μg ml−1) or with 10 μg ml−1 of a closely related scrambled control peptide that was used in our previous experiments4 (SP) in the presence or absence of growth factors. After 24 h or 48 h, BrdU assay and MTS assay were used for measurements of cell proliferation and viability, respectively. A. Left, pretreatment with L-5F has no effect on proliferation of HUVECs at 24 h. Middle, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated proliferation of HUVECs at 24 h. Right, pretreatment with L-5F dose-dependently inhibits bFGF-stimulated proliferation of HUVECs at 24 h. B. Left, pretreatment with L-5F has no effect on viability of HUVECs at 24 h. Middle, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated viability of HUVECs at 24 h. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated viability of HUVECs at 24 h. C. Left, pretreatment with L-5F has no effect on proliferation of HUVECs at 48 h. Middle, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated proliferation of HUVECs at 48 h. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated proliferation of HUVECs at 48 h. D. Left, pretreatment with L-5F has no effect on viability of HUVECs at 48 h. Middle, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated viability of HUVECs at 48 h. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated viability of HUVECs at 48 h. E. Left, post-treatment with L-5F dose-dependently inhibits VEGF-stimulated viability of HUVECs at 48 h. Right, post-treatment with L-5F dose-dependently inhibits bFGF-stimulated viability of HUVECs at 48 h. #, p < 0.05, compared to the corresponding control group. ##, p < 0.01, compared to the corresponding control group. *, p < 0.05, compared to the corresponding VEGF- or bFGF-treated groups. **, p < 0.01, compared to the corresponding VEGF- or bFGF-treated groups. n = 4 for each group. SP10, a scrambled peptide used at 10 μg ml−1. Pretreatment, cells were treated with L-5F for 5 min, and then VEGF (10 ng ml−1) or bFGF (5 ng ml−1) were added. Post-treatment, cells were treated with VEGF (10 ng ml−1) or bFGF (5 ng ml−1) for 1 h and then L-5F was added. | ||
L-5F inhibits VEGF/bFGF-induced migration, invasion, and tube formation of HUVECs
Cell migration and invasion are two important steps for angiogenesis. We performed a wound healing migration assay and a transwell assay to determine the effects of L-5F on VEGF/bFGF-induced migration and invasion of HUVECs. L-5F inhibited the migration and invasion of HUVECs in a dose-dependent manner (Fig. 3A and B). Two positive controls, U0126 (a MEK1/2 inhibitor) and LY294002 (a PI3 kinase inhibitor), inhibited VEGF/bFGF-induced migration and invasion of HUVECs, as previously reported.15,16 Treatment with a closely related, scrambled control peptide (SP) had no effect on cell migration and invasion. We next examined the effect of L-5F on the differentiation of HUVECs. We performed tube formation assays in the presence of different concentrations of L-5F peptide with or without VEGF/bFGF. As shown in Fig. 3C, L-5F, U0126, and LY294002, but not the closely related, scrambled control peptide (SP), dramatically inhibited VEGF/bFGF-stimulated tube formation of HUVECs, suggesting that L-5F affects differentiation in vitro. | ||
| Fig. 3 L-5F inhibits VEGF- and bFGF-stimulated migration, invasion, and tube formation of HUVECs. HUVECs were treated with vehicle or different concentrations of L-5F (1, 3, and 10 μg ml−1) or with or with 10 μg ml−1 of a closely related scrambled control peptide that was used in our previous experiments4 (SP) in the presence or absence of growth factors. A. Left, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated migration of HUVECs at 16 h. VEGF was used at 10 ng ml−1. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated migration of HUVECs at 16 h. bFGF was used at 5 ng ml−1. B. Left, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated invasion of HUVECs at 5 h. VEGF was used at 20 ng ml−1. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated invasion of HUVECs at 5 h. bFGF was used at 20 ng ml−1. C. Left, pretreatment with L-5F (but not SP) dose-dependently inhibits VEGF-stimulated tube formation of HUVECs at 8 h. VEGF was used at 50 ng ml−1. Right, pretreatment with L-5F (but not SP) dose-dependently inhibits bFGF-stimulated tube formation of HUVECs at 8 h. bFGF was used at 50 ng ml−1. #, p < 0.05, compared to the corresponding control group. ##, p < 0.01, compared to the corresponding control group. *, p < 0.05, compared to the corresponding VEGF- or bFGF-treated groups. **, p < 0.01, compared to the corresponding VEGF- or bFGF-treated groups. n = 4 for each group. SP10, a scrambled peptide used at 10 μg ml−1. U0126, a MEK1/2 inhibitor used at 10 μM. LY294002, a PI3 kinase inhibitor used at 10 μM. Pretreatment, cells were treated with L-5F for 5 min, and then VEGF or bFGF were added. | ||
L-5F inhibits VEGF/bFGF-induced activation of VEGFR2 and FGFR1, and phosphorylation of downstream signaling molecules
VEGF and bFGF produce angiogenic effects in endothelial cells by signaling through cell surface receptor tyrosine kinases. VEGFR2 is a major receptor for VEGF-induced signaling in endothelial cells,17 and phosphorylation of FGFR1 is important for bFGF signaling pathway.18 Our data showed that while L-5F had no effect on basal VEGFR2 and FGFR1 kinase activities (data not shown), it abolished VEGF/bFGF-stimulated activation of both receptors (Fig. 4A). Moreover, L-5F treatment inhibited phosphorylation of signaling molecules downstream of VEGFR2 and FGFR1, including Akt and extracellular-signal-regulated kinase-1/2 (ERK1/2) (Fig. 4B). | ||
