Antibody-based targeting of interferon-alpha to the tumor neovasculature: a critical evaluation†
Katharina
Frey
a,
Andjelija
Zivanovic
a,
Kathrin
Schwager
b and
Dario
Neri
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Wolfgang-Pauli-Str. 10, CH-8093 Zurich, Switzerland. E-mail: neri@pharma.ethz.ch; Fax: +41 44 633 13 58; Tel: +41 44 633 74 01
bPhilochem AG, c/o ETH Zurich, Wolfgang-Pauli-Str. 10, CH-8093 Zurich, Switzerland
First published on 11th January 2011
Abstract
The antibody-mediated targeted delivery of cytokines, growth factors and immunomodulators offers great potential for the therapy of cancer and other serious conditions. Interferon-alpha has long been used in the clinic for the treatment of patients with certain malignancies or with viral disease. Promising anticancer activity has recently been reported for two fusion proteins consisting of immunoglobulins bearing the interferon-alpha polypeptide at the C-terminal end of the molecule. Here we describe the design, production and characterization of a novel immunocytokine, in which murine interferon-alpha2 was sequentially fused with the tumor-targeting antibody fragment scFv(F8), specific to the alternatively-spliced EDA domain of fibronectin. The resulting fusion protein (F8-IFNa) could be produced to homogeneity and was shown to retain both antigen binding activity and interferon-alpha activity. Biodistribution studies in tumor-bearing mice with radioiodinated protein preparations confirmed the ability of F8-IFNa to selectively localize at the tumor site. However, using two different murine models of cancer (F9 teratocarcinomas and Cloudman S91 melanomas in immunocompetent mice), we could not detect a striking superiority for the therapeutic performance of F8-IFNa as compared to KSF-IFNa, a fusion protein of irrelevant specificity in the mouse which was used as negative control. In the paper, we present hypotheses why the antibody-based pharmacodelivery of interferon-alpha fails to eradicate tumors, in contrast to the situation observed by our group for other immunocytokines, which benefit from a selective localization at the tumor site.
Insight, innovation, integrationInterferon-alpha is a therapeutic protein widely used in the clinic for the treatment of patients with cancer or with viral infections. We describe for the first time that interferon-alpha can be selectively delivered to the tumor neovasculature by fusion to an antibody specific to a component of the modified subendothelial extracellular matrix. In spite of an excellent accumulation of the immunocytokine around tumor blood vessels, vascular targeting of interferon-alpha mediates only a modest tumor growth inhibition. The comparative analysis of a series of vascular targeting immunocytokines specific to splice isoforms of fibronectin allow the identification of immunomodulatory proteins which display an anticancer activity upon localization on the tumor subendothelial extracellular matrix, facilitating selection of candidates for clinical trials and tumor biology studies. |
Introduction
Interferons-alpha (IFNa) are α-helical cytokines belonging together with interferon-β to the type I interferon family, binding to and acting through the same heterodimeric receptor composed of IFNAR1 and IFNAR2 subunits.1,2 Both human3 and murine4genomes contain more than 20 different intronless IFNa genes which encode for at least 13 functional protein subtypes. Murine and human IFNa elicit their biological functions in a species specific manner, and murine IFNa does not act on human IFNAR receptor, e.g. on human tumor xenografts. IFNa are pleiotropic acting cytokines, exerting multiple effects on cellular functions. Despite its antiviral activity, antitumor activity of IFNa can be divided into the direct antitumoral effects on the tumor cell and into the indirect immunomodulatory effects on the host's immune system. Direct antitumor effects include the inhibition of tumor cell proliferation5 and the delivery of proapoptotic signals,2,6–8 downregulation of oncogene expression, induction of tumor-suppressor genes, upregulation of MHC class I,6 but also inhibition of tumor-induced angiogenesis.9 However, immunostimulatory effects of IFNa also contribute to the anticancer activity,2,10,11 including the generation and long-term survival of memory T cells, activation of NK cells and CTLs, differentiation and activation of dendritic cells (DC) and B cells,12,13 expression of various cytokines and chemokines, and induction of Th1 polarization. In general, IFNa is a potent mediator to connect innate and adaptive immunity in both mice and humans.12,14In 1986, IFNa was the first cytokine to become approved for clinical use, namely for the treatment of hairy cell leukemia. Even today, IFNa is still widely used for the treatment of certain malignancies, such as e.g. hepatitis C, hematological diseases and metastatic melanoma,15,16 also in combinations with other anticancer drugs that have recently been developed (e.g. in kidney cancer). Cytokines are key mediators of innate and cellular immunity and often display potent antitumoral and antimetastatic activity. However, the administration of most cytokines which have been used in the clinical setting is often limited due to the short half-life and severe toxicity, even at single-digit milligram doses, preventing dose escalation and hampering their development as anticancer drugs. Various IFNa2b regimens have been investigated in the clinic which can be classified as high dose (20 MUnits m−2), intermediate dose (10 MUnits m−2) or low dose treatment (3 MUnits m−2). Typically, an intense induction phase at the described dose is administered i.v. for one month, followed by a maintenance phase (half the dose, subcutaneous administration).6,15,17 The most significant side effects include short term fever, malaise, and more prominent long term liver toxicity, granulocytopenia, anemia, neurological disorders and fatigue.17
Antibodies can be used to deliver cytokines to the tumor environment or to other sites of disease (e.g., rheumatoid arthritis), thus sparing normal tissues.18–20 The targeting of the tumor neovasculature is particularly attractive, because of the dependence of cancer on new blood vessels and because of the accessibility of these structures from the blood stream.19 Our group has recently developed and tested in animal models several immunocytokines, based on the clinical-stage tumor targeting antibodies L19, F16 and F8. These antibodies were typically used in scFv format21 and were fused to several cytokines and growth factors, including interleukin-2,22–25 interleukin-10,26,27 interleukin-12,28,29 interleukin-15,30GM-CSF,30TNF,28,31–34 VEGFs28 and to interferon-gamma.35 Four of these fusion proteins are currently being investigated in Phase I and Phase II clinical trials in patients with cancer (L19-IL2, L19-TNF, F16-IL2, www.philogen.it)36 or with rheumatoid arthritis (F8-IL10).
The F8 and L19 antibodies recognize the alternatively spliced EDA and EDB domains of fibronectin, respectively,37,38 while the F16 antibody is specific to the alternatively spliced A1 domain of tenascin-C.39 These components of the modified extracellular matrix are virtually undetectable in normal adult tissues (exception made for the placenta, endometrium in the proliferative phase and some vessels in the ovaries26,39), but can be strongly overexpressed in solid tumors,40,41 lymphomas,42angiogenesis-related ocular disorders,43 arthritis,26,44 psoriasis,45 endometriosis (Schwager et al., manuscript in preparation) and atherosclerosis.46,47 Furthermore, expression of EDA (but not of EDB) has been reported for liver injury,48,49 fibrosis50 and liver cirrhosis51,52 making link to viral diseases such as hepatitis infections. Recently, also enhanced circulating levels of EDA have been correlated with the degree of liver fibrosis.53
The group of Sherie Morrison has recently described the fusion of full immunoglobulins in IgG format with murine interferon-alpha1. The resulting fusion proteins were specific to Her2/neu54 and CD20,55antigens expressed in breast cancer or B cell lymphoma, respectively. Both IFNa-antibody fusion proteins displayed longer half-lives and an anticancer activity on murine B cell lymphomas, transfected with the human antigens. Both immunocytokines exhibited antiproliferative and proapoptotic activity that led to an increased survival in mice.