| Fig. 4 L-5F inhibits the activation of VEGFR2 /FGFR1 and their downstreamsignaling molecules.A. L-5F abolishes VEGF- and bFGF-induced increase of kinase activity of their corresponding receptors. HUVECs were cultured in 6-well plates. Following an overnight starvation, cells were treated with various concentrations of L-5F (1, 3, and 10 μg ml−1) for 2 h, and VEGF or bFGF were added at a final concentration of 80 ng ml−1 for 5 min. Cell lysates were collected and kinase activity was analyzed as described under Materials and Methods. Left, effect of L-5F on VEGF-induced VEGFR2 kinase activity. Right, effect of L-5F on bFGF-induced FGFR1 kinase activity. #, p < 0.05, compared to the corresponding control group. *, p < 0.05, compared to the corresponding VEGF- or bFGF-treated group. n = 3 for each group. B. L-5F inhibits VEGF- and bFGF-induced phosphorylation of downstream signaling molecules. HUVECs were cultured in 6-well plates. After starvation, cells were treated with various concentrations of L-5F (1, 3, and 10 μg ml−1) for 2 h, and VEGF or bFGF was added for various time points. For detection of phosphorylation of ERK, cells were stimulated with VEGF at 10 ng ml−1 or bFGF at 5 ng ml−1 for 15 min. For detection of phosphorylation of Akt, cells were stimulated with VEGF at 10 ng ml−1 or bFGF at 5 ng ml−1 for 20 min. Top, western blotting images for VEGF/bFGF-induced phosphorylation of ERK1/2 and Akt. Middle, ratio of p-ERK1/2 to ERK1/2 signal intensities. Bottom, ratio of p-Akt to Akt signal intensities. | ||
L-5F treatment reduces VEGF levels in vivo
We next examined whether L-5F treatment affects VEGF levels in vivo. Tumor tissues and serum samples from experiments described under Fig. 1 were analyzed for VEGF levels using three different approaches, and these levels were found to be significantly reduced in tissues and the circulation of mice that received L-5F when compared to control mice (Fig. 5A and B). | ||
| Fig. 5 L-5F inhibits VEGF production in vivo. Flank tumors were established in wild-type C57BL6/J mice as described under Materials and Methods. After treatment with ABCT buffer or L-5F (10 mg kg−1, s.c., daily injection) for 5 weeks, the tumor tissues and serum were collected and used for further analysis. A. IHC staining analysis for VEGF expression in tumor tissues. B. ELISA analysis of VEGF levels in mouse serum. #, p < 0.05, compared to the corresponding vehicle-treated group. n = 6 for each group. | ||
L-5F inhibits LPA-induced tumor cell growth and VEGF production in both mouse and human ovarian cancer cell lines
We have previously reported that lysophosphatidic acid (LPA), a bioactive phospholipid, is reduced in mice treated with L-5F.4LPA induces both ovarian cancer cell proliferation and VEGF production.19,20 We next examined whether L-5F can inhibit LPA-induced cell viability and VEGF production in two human ovarian cancer cell lines, SKOV3 and OV2008. L-5F dose-dependently inhibited exogenously added LPA-induced viability of SKOV3 and OV2008 cells at 24 h after LPA treatment (Fig. 6A). Moreover, L-5F treatment resulted in significantly decreased VEGF levels in supernatants of both SKOV3 and OV2008 cells in the presence and absence of exogenously added LPA (Fig. 6B). However, the inhibitory effect of L-5F on LPA-induced VEGF production was only observed when cells were treated with LPA and L-5F at the same time, but not when cells were pretreated with L-5F, indicating that L-5F binds to LPA and inhibits its effects (data not shown). Similarly, L-5F not only inhibited exogenously added LPA-induced cell viability, but also dose-dependently decreased basal and exogenously added LPA-induced VEGF production in ID8 cells (Fig. 6A and B). Consistent with the ELISA data, we observed a dose-dependent inhibitory effect of L-5F on exogenously added LPA-stimulated VEGF mRNA expression (Fig. 6C). | ||
| Fig. 6 L-5F inhibits cell proliferation and VEGF production in mouse and human ovarian cancer cell lines in the presence or absence of LPA. A. Cells were plated in 96-well plate at 2000 cells well−1. Following an overnight starvation, cells were treated with various concentrations of L-5F (1, 3, and 10 μg ml−1) in the presence or absence of LPA at 20 μM for 24 h. Then, cells were subjected to MTS assay for measurement of cell viability. Left, L-5F inhibits LPA-stimulated viability of SKOV3 cells. Middle, L-5F inhibits LPA-stimulated viability of OV2008 cells. Right, L-5F inhibits LPA-stimulated viability of ID8 cells. B. Cells were plated in 6-well plates at 2 × 105cells well−1. After starvation, cells were treated with various concentrations of L-5F (1, 3, and 10 μg ml−1) in the presence or absence of LPA at 20 μM for 12 h and then, supernatants were collected for ELISA assay. Left, L-5F decreases VEGF levels in supernatants of SKOV3 cells. Middle, L-5F decreases VEGF levels in supernatants of OV2008 cells. Right, L-5F decreases VEGF levels in supernatants of ID8 cells. C. Cells were plated in 6-well plates at 2 × 105cells well−1. After starvation, cells were treated with various concentrations of L-5F (1, 3, and 10 μg ml−1) in the presence or absence of LPA at 20 μM for 4 h and then, total RNA was collected for Q-PCR assay. Left, L-5F decreases LPA-stimulated increase of VEGF mRNA level in OV2008 cells. Right, L-5F decreases LPA-stimulated increase of VEGF mRNA level in ID8 cells. #, p < 0.05, compared to the corresponding control group. ##, p < 0.01, compared to the corresponding control group. *, p < 0.05, compared to the corresponding LPA-treated groups. **, p < 0.01, compared to the corresponding LPA-treated groups. n = 4 for each group. | ||