In this article, we describe the cloning, production and characterization of the novel immunocytokine F8-IFNa: a fusion protein consisting of scFv(F8) sequentially fused with murine interferon-alpha2. We used a 5-amino acid linker between VH and VL in the scFv antibody fragment in order to drive the formation of a stable non-covalent homodimeric fusion protein.22,26,56 We preferred the use of antibody fragments rather than full immunoglobulin, since the Fc portion of an IgG could cross-link interferon-alpha onto Fcγ receptor-positive cells in a non-specific fashion and since we have previously observed for other biological modulators that small immunocytokines have rapid blood clearance profiles which are comparable to the ones of the unmodified parent cytokine.22–24,29 In turn, as a consequence of rapid blood clearance, the toxicity profile of cytokines and small immunocytokines is usually comparable, while the antibody-mediated disease seeking activity can deliver a substantial increase in therapeutic index.
Results
Cloning, production and in vitro characterization of F8-IFNa and KSF-IFNa
The fusion proteins F8-IFNa and KSF-IFNa consist of a scFv antibody sequentially fused with murine interferon-alpha2. The choice of a 5-amino acid linker between VH and VL of the antibody drives the formation of stable non-covalent homodimers.22,26,56 The fully human F8 antibody recognizes both the human and murine EDA domain of fibronectin with comparable affinity,38 whereas the KSF antibody is specific to hen egg lysozyme and was used as a negative control antibody. Fig. 1A illustrates the cloning strategy chosen for the expression of the two immunocytokines in stably transfected mammalian cells, while Fig. 1B presents a schematic representation of the domain assembly into a non-covalent homodimeric structure, held together by interactions at the VH-VL interface. In SDS-PAGE analysis, the immunocytokines ran with an apparent molecular weight of ∼50 kDa in both reducing and non-reducing conditions (Fig. 1C), while a gel filtration analysis confirmed the stable homodimeric nature of the products (Fig. 1D). As expected, the BIAcore analysis of F8-IFNa confirmed high affinity binding to the cognate antigen, with slow dissociation kinetics (apparent KD 1.3 nM) (Fig. 1E). When tested in an activity assay based on the inhibition of proliferation6,7 of Cloudman S91 melanoma cells, both F8-IFNa and KSF-IFNa revealed a biological activity comparable to the one of recombinant murine interferon-alpha2, with half-maximal activity in the 100 pM concentration range (Fig. 1F). | ||
| Fig. 1 Cloning, expression and characterization of F8-IFNa and KSF-IFNa. (A) Schematic representation of the cloning strategy of F8-IFNa and KSF-IFNa fusion proteins. SP, secretion sequence peptide; VH, VL, variable heavy and light chain. F8 antibody is specific to EDA of human and murine fibronectin, KSF antibody is specific to hen egg lysozyme and used as negative control. (B) Domain assembly of an IFNa-immunocytokine diabody due to a short linker between VH and VL. (C) SDS-PAGE analysis of purified F8-IFNa and KSF-IFNa: M, molecular marker; 1, F8-IFNa; 2, KSF-IFNa; nr, r, non-reducing and reducing conditions (molecular weight F8-IFNa and KSF-IFNa in monomeric form is 44.7 kDa). (D) Gel filtration analysis of affinity-purified F8-IFNa. The peak eluting at a retention volume of 13.6 ml corresponds to the noncovalent homodimeric form of F8-IFNa (90 kDa). KSF-IFNa (not shown) had similar profiles and retention times. Arrows indicate standard proteins (11 ml: Ferritin 440 kDa; 14.1 ml: BSA 67 kDa; 15.4 ml: β-lactoglobulin 35 kDa). (E) BIAcore analysis of F8-IFNa on EDA coated chip. (F) Growth inhibition bioactivity assay on murine Cloudman S91 melanoma cells. F8-IFNa and KSF-IFNa displayed biological activity comparable with the one of recombinant murine IFNa (mean of 9 replicates ± SD). | ||
Biodistribution studies
The tumor targeting properties of F8-IFNa and of KSF-IFNa were studied by quantitative biodistribution analysis with radioiodinated protein preparations in immunocompetent mice bearing either murine F9 teratocarcinomas or murine Cloudman S91 melanomas (Fig. 2 and 3). Both tumor models exhibited an intense staining with the F8 antibody, with a prominent perivascular pattern of EDA expression (Fig. 2A and 3A). As for other immunocytokines, F8-IFNa preferentially localized at the tumor site, as evidenced by the analysis of the percent of injected dose per gram of tissue (%ID/g) in various organs 24 h after intravenous administration of the 125I-labeled immunocytokines. In the F9 setting, tumor uptake values for F8-IFNa expressed as %ID/g were above 8%ID/g at 24 h, with a stable tumor retention over time and with tumor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :blood ratios of 10 at 24 h and 21 and 48 h (Fig. 2B). The immunocytokine KSF-IFNa of irrelevant specificity in the mouse, which we used as negative control, did not show a preferential accumulation in the tumor 24 h after injection, with low levels in the neoplastic mass (0.4%ID/g) and with tumor:blood = 1.2. In order to confirm localization of F8-IFNa around the tumor neovasculature following intravenous administration, tumors from the biodistribution studies in F9 teratocarcinoma were excised and sections were studied by immunofluorescence, using anti-interferon-alpha antibodies as detection reagent. As expected, a selective localization of F8-IFNa, but not of KSF-IFNa, could be observed around tumor blood vessels (Fig. 2C).
:blood ratios of 10 at 24 h and 21 and 48 h (Fig. 2B). The immunocytokine KSF-IFNa of irrelevant specificity in the mouse, which we used as negative control, did not show a preferential accumulation in the tumor 24 h after injection, with low levels in the neoplastic mass (0.4%ID/g) and with tumor:blood = 1.2. In order to confirm localization of F8-IFNa around the tumor neovasculature following intravenous administration, tumors from the biodistribution studies in F9 teratocarcinoma were excised and sections were studied by immunofluorescence, using anti-interferon-alpha antibodies as detection reagent. As expected, a selective localization of F8-IFNa, but not of KSF-IFNa, could be observed around tumor blood vessels (Fig. 2C).
 | ||
| Fig. 2 Tumor targeting properties of F8-IFNa in F9 teratocarcinoma. (A) Immunohistochemical analysis of murine F9 tumors using the F8 antibody in SIP (small immunoprotein) format. F8 exhibited strong staining in F9 tumor sections. Scale bars = 100 μm. (B) Biodistribution study in subcutaneous F9 bearing 129/SvEv mice. Radiolabeled 125I-F8-IFNa or 125I-KSF-IFNa antibody preparations (20 μg) were injected i.v. and mice were sacrificed after 24 and 48 h (F8-IFNa) or after 24 h (KSF-IFNa). Organs were excised and radioactivity counted, expressing results as percent of injected dose per gram of tissue (%ID/g) ± SE. A high selective accumulation of F8-IFNa in F9 tumors could be observed over time with tumor:blood ratios of 10 at 24 h and 21 at 48 h. KSF-IFNa did not accumulate in the tumor with tumor:blood = 1.2 at 24 h. (C) Ex vivodetection of IFNa in F9 tumors treated with 20 μg F8-IFNa or KSF-IFNa. Biodistribution mice were sacrificed 24 h after injection. Tumor sections were stained with an anti-IFNa antibody (red) and with an anti-CD31 antibody (green) to detect vessels. Scale bars = 100 μm. | ||
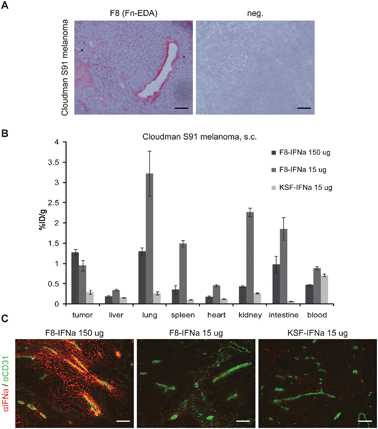 | ||
| Fig. 3 Tumor targeting properties of F8-IFNa and KSF-IFNa in Cloudman S91 melanoma. (A) Immunohistochemical analysis of murine Cloudman S91 tumors using the F8 antibody in SIP (small immunoprotein) format. F8 exhibited perivascular staining in Cloudman S91 tumor sections. Scale bars = 100 μm. (B) Biodistribution study in subcutaneous Cloudman S91 bearing DBA/2J mice. Radiolabeled 125I-F8-IFNa at high dose (150 μg) and low dose (15 μg) and 125I-KSF-IFNa (15 μg) antibody preparations were injected i.v. and mice were sacrificed after 24 h. Organs were excised and radioactivity counted, expressing results as percent of injected dose per gram of tissue (%ID/g) ± SE. Maximal tumor uptake values of F8-IFNa in Cloudman S91 tumors were lower compared to F9 tumors, and KSF-IFNa did not accumulate in Cloudman S91, with tumor:blood ratios of 1% (low dose F8-IFNa), 2.7 (high dose F8-IFNa), 0.4 (KSF-IFNa) at 24 h. (C) Ex vivodetection of IFNa in Cloudman S91 tumors treated with 15 μg or 150 μg F8-IFNa and 15 μg KSF-IFNa. Biodistribution mice were sacrificed 24 h after injection. Tumor sections were stained with an anti-IFNa antibody (red) and with an anti-CD31 antibody (green) to detect vessels. Scale bars = 100 μm. | ||
Tumor uptake values in the Cloudman S91 setting were lower (around 1%ID/g at 24 h), reflecting a lower amount of antigen in the tumor mass. Percent injected dose per gram values did not vary between the 15 μg and 150 μg doses of F8-IFNa. As anticipated, tumor:blood values where higher for F8-IFNa (tumor:blood = 1.1 for 15 μg; 2.7 for 150 μg) compared to KSF-IFNa (0.4 for 15 μg). However, antibody uptake in normal organs was higher with the F8 immunocytokine, compared to the KSF immunocytokine (Fig. 3B). The administration of the higher dose of F8-IFNa (150 μg) resulted in a marked decrease of F8-IFNa uptake in normal organs. Similar to the situation encountered in the F9 teratocarcinoma targeting studies, a preferential localization around the tumor neovasculature could be detected for F8-IFNa (but not for KSF-IFNa) in Cloudman S91 tumors, with increasing amounts of localized protein when 150 μg immunocytokine were administered rather than 15 μg (see also below) (Fig. 3C).
Therapeutic activity of F8-IFNa and KSF-IFNa
The therapeutic activity of F8-IFNa and of KSF-IFNa were studied in syngeneic immunocompetent mouse models of F9 and Cloudman S91 tumors (150 μg of fusion protein injected three times every 48 h; number of mice = 5). Treatment with 150 μg F8-IFNa and KSF-IFNa was well tolerated over time as documented by body weight changes (data not shown). In order to assess antitumoral activity on small and large neoplastic masses, therapy was started when tumors had reached a size of 100 mm3 or 300 mm3 (Fig. 4 and 5). For small F9 tumors, a significant tumor growth retardation of IFNa-immunocytokines compared to saline treatment (p = 0.0079, day 13) could be observed, which was however not different between F8-IFNa and KSF-IFNa (Fig. 4A). By contrast, a moderate superiority of the targeted version of IFNa could be observed with the larger F9 tumors (F8-IFNavs. KSF-IFNa p = 0.114, day 10) (Fig. 4B). In both cases, tumors could not be eradicated by the use of interferon-alpha fusion proteins as single agent. | ||
| Fig. 4 Therapeutic activity of F8-IFNa in murine F9 teratocarcinomas. Immunocompetent 129/SvEv mice bearing s.c. F9 teratocarcinomas were treated i.v. with 150 μg F8-IFNa (targeting EDA) or 150 μg KSF-IFNa (specific to an irrelevant antigen) or PBS as control (5 mice per group). Treatment was performed three times every 48 h (arrows). Therapy was started in mice with small tumors of 100 mm3 (A) and in mice with large tumors of 300 mm3 (B). Data represent mean tumor volumes (±SE). Tumor growth curves were stopped when the first tumor per group reached 2000 mm3. | ||
 | ||
| Fig. 5 Therapeutic activity of F8-IFNa in murine Cloudman S91 melanomas. Immunocompetent DBA/2J mice bearing s.c. Cloudman S91 melanomas were treated i.v. with 150 μg F8-IFNa (targeting EDA) or 150 μg KSF-IFNa (specific to an irrelevant antigen) or PBS as control (5 mice per group). Treatment was performed three times every 48 h (arrows). Therapy was started in mice with small tumors of 100 mm3 (A) and in mice with large tumors of 300 mm3 (B). Data represent mean tumor volumes (±SE). Tumor growth curves were stopped when the first tumor per group reached 2000 mm3. | ||
In mice bearing Cloudman S91 melanomas, tumor growth retardation could be observed, with no statistically significant difference between F8-IFNa and KSF-IFNa (small tumors day 41: p = 0.222; large tumors day 29: p = 0.547) (Fig. 5).
In parallel to the therapy studies, additional F9 and Cloudman S91 tumor bearing mice were injected three times with the immunocytokines at the 150 μg dose, sacrificed 2 or 3 days after the last injection, and tumors were analyzed for antibody localization in the tumor mass. Similar to what was presented in Fig. 3C, a preferential accumulation of IFNa around tumor blood vessels was only observed with F8-IFNa but not with KSF-IFNa (Fig. 6).
 | ||
| Fig. 6 Ex vivo detection of IFNa after therapy with F8-IFNa, KSF-IFNa and PBS in F9 teratocarcinoma (A) and Cloudman S91 melanoma (B). Tumor-bearing mice were injected i.v. three times every 48 h with 150 μg F8-IFNa or 150 μg KSF-IFNa or PBS. F9 bearing mice were sacrificed 3 days after last injection and Cloudman S91 mice were sacrificed 2 days after last injection. Tumor sections were stained with an anti-IFNa antibody (red) and anti-CD31 antibody (green) to detect vessels. Scale bars = 100 μm. | ||
Tumor masses were also studied in terms of leukocyte infiltration at the end of therapeutic treatment. In general, more immune cells could be detected in Cloudman S91 melanomas compared to F9 teratocarcinomas (Fig. 7). However, a lower degree of leukocyte infiltration in the F9 setting was observed in this study, compared to our previous reports with other targeted immunocytokines.23,29,35,57 An increase in the infiltration of Asialo GM1-positive cells (i.e., mainly NK cells) was observed for F8-IFNa both in F9 and in Cloudman S91 tumors (Fig. 7). An increase of CD45 (leukocytes), F4/80 (macrophages) and I-A/I-E (all MHC class II: B, DC, macrophages) positive cells was observed with F8-IFNa in the case of F9 tumors, but less prominently with Cloudman S91 melanomas. The cellular infiltration was clearly enhanced as a result of F8-IFNa treatment, compared to the results observed in the KSF-IFNa and in the saline groups of treated mice (Fig. 7).