Discussion
Our previous results showed that apoA-I inhibits tumor growth and improves survival in a mouse ovarian cancer model.4 We have also demonstrated that apoA-I mimetic peptides L-4F, D-4F, and L-5F are effective anti-tumorigenic molecules.4 However, the details of the mechanism(s) by which apoA-I mimetic peptides inhibit tumor growth have not previously been addressed. In this study, we provide evidence, for the first time, that L-5F, an apoA-I mimetic peptide, inhibits angiogenesis. Angiogenesis is a complex process requiring multiple essential steps, including cell proliferation, migration, invasion, and tube formation. We show that L-5F (i) reduced tumor angiogenesisin vivo (Fig. 1) and (ii) significantly inhibited VEGF/bFGF-stimulated proliferation, migration, invasion, and tube formation of HUVECs (Fig. 2 and 3). The mechanism by which apoA-I mimetic peptides inhibit inflammatory responses in a number of pro-inflammatory models5 is related to their remarkable ability to bind21 and remove22 bioactive lipids from the circulation. We previously demonstrated that (i) apoA-I mimetic peptides bind with great affinity to the tumorigenic bioactive lipid lysophosphatidic acid (LPA) and (ii) apoA-I mimetic peptide treatment significantly reduced LPA levels in mice concurrent with reduced tumor load.4 In the current report, we demonstrate that L-5F inhibits angiogenesis by mechanisms that may involve the removal of LPA-like bioactive lipids from the tumor environment. Based on our results, we propose a potential mechanism of action for L-5F and other apoA-I mimetic peptides on angiogenesis (Fig. 7).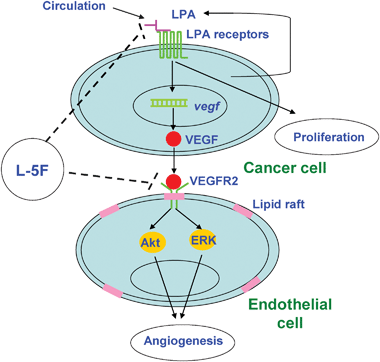 | ||
| Fig. 7 A model for the proposed mechanism of action of L-5F-mediated inhibition of tumor angiogenesis.LPA from the circulation or produced by ovarian tumors binds to LPA receptors and stimulates production and secretion of pro-angiogenic factors, including VEGF. VEGF binds to VEGFR2, activates downstream signaling molecules, and leads to angiogenesis. L-5F inhibits LPA-induced VEGF production, thus blocking VEGF-induced activation of VEGFR2 and downstream signaling pathways, and as a result, tumor angiogenesis is suppressed. | ||
Our previous results show that L-5F inhibits proliferation and viability of both human and mouse ovarian cells.4 In the present study we demonstrate that L-5F has no effect on proliferation and viability of HUVECs in the basal state, but that it abolishes VEGF/bFGF-stimulated growth (Fig. 2), suggesting that L-5F's effect on cell growth and viability is specific to cancer and growth factor induced cells. It is known that apoA-I mimetic peptides exert their effects, in large part, by binding pro-inflammatory lipids.5,21LPA is a bioactive lysophospholipid that elicits multiple cellular responses, including proliferation, migration, invasion, and production of growth factors. Xu et al.23 reported that plasma levels of LPA were dramatically elevated in patients with ovarian cancer (Stage I: 5.6 ± 2.99 μM, Stage II, III, and IV: 8.1 ± 1.87 μM, Recurrent: 11.5 ± 3.16 μM), compared to healthy controls (0.6 ± 0.19 μM). Various LPA acyl species were elevated 4–8 fold in malignant effusions from patients with ovarian cancer, and the greatest increase was observed on LPA 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4,24 which was used in the present study. In another study Xiao et al.25 reported that the mean level of total LPA in ascites samples from patients with ovarian cancer was 18.9 μM compared to 2.9 μM in nonmalignant samples. Our data showed that LPA at 20 μM increased secretion of VEGF from ovarian cancer cell lines (Fig. 6), which is consistent with the previous reports.20,26 Although the increase in VEGF protein was modest, a two-fold to four-fold increase in VEGF leads to considerable tumorigenic competence in colorectal cancer.20,27L-5F significantly suppressed LPA-stimulated proliferation of ovarian cancer cells and production of VEGF (Fig. 6), which may be mediated by strong binding between LPA and apoA-I mimetic peptides, including L-5F.4 Based on the similarity of apoA-I mimetic peptides on lipid-associating properties,21 binding to LPA and inhibiting its function may be considered as one of the common mechanisms responsible for the anti-angiogenic effects of apoA-I and apoA-I mimetic peptides.
:4,24 which was used in the present study. In another study Xiao et al.25 reported that the mean level of total LPA in ascites samples from patients with ovarian cancer was 18.9 μM compared to 2.9 μM in nonmalignant samples. Our data showed that LPA at 20 μM increased secretion of VEGF from ovarian cancer cell lines (Fig. 6), which is consistent with the previous reports.20,26 Although the increase in VEGF protein was modest, a two-fold to four-fold increase in VEGF leads to considerable tumorigenic competence in colorectal cancer.20,27L-5F significantly suppressed LPA-stimulated proliferation of ovarian cancer cells and production of VEGF (Fig. 6), which may be mediated by strong binding between LPA and apoA-I mimetic peptides, including L-5F.4 Based on the similarity of apoA-I mimetic peptides on lipid-associating properties,21 binding to LPA and inhibiting its function may be considered as one of the common mechanisms responsible for the anti-angiogenic effects of apoA-I and apoA-I mimetic peptides.