 | ||
| Fig. 7 Immunofluorescence analysis of tumor-infiltrating immune cells in F9 teratocarcinoma and Cloudman S91 melanoma. Immunocompetent tumor-bearing mice were injected i.v. three times every 48 h with 150 μg F8-IFNa or 150 μg KSF-IFNa or PBS. F9 bearing mice were sacrificed 3 days after last injection and Cloudman S91 mice were sacrificed 2 days after last injection. F9 tumor sections (A) and Cloudman S91 tumor sections (B) were stained for NK cells (Asialo GM1), leukocytes (CD45), macrophages (F4/80) and MHC class II positive cells, e.g.B cells, DC, macrophages (I-A/I-E). Panel (A) and (B) show representative immunofluorescence images. Scale bars = 100 μm. F9 tumor sections (C) and Cloudman tumor sections (D) were evaluated for area percentage positive staining using ImageJ (average of three pictures). | ||
Discussion
In this article, we reported the generation and in vivo characterization of an immunocytokine based on the fusion of the human anti-EDA antibody scFv(F8) to murine IFNa2 (F8-IFNa), in comparison to an antibody fusion protein KSF-IFNa of irrelevant specificity in the mouse, chosen as negative control. Both immunocytokines were successfully cloned, expressed and purified to homogeneity. They retained antigen-binding properties as well as the full biological activity of IFNa in vitro. While F8-IFNa demonstrated a high specific accumulation in the tumor with high tumor:blood values, especially in F9 teratocarcinomas which express high levels of EDA domain of fibronectin, KSF-IFNa did not exhibit a preferential localization at the tumor site, as expected. The accumulation of F8-IFNa in the tumor was stable over time. An antitumor effect could be observed in vivo in two solid tumor models (syngeneic F9 teratocarcinoma and Cloudman S91 melanoma), which was superior compared to saline treatment. In the majority of the therapeutic schemes tested in vivo, targeting of F8-IFNa did not result in an improved performance compared to untargeted KSF-IFNa at corresponding doses. However, the two therapeutic proteins exhibited differences at the level of leukocyte recruitment into the tumor mass.
Morrison and collaborators54,55 have reported on the development of murine IFNa1 antibody fusion proteins in the IgG format. These immunocytokines, which targeted human Her2/neu or CD20 on murine cell lymphomas transduced with the human antigen, were shown to improve survival and, in some settings, induce complete tumor eradication when injected directly after tumor implantation or in small established tumors. The therapeutic performance of the antibody-IFNa1 fusions was superior to the one of antibody-IFNa1 of irrelevant specificity in the mouse or to recombinant IFNa. The major effects in their tumor models can be attributed to direct antitumoral effects such as antiproliferative and apoptotic activity of IFNa1,54,55 also demonstrated by therapy failure after IFNAR knockout in the cell line.55 Recently, Rossi et al. described an antibody fusion protein with human IFNa2b in human B cell lymphoma xenograft, with the aim to elicit a direct antitumor action with no contribution of the host's immune system.58
In our study, we have used two syngeneic immunocompetent animal models of solid cancer, which express different amounts of EDA antigen, in order to mimic as closely as possible the potential therapeutic action of F8-IFNa in the clinical setting. In contrast to the previous studies, the cancer cell lines used in our study were not transfected with a human antigen (which could be immunogenic in the mouse setting, thus influencing therapy outcome). In vivo, they gave rise to tumors which express EDA at levels comparable to the ones observed in clinical specimens.
F8-IFNa achieved a high density in vivo on the subendothelial extracellular matrix of tumor blood vessels. The modest tumor growth inhibition observed in the studies indicates that tumor endothelial cells are not affected by a high local concentration of interferon-alpha. At the same time, a direct effect on tumor cells can be excluded, as only endothelial cells and perivascular tumor cells were likely to be in contact with F8-IFNain vivo for a prolonged period of time. While a strong antitumoral performance of targeted IFNa antibody fusion proteins could be observed for disseminated disease,54,55,58 it appears that in solid cancer types localization of IFNa is not sufficient for efficient tumor eradication. In lymphoma models, the individual tumor cell stays in persistent contact with the IFNa immunocytokine while in solid disease only perivascular tumor cells and tumor endothelium can be targeted. Although the mode of antibody binding on the tumor subendothelial extracellular matrix might be sufficient for more potent immunocytokines consisting of IL2,23–25IL1228,29 or TNF,28,31,32 the eradication of the vast neoplastic mass via targeted IFNa could not be achieved. In spite of the fact that only a partial tumor growth rate inhibition was observed with F8-IFNa, a systematic and unbiased evaluation of different payloads with the same antibody as targeting moiety represents the first essential step towards a better integrative biology understanding of pharmacodelivery options with clinical-stage monoclonal antibodies.19
The lymphoma cells used by Morrison et al. are extremely sensitive to IFNa,59 as observed in antiproliferative activity assays.54 The IC50 of Cloudman S91 melanoma is 10 fold higher (Fig. 1F) than the one for the described lymphoma model. It is likely that both in vivolocalization performance and tumor cell sensitivity to interferon-alpha may contribute to therapy.
Murine IFNa1 and IFNa2 share similar antitumoral activity in vitro.4 More potent murine IFNa subtypes4 (e.g.mIFNa11) could be considered for future studies. It was surprising to observe a modest anti-tumor activity for F8-IFNa, which was used at 150 μg doses and which accumulated selectively on F9 and Cloudman S91 tumors, while in the experiments of the Morrison group only 10 μg doses of immunocytokines were used. It is difficult to know whether the differential therapeutic outcome was due to the longer circulation time of IgG-based immunocytokines or rather to an intrinsically high sensitivity towards interferon-alpha of stably-transfected murine tumor cells carrying a potentially immunogenic human tumor antigen.
For proinflammatory cytokines such as IL2,23–25IL1228,29 and TNF,28,31,33 a strong antitumor activity has been documented as a result of the antibody-mediated delivery of the bioactive moiety to the tumor neovasculature. By contrast, a comparable therapeutic benefit could not be observed for the F8-IFNa fusion protein. While IL2, IL12 or TNF strongly recruit NK cells, cytotoxic T cells and promote cytotoxic and inflammatory responses, IFNa has a more indirect function on the cytotoxic effector cells leading to a milder direct antitumoral response and overall boost of the host immune system.2,6,10 In F9 teratocarcinomas, F8-IFNa enhanced the recruitment of NK cells, leukocytes and MHC class II positive cells (B, DC, macrophages), however, the extent of infiltration was low (about 4–8%) compared to e.g.F8-IL2 in kidney cancer xenograft,22 where about 60% of positive area has been reached (Fig. 7). A less prominent discrimination, but at higher levels of about 20–40% positive area, of tumor-infiltrating immune cells was observed for Cloudman S91 tumor masses, thus correlating to no difference in the tumor growth curves of F8-IFNa and KSF-IFNa (Fig. 5). Even though targeting of IFNa to the subendothelial matrix in solid tumors was successful and led to an increase in immune cell infiltrates, complete tumor responses could not be obtained. By contrast, with other immunocytokines (e.g., those based on IL2 as therapeutic payload24,25,57), a potent immune cell infiltration in the neoplastic mass could lead to tumor eradication.
In previous immunohistochemical and biodistribution studies performed with the F8 antibody, we have shown that the majority of tumor blood vessels strongly express the EDA domain of fibronectin, both in small murine tumors (0.1–1 gram) and in larger human cancer specimens.22,38,42,60 Importantly, the vast majority of tumor blood vessels can be reached in vivo by the F8 antibody, as evidenced by microautoradiographic studies following intravenous administration of radioiodinated antibody preparations.38 The EDA domain of fibronectin does not appear to have an essential function in the mouse, since EDA−/− mice exhibit normal developmental patterns.61 However, simultaneous deletion of EDA and EDB exons leads to embryonic lethality at E10.5, with ca. 80% penetrance due to mixed genetic background, which can be attributed to strong cardiovascular defects, reduced α-SMA positive cells and aberrant endothelial tube formation.61,62 In our laboratory, therapy studies performed with the F8 antibody alone (i.e., in the absence of a toxic payload and in various antibody formats, such as IgG) could not induce any detectable anti-tumor activity, reinforcing the concept that EDA is a good target for pharmacodelivery applications but may not be a good target for biological inhibition strategies.
From a safety perspective, repeated high-dose administrations of 150 μg F8-IFNa were very well tolerated in mice. As no statistically significant weight loss was observed at this dose, other therapeutic regimens and higher doses could be considered for future studies. While the antitumor activity was modest in the presented tumor models, F8-IFNa can now be studied in other preclinical settings, e.g. in mouse models of viral infection (e.g., liver cirrhosis), for which expression of the EDA domain of fibronectin has been documented.48–52
Conclusion
Interferon-alpha has a long record in clinical practice for the treatment of antiviral diseases and different cancer types but its clinical use is limited by systemic toxicities. The targeted delivery of IFNa-antibody fusion proteins to the neovasculature at the site of disease while sparing healthy tissue and thus reducing adverse events is a very attractive strategy, also in the combined setting with other drugs for different malignancies. Recent clinical trials with different immunocytokines (L19-IL2, F16-IL2, L19-TNF, F8-IL10) have proven a therapeutic benefit and encourages us to continue our investigations on the development of novel antibody-based vascular targeting.Experimental
Cell lines and animals
The tumor cell lines used were the murine teratocarcinoma cell line F9 and the murine melanoma cell line Cloudman S91-M3. F9 cells were cultured on 0.1% gelatin-coated tissue flasks in DMEM supplemented with 10% FCS. The cell line Cloudman S91-M363 was kindly provided by Dr Silvio Hemmi (University of Zurich, Switzerland) and cultured in DMEM supplemented with 10% FCS. CHO–S cells (Invitrogen) were cultured adherent in RPMI supplemented with 10% FCS, 2 mM Glutamine or in suspension in PowerCHO-2CD (Lonza), 8 mM Glutamine, HT supplement.Female 129/SvEv mice were obtained from Taconic (Denmark) and female DBA/2J mice were from Janvier (France).
Cloning of F8-IFNa and KSF-IFNa
For cloning of F8-IFNa and KSF-IFNa, the gene for mature murine IFNa2 (aa 24-190) was amplified from a commercial cDNA of mouse spleen and thymus (Clontech) by PCR using the primer pair 5′-TGCGATCTGCCTCACACTTATAACCTC-3′ and 5′-CTCCTTCTCTTCACTCAGTCTTGGCAG-3′. The resulting fragment was further amplified with the primer 5′-TCCGGATCCGCGGGAGGTTCAGGTGGATGCGATCTGCCTCACACTTATAACC-3′, appending a part of the 9-amino acid linker (SGSAGGSGG) at the N-terminus, used later for the fusion of the cytokine to the diabody, and primer 5′-TTTTCCTTTTGCGGCCGCTCATTACTCCTTCTCTTCACTCAGTCTTGGC-3′ introducing C-terminally a stop codon and NotI restriction site.The gene for the F8 diabody,38 specific to EDA of fibronectin, was amplified in a diabody format from a previously described immunocytokine F8-IL222 with a secretion sequence peptide using the primers 5′-CCCAAGCTTGTCGACCATGGGCTGGAGC-3′ and 5′-ACCTCCCGCGGATCCGGATTTGATTTCCACCTTGGTCCCTTG-3′, introducing an N-terminal HindIII restriction site and C-terminally a complementary part of the 9-amino acid linker. The F8 diabody and mIFNa2 fragment was assembled by PCR and cloned via HindIII/NotI restriction sites into the mammalian expression vector pcDNA3.1(+) (Invitrogen). The negative control antibody KSF, specific to hen egg lysozyme, has been isolated by phage display from the ETH2-GOLD library and was amplified and assembled as mIFNa2-fusion protein as described for F8.
Expression, purification and characterization of IFNa-antibody fusion proteins
Adherent CHO–S cells (Invitrogen) were stably transfected by electroporation with the plasmids pcDNA3.1-F8/KSF-IFNa. Selection was carried out in the presence of G418 (0.5 g l−1). Clones of G418-resistant cells were screened for the expression of the fusion protein by ELISA using recombinant EDA of human fibronectin or hen egg lysozyme as antigen and proteinA-HRP for detection (GE Healthcare). The best expressing clone was adapted to grow in suspension in PowerCHO-2CD protein free medium for large-scale production of F8-IFNa and KSF-IFNa. The fusion proteins were purified from the cell culture medium by proteinA affinity chromatography. The size of the fusion proteins was analyzed under reducing and non-reducing conditions by SDS-PAGE and under native conditions by FPLC gel filtration on a Superdex200 10/300GL size exclusion column (GE Healthcare). The apparent KD value of the F8-IFNa was determined by BIAcore on an EDA antigen coated sensor chip.Bioactivity assay
The biological activity of murine IFNa2 was determined by its ability to inhibit proliferation on tumor cells. Cloudman S91 cells (5000 c/well) were seeded in 96-well plates. Upon adherence after 24 h, the culture medium was exchanged for medium supplemented with varying concentrations of murine IFNa2 (ebioscience), F8-IFNa2 or KSF-IFNa2, with concentrations ranging from 150 nM to 36 fM of IFNa2-equivalents. After incubation at 37 °C for 72 h, inhibition of cell proliferation was determined with the Cell Titer Aqueous One Solution (Promega) following the manufacturer's protocol, and calculated as: inhibition of proliferation (%) = 100 − (OD490treated − OD490medium)/(OD490untreated − OD490medium) × 100%.Immunohistochemistry on tumor sections
Cryostat sections (10 μm) of frozen syngeneic F9 teratocarcinoma or Cloudman S91 melanoma specimens were fixed in ice-cold acetone, rehydrated in TBS (50 mM Tris, 100 mM NaCl, 0.01% Aprotinin, pH 7.4) and blocked with 20% FCS. Primary biotinylated F8 antibody in SIP format38 (small immunoprotein; 2 μg ml−1 final concentration) was added and detected with a streptavidin-alkaline phosphatase complex (Biospa) and Fast Red TRSalt substrate (Sigma). Sections were counterstained with Gill's hematoxylin solution No. 2 (Sigma), mounted with glycergel (Dako) and analyzed with an Axiovert S100TV microscope (Zeiss).Quantitative biodistribution studies