VEGF is a potent stimulator of angiogenesis associated with physiologic processes such as wound healing and with pathological conditions including ischemic diseases and tumor growth. VEGF is required for endothelial cell proliferation, survival, migration, and tube formation, and more recently VEGF has been implicated in the recruitment and vascularization of dendritic cells, including plasmacytoid dendritic cell and myeloid dendritic cell, from the bone marrow and peripheral tissues at sites of tumor growth.28 Our in vivo data showed that VEGF levels were reduced in both tumor tissue and the circulation after treatment with L-5F (Fig. 5). In light of our previous findings that LPA levels were reduced in mice that received apoA-I mimetic peptides,4 it is conceivable that LPA is at least in part responsible for the VEGF levels in the tumor milieu in the experimental mice. Our in vitro data further strengthen this hypothesis since L-5F inhibits LPA-stimulated cell viability and VEGF secretion from ovarian cancer cell lines (Fig. 6). Since L-5F treatment reduced basal VEGF levels in supernatants of ovarian cancer cells (Fig. 6B), it maybe that cancer cells (but not HUVEC) produce a continuous level of LPA (or other bioactive lipids) that induces basal levels of VEGF. Alternatively, alterations in membrane lipid structure in the presence of L-5F may modify the VEGF-producing capacity of these cells. In either case, it appears that L-5F treatment reduces basal VEGF production concurrent with decrease in viability (Fig. 6) in cancer cells. To further understand the molecular mechanism of L-5F-mediated anti-angiogenesis effect, we examined whether L-5F inhibits the activation of VEGFR2 signaling pathway. We found that L-5F had no effect on basal VEGFR2 kinase activity (data not shown), but completely abolished VEGF-stimulated increase of kinase activity (Fig. 4A). Western blot analysis also showed that L-5F pretreatment prevented VEGF-induced phosphorylation of downstream signaling molecules (Fig. 4B).
Inhibitors of angiogenesis can suppress tumor growth in preclinical models and have entered the clinic as prospective anticancer therapeutics.29 Formation of new vessels is caused by coordination of multiple growth factors. Since beacizumab, a humanized anti-VEGF monoclonal antibody, was approved for the treatment of metastatic colorectal cancer in 2004, VEGF-targeted therapy has become an important treatment option for the management of a number of human malignancies.30 However, resistance to VEGF-targeted therapy has developed in the vast majority of patients.31,32 As a result, VEGF-targeted monotherapy is not enough, and combinational therapy and inhibition of multiple targets are required. Based on this fact, we also tested whether L-5F inhibits bFGF-stimulated angiogenesis. Same to its effect on VEGF signaling pathway, L-5F blocked bFGF-induced activation of FGFR1 and phosphorylation of downstream signaling molecules (Fig. 4). Consistent with these data, we observed significant inhibitory effects of L-5F on bFGF-induced proliferation, viability, migration, invasion, and tube formation of endothelial cells (Fig. 2 and 3). The fact that L-5F blocks the activation of both VEGFR2 and FGFR1 suggests that L-5F's effects are not specific to one receptor. On the other hand, the consistency of effects of pretreatment and post-treatment of L-5F on proliferation of endothelial cells and usage of a closely related, scrambled control peptide exclude the possibility of non-specific blockade of L-5F on membrane functions. So, how does L-5F work to prevent activation of VEGFR2 and FGFR1? Previous reports show that apoA-I mimetic peptides can bind several bioactive lipids5,21 with very high affinity that are important for the maintenance of structure and function of the cellular membrane, as reviewed by Phillips et al., in 2009.33 We hypothesize that L-5F and apoA-I mimetic peptides alter lipid composition/structure of cellular membranes and may thus lead to alterations in function of membrane receptors, such as VEGFR2 and FGFR1. In support of this hypothesis, recent studies by Yvan-Charvet et al. report that ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation.34 The authors suggest that the membrane cholesterol content modulates the growth factor receptor mediated response of these cells and dictates their proliferation capacity.
Conclusion
Angiogenesis is critical for development of solid tumors and a number of diseases including diabetic retinopathy and obesity. Our studies show that L-5F, an apoA-I mimetic peptide, inhibits production of VEGF and VEGF/bFGF-stimulated angiogenesis, suggesting that L-5F and other apoA-I mimetic peptides, may serve as a novel class of anti-angiogenesis agents.Experimental
Cells, cell culture, reagents
HUVECs were a generous gift from Dr Gautam Chaudhuri's laboratory (Department of Obstetrics and Gynecology, UCLA). HUVECs were cultured in endothelial cell growth medium-2 (EGM-2) (Clonetics, Walkersville, MD) and were used between passages 4–6 for all experiments. ID8 cells were cultured in complete media consisting of Dulbecco's Modified Eagles Medium (DMEM) with high glucose and L-glutamine (2 mM), supplemented with 4% fetal bovine serum (FBS), penicillin (100 U ml−1), streptomycin (100 μg ml−1), and 1 × ITS liquid media supplement (10 μg ml−1insulin, 5 μg ml−1transferin, and 5 ng ml−1sodium selenite) (Sigma-Aldrich, St. Louis, MO). SKOV3 cells were cultured in complete media consisting of DMEM with high glucose and L-glutamine (2 mM), 10% FBS, penicillin (100 U ml−1), and streptomycin (100 μg ml−1). OV2008 cells were cultured in RPMI 1640 media with 10% FBS, penicillin (100 U ml−1), streptomycin (100 μg ml−1), 1 × MEM non-essential amino acid solution (Invitrogen, Carlsbad, CA), and Insulin (0.25 U ml−1) (Invitrogen). L-5F peptides were synthesized by Peptisyntha Inc. (Peptisyntha Inc., Torrance, CA), dissolved in ABCT buffer (50 mM ammonium bicarbonate, pH 7.0, containing 0.1 mg ml−1 Tween-20) at 1 mg ml−1 freshly, and diluted to the required concentrations before use. U0126 and LY294002 were purchased from Cell Signaling Technology. (Cell Signaling Technology, Inc., Danvers, MA). LPA (Avanti Polar Lipids, Inc., Alabaster, AL) in chloroform was dried as recommended by the manufacturer, dissolved in ethanol at a concentration of 20 mM as a stock solution, and diluted to the required concentrations in the corresponding cell culture media before use.ApoA-I mimetic peptides
L-4F (Ac-D-W–F–K-A-F–Y-D-K–V-A-E-K–F–K-E-A-F-NH2), L-5F: Ac-D-W-L-K-A-F–Y-D-K–V–F-E-K–F–K-E-F–F-NH2 and a scrambled peptide (designated as SP) containing the same amino acids as in the 4F peptides but arranged in a sequence (Ac-D-W–F-A-K-D-Y–F–K–K-A-F–V-E-E-F-A-K-NH2) that prevents the formation of a class A amphipathic helix were all synthesized from all L-amino acids.Cell proliferation and viability assays
Briefly, cells were plated at ∼2000 cells per well in 96-well plate, grown in complete medium for overnight, starved overnight in serum-free basal DMEM (for ID8 and SKOV3), RPMI-1640 medium (for OV2008) or endothelial cell basal medium-2 (EBM-2) supplemented with 0.1% charcoal-stripped FBS (Invitrogen) (for HUVECs), and treated with L-5F (1, 3, or 10 μg ml−1) in the presence or absence of VEGF (10 ng ml−1) (EMD Biosciences, Gibbstown, NJ), or bFGF (5 ng ml−1) (Millipore Corporate, Billerica, MA), or LPA (20 μM) for 24 or 48 h. The CellTiter96 Aqueous One solution cell proliferation assay (MTS assay) (Promega Corporation, Madison, WI) and BrdU cell proliferation assay (EMD Biosciences, Gibbstown, NJ) were used for measurements of viability and cell proliferation, respectively. The experiments were repeated twice with quadruplicate measurements in each experiment.Migration assay
HUVECs were allowed to grow to full confluence in 24-well plates precoated with 1.5% gelatin (Sigma-Aldrich) and then starved overnight in EBM-2 medium supplemented with 0.1% charcoal-stripped FBS. Monolayer HUVECs were wounded by scratching with 200 μl pipette tip. After washing twice with 1× phosphate buffered saline (PBS), cells were treated with L-5F (1, 3, or 10 μg ml−1) in the presence or absence of VEGF (10 ng ml−1) or bFGF (5 ng ml−1) for 16 h. The migrated cells were imaged and quantified by manual counting, and percentage inhibition was expressed using untreated wells at 100%. The experiment was repeated twice with quadruplicate measurements in each experiment.Transwell migration assay
The 96-transwell with 8-μm pore size (Corning Incorporated, Corning, NY) was precoated with 1.5% gelatin for 30 min at 37 °C. After washing the transwells twice with 1 × PBS, the bottom chambers were filled with EBM-2 medium supplemented with 0.1% charcoal-stripped FBS and VEGF (20 ng ml−1) or bFGF (20 ng ml−1), and the top chambers were seeded with starved HUVECs (1 × 104cells per well) in EBM-2 medium supplemented with 0.1% charcoal-stripped FBS. The top and bottom chambers contained the same series of concentrations of L-5F (1, 3, or 10 μg ml−1). After incubation for 5 h, cells on the top surface of the membrane were scraped off with a cotton swab. Cells on the bottom side of the membrane (migrated cells) were fixed with methanol for 20 min, stained with 0.1% crystal violet for 15 min, and washed with 1 × PBS for twice. Invading cells were imaged and quantified by manual counting, and percentage inhibition of invading cells were quantified and expressed on the basis of untreated cells representing 100%. The experiment was repeated twice with quadruplicate measurements in each experiment.Tube formation assay
Growth-factor-reduced Matrigel (BD Biosciences, San Jose, CA) were thawed at 4 °C for overnight and diluted with equal volume of serum-free EBM-2 medium. The final concentration of Matrigel was 5 mg ml−1. Then, each well of prechilled 96-well plates was coated with 50 μl diluted Matrigel and incubated at room temperature for 45 min. Starved HUVECs (1 × 104cells well−1) were plated in EBM-2 medium supplemented with 0.1% charcoal-stripped FBS and treated with different concentrations of L-5F (1, 3, or 10 μg ml−1) in the presence or absence of VEGF (50 ng ml−1) or bFGF (50 ng ml−1). Tubular structures were imaged and counted after 8 h, and inhibition percentage was expressed using untreated wells as 100%. The tubule structures were scored following the same rule as published before:35 0, no real tubes; 1, some poorly formed tubes; 2, some formed tubes; 3, network of tubes both formed and poorly formed; 4, network of formed tubes; and 5, network of well formed tubes. The experiment was repeated twice with quadruplicate measurements in each experiment.Measurement of VEGF production