To evaluate the in vivo targeting performance, purified F8-IFNa and KSF-IFNa were radioiodinated with 125I and Chloramine T hydrate (0.25 μg/μg protein, Sigma) and purified on a PD-10 column (GE Healthcare). For F9 teratocarcinoma, radiolabeled F8-IFNa or KSF-IFNa (20 μg antibody, 12 μCi /mouse) was injected intravenously into 129/SvEv mice (n = 7) bearing subcutaneous F9 tumors. Mice were sacrificed 24 h (F8-IFNa, KSF-IFNa) and 48 h (F8-IFNa) after injection. For Cloudman S91 melanoma, radiolabeled F8-IFNa at low dose (15 μg protein, 10 μCi/mouse) or high dose (135 μg cold protein + 15 μg hot protein = 150 μg total, 10 μCi/mouse) and KSF-IFNa (15 μg protein, 10 μCi/mouse) were injected intravenously into Cloudman S91 tumor bearing DBA/2J mice (n = 5), which were sacrificed 24 h after injection. Organs were weighed and radioactivity was counted using a Packard Cobra γ counter. Radioactivity content of representative organs was expressed as percentage of the injected dose per gram of tissue (%ID/g ± standard error).Ex vivo detection of IFNa fusion proteins by immunofluorescence
In order to detect the accumulation of interferon-alpha antibody fusion proteins in tumor tissue after intravenous injection, mice from the F9 biodistribution (injected with 20 μg F8-IFNa or 20 μg KSF-IFNa) and Cloudman S91 biodistribution (injected with 15 μg, 150 μg F8-IFNa or 15 μg KSF-IFNa) were sacrificed 24 h after injection. For ex vivodetection of IFNa antibody fusion proteins after F9 and Cloudman S91 therapy, mice were injected three times (day 1, 3, 5) with either 150 μg F8-IFNa or 150 μg KSF-IFNa or PBS. F9 tumor bearing mice were sacrificed three days, while Cloudman S91 bearing mice were sacrificed two days after the last injection. Tumors were excised, embedded in cryoembedding medium (Thermo Scientific) and stored at −80 °C.Cryostat sections (10 μm) of tumors were fixed in ice-cold acetone, rehydrated with PBS and blocked with 10% normal-goat-serum/10% normal-donkey-serum in PBS. Rabbit-a-mouse-IFNa antibody (Merck) and rat-a-mouse-CD31 (BD Biosciences) were used for costaining of IFNa antibody fusion proteins and endothelial cells from blood vessels, respectively. Anti-Rabbit-Alexa594 and anti-Rat-Alexa488 (Invitrogen) coupled secondary antibodies were used for detection. Slides were mounted with fluorescent mounting medium (Dako) and analyzed with an Axioskop2 mot plus microscope (Zeiss).
Syngeneic tumor mouse models in immunocompetent mice
Tumor bearing mice were obtained by subcutaneous injection of F9 teratocarcinoma cells (3 × 107) in the flank of 10-week-old female 129/SvEv mice or by s.c. injection of Cloudman S91 melanoma cells (0.5 × 106) in the flank of 8–10 week-old female DBA/2J mice. When tumors reached a size of either 100 mm3 or 300 mm3, mice were randomly grouped (n = 5) into groups of small tumors (100 mm3) and large tumors (300 mm3) and treatment was started. Mice were injected into the lateral tail vein with either 150 μg F8-IFNa or 150 μg KSF-IFNa or PBS for three injections every 48 h.Mice were monitored daily, tumor volumes were measured three to four times per week with a digital caliper and calculated using the formula: volume = length × width2 × 0.5. Animals were sacrificed when tumor volumes reached 2000 mm3. Experiments were performed under a project license granted by the Veterinäramt des Kantons Zürich, Switzerland (169/2008).
Immunofluorescence of tumor infiltrating immune cells
F9 and Cloudman S91 bearing tumor mice, treated with 150 μg F8-IFNa, 150 μg KSF-IFNa and PBS during therapy (3 i.v. injections every 48 h), were sacrificed 3 days (F9) or 2 days (Cloudman S91) after last treatment. Immunofluorescent staining of 10 μm-tumor sections was performed using the antibodies: F4/80 (macrophages, Abcam), CD45 (leukocytes, BD Biosciences), Asialo GM1 (NK cells, Wako Pure Chemical Industries), I-A/I-E (MHC II positive cells, e.g. DC, macrophages, B cells, BD Biosciences); and detected with AlexaFluor488 coupled secondary antibodies. Images were obtained with an Axioskop2 mot plus microscope (Zeiss), the staining areas analyzed using ImageJ software and expressed as percentage of measurement area.Statistical analysis
Differences in tumor volume between therapeutic groups were compared using Mann-Whitney U test with p<0.05 considered to be significant.Acknowledgements
Katharina Frey and Dr Dario Neri are members of the Cancer Network Zurich and thank Dr Silvio Hemmi for providing Cloudman S91 melanoma cells. Financial contributions from the Swiss National Science Foundation, the ETH Zürich, the European Union (ADAMANT Project), the Swiss Cancer League, the Swiss-Bridge Foundation and the Stammbach Foundation are gratefully acknowledged.References
- E. C. Borden, G. C. Sen, G. Uze, R. H. Silverman, R. M. Ransohoff, G. R. Foster and G. R. Stark, Interferons at age 50: past, current and future impact on biomedicine, Nat. Rev. Drug Discovery, 2007, 6, 975–90 CrossRef CAS.
- A. N. Theofilopoulos, R. Baccala, B. Beutler and D. H. Kono, Type I interferons (alpha/beta) in immunity and autoimmunity, Annu. Rev. Immunol., 2005, 23, 307–36 CrossRef CAS.
- S. Pestka, C. D. Krause and M. R. Walter, Interferons, interferon-like cytokines, and their receptors, Immunol. Rev., 2004, 202, 8–32 CrossRef CAS.
- V. van Pesch, H. Lanaya, J. C. Renauld and T. Michiels, Characterization of the murine alpha interferon gene family, J. Virol., 2004, 78, 8219–28 CrossRef CAS.
- N. Tiefenbrun, D. Melamed, N. Levy, D. Resnitzky, I. Hoffman, S. I. Reed and A. Kimchi, Alpha interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest, Mol. Cell Biol., 1996, 16, 3934–44 CAS.
- P. Tagliaferri, M. Caraglia, A. Budillon, M. Marra, G. Vitale, C. Viscomi, S. Masciari, P. Tassone, A. Abbruzzese and S. Venuta, New pharmacokinetic and pharmacodynamic tools for interferon-alpha (IFN-alpha) treatment of human cancer, Cancer Immunol. Immunother., 2005, 54, 1–10 CrossRef CAS.
- M. Chawla-Sarkar, D. J. Lindner, Y. F. Liu, B. R. Williams, G. C. Sen, R. H. Silverman and E. C. Borden, Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis, Apoptosis, 2003, 8, 237–49 Search PubMed.
- J. Rodriguez-Villanueva and T. J. McDonnell, Induction of apoptotic cell death in non-melanoma skin cancer by interferon-alpha, Int. J. Cancer, 1995, 61, 110–4 CrossRef CAS.
- Y. A. Sidky and E. C. Borden, Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses, Cancer Res., 1987, 47, 5155–61 CAS.
- L. Bracci, E. Proietti and F. Belardelli, IFN-alpha and novel strategies of combination therapy for cancer, Ann. N. Y. Acad. Sci., 2007, 1112, 256–68 CrossRef CAS.
- M. Ferrantini, I. Capone and F. Belardelli, Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use, Biochimie, 2007, 89, 884–93 CrossRef CAS.
- C. A. Biron, Interferons alpha and beta as immune regulators—a new look, Immunity, 2001, 14, 661–4 CrossRef CAS.
- S. M. Santini, C. Lapenta, M. Logozzi, S. Parlato, M. Spada, T. Di Pucchio and F. Belardelli, Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice, J. Exp. Med., 2000, 191, 1777–88 CrossRef CAS.
- G. P. Dunn, C. M. Koebel and R. D. Schreiber, Interferons, immunity and cancer immunoediting, Nat. Rev. Immunol., 2006, 6, 836–48 CrossRef CAS.
- J. M. Kirkwood, J. G. Ibrahim, V. K. Sondak, J. Richards, L. E. Flaherty, M. S. Ernstoff, T. J. Smith, U. Rao, M. Steele and R. H. Blum, High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190, J. Clin. Oncol., 2000, 18, 2444–58 CAS.
- J. M. Kirkwood, A. A. Tarhini, M. C. Panelli, S. J. Moschos, H. M. Zarour, L. H. Butterfield and H. J. Gogas, Next generation of immunotherapy for melanoma, J. Clin. Oncol., 2008, 26, 3445–55 CrossRef CAS.
- A. Hauschild, H. Gogas, A. Tarhini, M. R. Middleton, A. Testori, B. Dreno and J. M. Kirkwood, Practical guidelines for the management of interferon-alpha-2b side effects in patients receiving adjuvant treatment for melanoma: expert opinion, Cancer, 2008, 112, 982–94 CrossRef CAS.
- P. J. Carter, Potent antibody therapeutics by design, Nat. Rev. Immunol., 2006, 6, 343–57 CrossRef CAS.
- D. Neri and R. Bicknell, Tumour vascular targeting, Nat. Rev. Cancer, 2005, 5, 436–46 CrossRef CAS.
- D. Schrama, R. A. Reisfeld and J. C. Becker, Antibody targeted drugs as cancer therapeutics, Nat. Rev. Drug Discovery, 2006, 5, 147–159 CrossRef CAS.
- J. S. Huston, D. Levinson, M. Mudgett-Hunter, M. S. Tai, J. Novotny, M. N. Margolies, R. J. Ridge, R. E. Bruccoleri, E. Haber and R. Crea, et al., Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5879–83 CrossRef CAS.
- K. Frey, C. Schliemann, K. Schwager, R. Giavazzi, M. Johannsen and D. Neri, The immunocytokine F8-IL2 improves the therapeutic performance of sunitinib in a mouse model of renal cell carcinoma, J. Urol., 2010, 184, 2540–8 Search PubMed.
- B. Carnemolla, L. Borsi, E. Balza, P. Castellani, R. Meazza, A. Berndt, S. Ferrini, H. Kosmehl, D. Neri and L. Zardi, Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix, Blood, 2002, 99, 1659–65 CrossRef.
- J. Marlind, M. Kaspar, E. Trachsel, R. Sommavilla, S. Hindle, C. Bacci, L. Giovannoni and D. Neri, Antibody-mediated delivery of interleukin-2 to the stroma of breast cancer strongly enhances the potency of chemotherapy, Clin. Cancer Res., 2008, 14, 6515–24 CrossRef CAS.
- C. Schliemann, A. Palumbo, K. Zuberbuhler, A. Villa, M. Kaspar, E. Trachsel, W. Klapper, H. D. Menssen and D. Neri, Complete eradication of human B-cell lymphoma xenografts using rituximab in combination with the immunocytokine L19-IL2, Blood, 2009, 113, 2275–83 CrossRef CAS.
- K. Schwager, M. Kaspar, F. Bootz, R. Marcolongo, E. Paresce, D. Neri and E. Trachsel, Preclinical characterization of DEKAVIL (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis, Arthritis Res. Ther., 2009, 11, R142 CrossRef.
- E. Trachsel, F. Bootz, M. Silacci, M. Kaspar, H. Kosmehl and D. Neri, Antibody-mediated delivery of IL-10 inhibits the progression of established collagen-induced arthritis, Arthritis Res. Ther., 2007, 9, R9 CrossRef.
- C. Halin, V. Gafner, M. E. Villani, L. Borsi, A. Berndt, H. Kosmehl, L. Zardi and D. Neri, Synergistic therapeutic effects of a tumor targeting antibody fragment, fused to interleukin 12 and to tumor necrosis factor alpha, Cancer Res., 2003, 63, 3202–10 CAS.
- C. Halin, S. Rondini, F. Nilsson, A. Berndt, H. Kosmehl, L. Zardi and D. Neri, Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature, Nat. Biotechnol., 2002, 20, 264–9 CrossRef CAS.
- M. Kaspar, E. Trachsel and D. Neri, The antibody-mediated targeted delivery of interleukin-15 and GM-CSF to the tumor neovasculature inhibits tumor growth and metastasis, Cancer Res., 2007, 67, 4940–8 CrossRef CAS.
- L. Borsi, E. Balza, B. Carnemolla, F. Sassi, P. Castellani, A. Berndt, H. Kosmehl, A. Biro, A. Siri, P. Orecchia, J. Grassi, D. Neri and L. Zardi, Selective targeted delivery of TNFalpha to tumor blood vessels, Blood, 2003, 102, 4384–92 CrossRef CAS.
- E. Balza, B. Carnemolla, L. Mortara, P. Castellani, D. Soncini, R. S. Accolla and L. Borsi, Therapy-induced antitumor vaccination in neuroblastomas by the combined targeting of IL-2 and TNFalpha, Int. J. Cancer, 2010, 127, 101–10 CAS.
- E. Balza, L. Mortara, F. Sassi, S. Monteghirfo, B. Carnemolla, P. Castellani, D. Neri, R. S. Accolla, L. Zardi and L. Borsi, Targeted delivery of tumor necrosis factor-alpha to tumor vessels induces a therapeutic T cell-mediated immune response that protects the host against syngeneic tumors of different histologic origin, Clin. Cancer Res., 2006, 12, 2575–82 CrossRef CAS.
- L. Mortara, E. Balza, F. Sassi, P. Castellani, B. Carnemolla, A. De Lerma Barbaro, S. Fossati, G. Tosi, R. S. Accolla and L. Borsi, Therapy-induced antitumor vaccination by targeting tumor necrosis factor alpha to tumor vessels in combination with melphalan, Eur. J. Immunol., 2007, 37, 3381–92 CrossRef CAS.
- C. Ebbinghaus, R. Ronca, M. Kaspar, D. Grabulovski, A. Berndt, H. Kosmehl, L. Zardi and D. Neri, Engineered vascular-targeting antibody-interferon-gamma fusion protein for cancer therapy, Int. J. Cancer, 2005, 116, 304–13 CrossRef CAS.
- M. Johannsen, G. Spitaleri, G. Curigliano, J. Roigas, S. Weikert, C. Kempkensteffen, A. Roemer, C. Kloeters, P. Rogalla, G. Pecher, K. Miller, A. Berndt, H. Kosmehl, E. Trachsel, M. Kaspar, V. Lovato, R. Gonzalez-Iglesias, L. Giovannoni, H. D. Menssen, D. Neri and F. de Braud, The tumour-targeting human L19-IL2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma, Eur. J. Cancer, 2010, 46(16), 2926–2935 CrossRef CAS.
- A. Pini, F. Viti, A. Santucci, B. Carnemolla, L. Zardi, P. Neri and D. Neri, Design and use of a phage display library. Human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel, J. Biol. Chem., 1998, 273, 21769–21776 CrossRef CAS.