ID8, OV2008, and SKOV3 cells were seeded in 6-well plate at 2 × 105cells per well20,26 and cultured overnight in normal cultured conditions, starved in serum-free media for overnight, and treated with L-5F (1, 3, or 10 μg ml−1) in the presence or absence of LPA at 20 μM for 12 h. Then, supernatants were collected and subjected to measurement of VEGF levels using the corresponding anti-human or anti-mouse ELISA kit (Raybiotech Inc., Norcross, GA). Concentrations were calculated by comparing the sample absorbance to standard curves. The experiment was repeated twice with quadruplicate measurements in each experiment.Quantitative real-time PCR
Total RNA was extracted from cells using a PureLink™ RNA Mini Kit (Invitrogen). The quantity and quality of RNA extracted were assessed using a SmartSpec™ 3000 Spectrophotometer (Bio-Rad). A High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) was applied to synthesize cDNA according to the manufacturer's instructions. PCR reactions were performed using the CFX96 Real-time PCR system (Applied Biosystems). The cycling conditions were as follows: 3 min at 95 °C followed by 40 cycles of: 95 °C, 10 s; 60 °C, 10 s; 72 °C, 30 s; followed by a final extension at 72 °C for 10 min. Each 25 μl reaction contained 0.4 μg cDNA, 12.5 μl SYBR Green qPCR SuperMix (Bio-Rad), 250 nM forward and reverse primers, and nuclease free water. Primers used were as follows: human VEGF, 5′- CGG CGA AGA GAA GAG ACA CA -3′ and 5′- GGA GGA AGG TCA ACC ACT CA -3′; mouse VEGF, 5′- TTA CTG CTG TAC CTC CAC CA -3′ and 5′- ACA GGA CGG CTT GAA GAT GTA -3′; human GAPDH, 5′- GGA AGG TGA AGG TCG GAG TCA -3′ and 5′- GTC ATT GAT GGC AAC AAT ATC CAC T -3′; mouse GAPDH, 5′- GCA CAG TCA AGG CCG AGA AT -3′ and 5′- GCC TTC TCC ATG GTG GTG AA- 3′. The experiment was repeated once with quadruplicate measurements in each experiment.Analysis of receptor kinase activity of VEGFR2 and FGFR1
HUVECs were cultured in 6-well plates, starved overnight in EBM-2 medium supplemented with 0.1% charcoal-stripped FBS, pretreated with L-5F (1, 3, or 10 μg ml−1) for 2 h, treated with 80 ng ml−1 VEGF or bFGF for 5 min, and cell lysates were collected for the analysis of receptor kinase activity. 0.6 μg and 1.2 μg of total protein were used for VEGFR2 kinase activity analysis and FGFR1 kinase activity analysis, respectively. The HTScan VEGFR2 kinase assay kit or the HTScan FGFR1 kinase assay kit (Cell Signaling Technology, Inc.) were used for determining the kinase activities and all experiments were performed according to manufacturer's protocol.Western blot analysis
Western blot analyses were performed as described previously.36Cell lysates were collected in a lysis buffer containing 0.1 M NaCl, 5 mM EDTA, 50 μM sodium orthovanadate, 1% Triton X-100, and protease inhibitor tablet (Roche Diagnostics) in 50 mM Tris buffer (pH 7.5), loaded onto 4–15% SDS gel, transferred to PVDF membrane, and incubated with the appropriate antibodies. The following antibodies were used: rabbit anti-pThr202/Tyr204-Erk (Cell Signaling Technology, Inc.), rabbit anti-Erk (Cell Signaling Technology, Inc.), rabbit anti-pSer473-Akt (Cell Signaling Technology, Inc.), rabbit anti-Akt (Cell Signaling Technology, Inc.). The signal strength was determined with Scion Image software.In vivo tumor model
Nine-week-old C57BL/6J female mice were given a 0.5 ml subcutaneous (s.c.) injection of 5 × 106ID8 cells prepared as a single cell suspension in PBS mixed with an equal volume of the cold Matrigel (BD Biosciences). Mice received L-5F (10 mg kg−1) or vehicle ABCT buffer by s.c. injection at a site distant from the site where the ID8 cells were injected daily for five weeks. After 5 weeks, the retro-orbital procedure was performed and mouse serum was collected using serum separator tubes (BD Biosciences). The mice were sacrificed for tumor collection and further analyses. For experiments involving L-4F (Supplemental Fig. 1‡), experimental details are identical to L-5F experiments except that the injections of L-4F and SP were started two weeks after the injection of ID8 cells in the flank.Micro-CT angiography
ID8-tumor-bearing mice received a 50 μl intraperitoneal injection of heparin (1000 U ml−1) 15 min before sacrifice. The mice were perfused with PBS for 10 min at a rate of 6 ml min−1 to remove blood. Mice were further perfused with 20 ml of MICROFIL (Flow Tech Inc., Carver, MA), at a rate of 2 ml min−1, and the infused mixture was allowed to polymerize at 4 °C overnight. Dissected tumors were immersed in 10% neutral buffered formalin, and scanned on a SCANCO μCT40 (SCANCO, USA, Wayne, PA) at 6 μm resolution. Scan parameters were as follows: current, 145 μA; voltage, 55 kVp; exposure time, 200 ms; 2000 views, 5 frames per view. Scan times were between 10.1 h and 12.9 h and respective file sizes were between 0.8 GB and 1.1 GB. The microCT-generated DICOM files were converted into a file format compatible with AltaViewer™ (Numira Biosciences Inc., Salt Lake City, UT), which provides both planar views and volume renderings of the samples. SCIRun (Scientific Computing Institute, University of Utah, Salt Lake City, UT) was used to generate pseudo-colored volume renderings.Immunohistochemistry (IHC) staining
Tumor tissues were fixed with 10% neutral buffered formalin and embedded with paraffin and sectioned. Tissue sections (5 μm) were deparaffinized with xylene, rehydrated with 100%, 90%, 70%, and 50% ethanol, treated with proteinase K (10 μg ml−1) (Sigma-Aldrich) for 30 min and with H2O2 (3%) (Sigma-Aldrich) for an additional 30 min. The sections were blocked with 10% normal goat serum and 4% BSA (for CD31 staining) or non-fat milk (for VEGF staining) prepared in PBS for 3 h, and immediately incubated with rat anti-mouse monoclonal CD31 antibody (1:25) (Abcam, Cambridge, MA) or rabbit anti-mouse polyclonal VEGF antibody (1:50) (Abcam) overnight at 4 °C. The sections were then incubated with corresponding biotinylated secondary antibodies (Vector laboratories, Inc., Burlingame, CA) for 60 min (for CD31 staining) or 30 min (for VEGF staining) at room temperature followed by incubation with Vectastain ABC Elite reagents (Vector laboratories, Inc.) to visualize the staining. Finally, sections were lightly counterstained with haematoxylin, dehydrated, and coverslipped with VECTAMOUNT solution (Vector laboratories, Inc.).
Statistics
The data are shown as mean ± S.D. for each group. We performed statistical analyses by unpaired t test. Results for all tests were considered significant if p < 0.05.Acknowledgements
We thank Ms Svetlana Roberts and Dr Gautam Chaudhuri (Dept. of OB/GYN, UCLA) for kindly providing HUVECs, and Dr Chintda Santiskulvong and Dr Oliver Dorigo (Dept. of OB/GYN, UCLA) for kindly providing SKOV3 and OV2008 cell lines. This work was supported by funds from the Womens Endowment; Carl and Roberta Deutsch Family Foundation; the Joan English Fund for Women's Cancer Research; the VA Merit I Award (R.F-E.), the Ovarian Cancer Coalition, the Helen Beller Foundation, USPHS grants HL-30568 (AMF, MN, and STR), HL-082823 (STR), HL-34343 (GMA) and the Laubich and M.K. Grey funds at UCLA.Disclosures
STR, MN, GMA, and AMF are principals in Bruin Pharma, and AMF is an officer in Bruin Pharma.
References
- K. R. Kozak, M. W. Amneus, S. M. Pusey, F. Su, M. N. Luong, S. A. Luong, S. T. Reddy and R. Farias-Eisner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12343–12348 CrossRef CAS.
- K. R. Kozak, F. Su, J. P. Whitelegge, K. Faull, S. Reddy and R. Farias-Eisner, Proteomics, 2005, 5, 4589–4596 CrossRef CAS.
- V. Nosov, F. Su, M. Amneus, M. Birrer, T. Robins, J. Kotlerman, S. Reddy and R. Farias-Eisner, Am. J. Obstet. Gynecol., 2009, 200, 639 .e1–e5.
- F. Su, K. R. Kozak, S. Imaizumi, F. Gao, M. W. Amneus, V. Grijalva, C. Ng, A. Wagner, G. Houg, G. Farias-Eisner, G. M. Anantharamaiah, B. J. Van Lenten, M. Navab, A. M. Fogelman, S. T. Reddy and R. Farias-Eisner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19997–20002 CrossRef CAS.
- M. Navab, G. M. Anantharamaiah, S. T. Reddy, S. Hama, G. Hough, V. R. Grijalva, N. Yu, B. J. Ansell, G. Datta, D. W. Garber and A. M. Fogelman, Arterioscler. Thromb. Vasc. Biol., 2005, 25, 1325–1331 CrossRef CAS.
- M. Navab, I. Shechter, G. M. Anantharamaiah, S. T. Reddy, B. J. Van Lenten and A. M. Fogelman, Arterioscler. Thromb. Vasc. Biol., 2010, 30, 164–168 CrossRef CAS.
- R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS.
- R. C. Jr. Bast, B. Hennessy and G. B. Mills, Nat. Rev. Cancer, 2009, 9, 415–428 CrossRef.
- T. A. Yap, C. P. Carden and S. B. Kaye, Nat. Rev. Cancer, 2009, 9, 167–181 CrossRef CAS.
- T. A. Olson, D. Mohanraj, L. F. Carson and S. Ramakrishnan, Cancer Res., 1994, 54, 276–280 CAS.
- P. J. Paley, K. A. Staskus, K. Gebhard, D. Mohanraj, L. B. Twiggs, L. F. Carson and S. Ramakrishnan, Cancer, 1997, 80, 98–106 CrossRef CAS.
- D. P. Barton, A. Cai, K. Wendt, M. Young, A. Gamero and S. De Cesare, Clin. Cancer Res., 1997, 3, 1579–1586 CAS.
- B. Davidson, I. Goldberg, W. H. Gotlieb, J. Kopolovic, G. Ben-Baruch, J. M. Nesland and R. Reich, Mol. Cell. Endocrinol., 2002, 187, 39–45 CrossRef CAS.
- A. Gadducci, P. Viacava, S. Cosio, D. Cecchetti, G. Fanelli, A. Fanucchi, G. Teti and A. R. Genazzani, Anticancer Res., 2003, 23, 3001–3008 CAS.