- A. Villa, E. Trachsel, M. Kaspar, C. Schliemann, R. Sommavilla, J. N. Rybak, C. Rosli, L. Borsi and D. Neri, A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo, Int. J. Cancer, 2008, 122, 2405–13 CrossRef CAS.
- S. S. Brack, M. Silacci, M. Birchler and D. Neri, Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C, Clin. Cancer Res., 2006, 12, 3200–8 CrossRef CAS.
- M. Pedretti, A. Soltermann, S. Arni, W. Weder, D. Neri and S. Hillinger, Comparative immunohistochemistry of L19 and F16 in non-small cell lung cancer and mesothelioma: two human antibodies investigated in clinical trials in patients with cancer, Lung Cancer, 2009, 64, 28–33 CrossRef.
- A. Berndt, R. Kollner, P. Richter, M. Franz, A. Voigt, L. Borsi, R. Giavazzi, D. Neri and H. Kosmehl, A comparative analysis of oncofetal fibronectin and tenascin-C incorporation in tumour vessels using human recombinant SIP format antibodies, Histochem. Cell Biol., 2010, 133, 467–75 CrossRef CAS.
- C. Schliemann, A. Wiedmer, M. Pedretti, M. Szczepanowski, W. Klapper and D. Neri, Three clinical-stage tumor targeting antibodies reveal differential expression of oncofetal fibronectin and tenascin-C isoforms in human lymphoma, Leuk. Res., 2009, 33, 1718–22 CrossRef CAS.
- M. Nicolo, A. Biro, F. Cardillo-Piccolino, P. Castellani, A. Giovannini, C. Mariotti, M. Zingirian, D. Neri and L. Zardi, Expression of extradomain-B-containing fibronectin in subretinal choroidal neovascular membranes, Am. J. Ophthalmol., 2003, 135, 7–13 CrossRef CAS.
- J. Kriegsmann, A. Berndt, T. Hansen, L. Borsi, L. Zardi, R. Brauer, P. K. Petrow, M. Otto, C. J. Kirkpatrick, S. Gay and H. Kosmehl, Expression of fibronectin splice variants and oncofetal glycosylated fibronectin in the synovial membranes of patients with rheumatoid arthritis and osteoarthritis, Rheumatol. Int., 2004, 24, 25–33 CrossRef CAS.
- E. Trachsel, M. Kaspar, F. Bootz, M. Detmar and D. Neri, A human mAb specific to oncofetal fibronectin selectively targets chronic skin inflammation in vivo, J. Invest. Dermatol., 2007, 127, 881–6 CrossRef CAS.
- C. M. Matter, P. K. Schuler, P. Alessi, P. Meier, R. Ricci, D. Zhang, C. Halin, P. Castellani, L. Zardi, C. K. Hofer, M. Montani, D. Neri and T. F. Luscher, Molecular imaging of atherosclerotic plaques using a human antibody against the extra-domain B of fibronectin, Circ. Res., 2004, 95, 1225–33 CrossRef CAS.
- M. Pedretti, Z. Rancic, A. Soltermann, B. A. Herzog, C. Schliemann, M. Lachat, D. Neri and P. A. Kaufmann, Comparative immunohistochemical staining of atherosclerotic plaques using F16, F8 and L19: Three clinical-grade fully human antibodies, Atherosclerosis, 2010, 208, 382–9 CrossRef CAS.
- M. L. Chang, J. C. Chen, C. R. Alonso, A. R. Kornblihtt and D. M. Bissell, Regulation of fibronectin splicing in sinusoidal endothelial cells from normal or injured liver, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18093–8 CrossRef CAS.
- W. R. Jarnagin, D. C. Rockey, V. E. Koteliansky, S. S. Wang and D. M. Bissell, Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis, J. Cell Biol., 1994, 127, 2037–48 CrossRef CAS.
- M. Odenthal, K. Neubauer, K. H. Meyer zum Buschenfelde and G. Ramadori, Localization and mRNA steady-state level of cellular fibronectin in rat liver undergoing a CCl4-induced acute damage or fibrosis, Biochim. Biophys. Acta, 1993, 1181, 266–72 CAS.
- G. K. Koukoulis, J. Shen, I. Virtanen and V. E. Gould, Immunolocalization of cellular fibronectins in the normal liver, cirrhosis, and hepatocellular carcinoma, Ultrastruct. Pathol., 1995, 19, 37–43 CrossRef CAS.
- S. Matsui, T. Takahashi, Y. Oyanagi, S. Takahashi, S. Boku, K. Takahashi, K. Furukawa, F. Arai and H. Asakura, Expression, localization and alternative splicing pattern of fibronectin messenger RNA in fibrotic human liver and hepatocellular carcinoma, J. Hepatol., 1997, 27, 843–53 CrossRef CAS.
- N. J. Hackl, C. Bersch, P. Feick, C. Antoni, A. Franke, M. V. Singer and I. A. Nakchbandi, Circulating fibronectin isoforms predict the degree of fibrosis in chronic hepatitis C, Scand. J. Gastroenterol., 2010, 45, 349–56 CrossRef CAS.
- T. H. Huang, K. R. Chintalacharuvu and S. L. Morrison, Targeting IFN-alpha to B cell lymphoma by a tumor-specific antibody elicits potent antitumor activities, J. Immunol., 2007, 179, 6881–8 CAS.
- C. Xuan, K. K. Steward, J. M. Timmerman and S. L. Morrison, Targeted delivery of interferon alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma, Blood, 2010, 115(14), 2864–2871 CrossRef CAS.
- P. Holliger, T. Prospero and G. Winter, “Diabodies”: small bivalent and bispecific antibody fragments, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6444–8 CrossRef CAS.
- M. Pedretti, C. Verpelli, J. Mårlind, G. Bertani, C. Sala, D. Neri and L. Bello, Combination of temozolomide with immunocytokine F16-IL2 for the treatment of glioblastoma, Br. J. Cancer, 2010, 103(6), 827–836 CrossRef CAS.
- E. A. Rossi, D. M. Goldenberg, T. M. Cardillo, R. Stein and C. H. Chang, CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas, Blood, 2009, 114, 3864–71 CrossRef CAS.
- T. Y. Basham, M. S. Kaminski, K. Kitamura, R. Levy and T. C. Merigan, Synergistic antitumor effect of interferon and anti-idiotype monoclonal antibody in murine lymphoma, J. Immunol., 1986, 137, 3019–24 CAS.
- J. N. Rybak, C. Roesli, M. Kaspar, A. Villa and D. Neri, The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases, Cancer Res., 2007, 67, 10948–57 CrossRef CAS.
- E. S. White, F. E. Baralle and A. F. Muro, New insights into form and function of fibronectin splice variants, J. Pathol., 2008, 216, 1–14 CrossRef.
- S. Astrof, D. Crowley and R. O. Hynes, Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin, Dev. Biol., 2007, 311, 11–24 CrossRef CAS.
- I. Peter, A. Mezzacasa, P. LeDonne, R. Dummer and S. Hemmi, Comparative analysis of immunocritical melanoma markers in the mouse melanoma cell lines B16, K1735 and S91-M3, Melanoma Res., 2001, 11, 21–30 CrossRef CAS.
Footnote |
| † Published as part of an Integrative Biology themed issue in honour of Mina J. Bissell: Guest Editor Mary Helen Barcellos-Hoff. |
| This journal is © The Royal Society of Chemistry 2011 |