- J. H. Ahn, J. S. Kim, H. K. Yu, H. J. Lee and Y. Yoon, J. Biol. Chem., 2004, 279, 21808–21814 CrossRef CAS.
- J. Redondo-Munoz, M. Jose Terol, J. A. Garcia-Marco and A. Garcia-Pardo, Blood, 2008, 111, 383–386 CrossRef CAS.
- A. K. Olsson, A. Dimberg, J. Kreuger and L. Claesson-Welsh, Nat. Rev. Mol. Cell Biol., 2006, 7, 359–371 CrossRef CAS.
- M. Mohammadi, I. Dikic, A. Sorokin, W. H. Burgess, M. Jaye and J. Schlessinger, Mol. Cell Biol., 1996, 16, 977–989 CAS.
- E. J. Goetzl, H. Dolezalova, Y. Kong, Y. L. Hu, R. B. Jaffe, K. R. Kalli and C. A. Conover, Cancer Res., 1999, 59, 5370–5375 CAS.
- Y. L. Hu, M. K. Tee, E. J. Goetzl, N. Auersperg, G. B. Mills, N. Ferrara and R. B. Jaffe, J. Natl. Cancer Inst., 2001, 93, 762–768 CrossRef CAS.
- B. J. Van Lenten, A. C. Wagner, C. L. Jung, P. Ruchala, A. J. Waring, R. I. Lehrer, A. D. Watson, S. Hama, M. Navab, G. M. Anantharamaiah and A. M. Fogelman, J. Lipid Res., 2008, 49, 2302–2311 CAS.
- S. Imaizumi, V. Grijalva, M. Navab, B. J. Van Lenten, A. C. Wagner, G. M. Anantharamiah, A. M. Fogelman and S. T. Reddy, Drug Metab. Lett., 2010, 4, 139–148 Search PubMed.
- Y. Xu, Z. Shen, D. W. Wiper, M. Wu, R. E. Morton, P. Elson, A. W. Kennedy, J. Belinson, M. Markman and G. Casey, J. Am. Med. Assoc., 1998, 280, 719–723 CrossRef CAS.
- D. L. Baker, P. Morrison, B. Miller, C. A. Riely, B. Tolley, A. M. Westermann, J. M. Bonfrer, E. Bais, W. H. Moolenaar and G. Tigyi, J. Am. Med. Assoc., 2002, 287, 3081–3082 CrossRef.
- Y. J. Xiao, B. Schwartz, M. Washington, A. Kennedy, K. Webster, J. Belinson and Y. Xu, Anal. Biochem., 2001, 290, 302–313 CrossRef CAS.
- M. M. Ptaszynska, M. L. Pendrak, R. W. Bandle, M. L. Stracke and D. D. Roberts, Mol. Cancer Res., 2008, 6, 352–363 CrossRef CAS.
- F. Okada, J. W. Rak, B. S. Croix, B. Lieubeau, M. Kaya, L. Roncari, S. Shirasawa, T. Sasazuki and R. S. Kerbel, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3609–3614 CrossRef CAS.
- J. R. Conejo-Garcia, F. Benencia, M. C. Courreges, E. Kang, A. Mohamed-Hadley, R. J. Buckanovich, D. O. Holtz, A. Jenkins, H. Na, L. Zhang, D. S. Wagner, D. Katsaros, R. Caroll and G. Coukos, Nat. Med., 2004, 10, 950–958 CrossRef CAS.
- R. Kerbel and J. Folkman, Nat. Rev. Cancer, 2002, 2, 727–739 CrossRef CAS.
- L. M. Ellis and D. J. Hicklin, Nat. Rev. Cancer, 2008, 8, 579–591 CrossRef CAS.
- L. M. Ellis and D. J. Hicklin, Clin. Cancer Res., 2008, 14, 6371–6375 CrossRef CAS.
- G. Bergers and D. Hanahan, Nat. Rev. Cancer, 2008, 8, 592–603 CrossRef CAS.
- R. Phillips, T. Ursell, P. Wiggins and P. Sens, Nature, 2009, 459, 379–85 CrossRef CAS.
- L. Yvan-Charvet, T. Pagler, E. L. Gautier, S. Avagyan, R. L. Siry, S. Han, C. L. Welch, N. Wang, G. J. Randolph, H. W. Snoeck and A. R. Tall, Science, 2010, 328, 1689–1693 CrossRef CAS.
- A. C. Lake, R. Vassy, M. Di benedetto, D. Lavigne, C. Le Visage, G. Y. Perret and D. Letourneur, J. Biol. Chem., 2006, 281, 37844–37852 CrossRef CAS.
- J. Gavard and J. S. Gutkind, Nat. Cell Biol., 2006, 8, 1223–1234 CrossRef CAS.
Footnotes |
| † Published as part of an Integrative Biology themed issue in honour of Mina J. Bissell: Guest Editor Mary Helen Barcellos-Hoff. |
| ‡ Electronic supplementary information (ESI) available: Supplemental Fig. 1. See DOI: 10.1039/c0ib00147c |
| § Departments of OB/GYN, Medicine, and Molecular and Medical Pharmacology, 650 Charles E. Young Drive South, MRL 3736, Los Angeles, CA 90095. Fax: +1 310-206-3605. Tel: +1 310-206-3915. |
| ¶ Department of OB/GYN, University of California Los Angeles, 650 Charles E. Young Drive South, CHS 24-127, Los Angeles, CA 90095. Fax: +1 310-206-3670. Tel: +1 310-794-1919. |
| This journal is © The Royal Society of Chemistry 2011 |