Computational analysis and structure predictions of CHH-related peptides from Litopenaeus vannamei†
G. Purna Chandra
Nagaraju
*a,
N. Siva
Kumari
b,
G. L. V.
Prasad
c,
B. Reddya
Naik
c and
D. W.
Borst
*a
aDepartment of Biology, University of Central Florida, Orlando, FL 32816, USA. E-mail: dborst@mail.ucf.edu; pganji@emory.edu; Fax: 407-823-5769; Tel: 407-823-1460
bDepartment of Marine Environmental Biology, Nicholls State University, Thibodaux, LA-70310, USA
cDepartment of Zoology, Osmania University, Hyderabad, AP-500 007, India
First published on 6th December 2010
Abstract
The crustaceans produce several related peptides that belong to the crustacean hyperglycemic hormone (CHH) family. While these peptides have similar amino acid sequences, they have diverse biological functions that must arise, in part, from differences in the 3D shape of these peptides. However, it is generally accepted that peptides with a high degree of sequence similarity also have a similar 3-D structure. We used the solution structure of one peptide in the crustacean hyperglycemic hormone family, the molt-inhibiting hormone of the kuruma prawn (Marsupenaeus japonicus), to predict the shape of the five known peptides related to CHH in the Pacific white shrimp, Litopenaeus vannamei. The high similarity of the 3-D structures of these peptides suggests a common fold for the entire family. Nevertheless, minor differences in the shape of these peptides were observed, which may be the basis for their different biological properties.
Insight, innovation, integrationCHH peptides were originally identified as regulators of carbohydrate homeostasis. It is now known that members of this CHH family regulate several physiological functions, such as molting, reproduction, osmoregulation, metamorphosis and metabolism. Different functions imply that they may have different 3-D structures, allowing them to bind to different receptor proteins. However, the clarification of a peptide 3-D structure is often difficult for technical reasons (obtaining enough peptide, creating diffracting crystals, etc.). We used homology modeling based on the Marja-MIH to predict the shape of the five type I CHH peptides that have been reported in the white shrimp L. vannamei. The high similarity of the 3-D structures of these peptides suggests a common fold for the entire family. Nevertheless, minor differences in the shape of these peptides were observed, which may be the basis for their different biological properties. |
1. Introduction
An important source of neuroendocrine peptides in decapod crustaceans is the X-organ-sinus gland complex (XO-SG). This complex is located in the eyestalk and produces many distinct neuropeptides. These peptides affect a wide variety of physiological activities either directly or indirectly by modulating other endocrine organs. In most decapods, Crustacean hyperglycemic hormone (CHH) is the most abundant peptide in the eyestalk. Many of the other eyestalk peptides have related amino acid sequences. Together, these neuropeptides form the CHH peptide family, which includes CHH, molt-inhibiting hormone (MIH), gonad/vitellogenic-inhibiting hormone (GIH/VIH), and mandibular organ-inhibiting hormone (MOIH). Additional hormones unrelated to this family are also produced by the XO-SG complex, and include the chromatotropins such as red pigment concentrating hormone (RPCH) and pigment dispersing hormone (PDH). There are several reviews of these peptides.1–4Peptides in the CHH family have a similar size (72–80 residues). They also show a considerable degree of sequence similarity, most notably the conservation of 6 cysteine residues at the same relative positions. The family can be divided into two subfamilies, those that are related to CHH (‘CHH-related’, or type I) and those that are related to MIH/MOIH/VIH (or type II). The two subfamilies differ substantially in the processing of the initial peptide leading to some mistaken identities. For example, several MIH or MIH-like peptides were identified in Litopenaeus vannamei by Lago-Lestónet al.5 and Sun.6 However, the amino acid sequences of their precursors indicate that they are CHH-like peptides, not MIH peptides. Two other eyestalk peptides were recently described that may be the MIH of L. vannamei.7
In some species, several of the type I CHH peptides are polymorphic and contain minor variations in primary sequence and/or isomerization of single residues.8–13 It has been proposed that type I CHH isoforms may be specialized for different physiological functions,14–17 but the significance of structural polymorphism for these different functions is not yet clear. In addition, recent studies have shown that type I CHH-peptides are produced in other, non-eyestalk tissues, including the stomach, the pericardial organ, hemocytes, and gills.18–23 These studies suggest that type I CHH variants have important additional roles in molting, reproduction and osmotic regulation. In short, we still have much to learn about the biochemistry and the biological function of the CHH peptide family, especially those members of the type I CHH subfamily.
Knowledge of the 3-dimensional (3-D) configuration of a protein or a peptide is an important aid to understand the function of the protein.24–26 High resolution structures of proteins, polypeptides, ligand–protein complexes and other macromolecular compounds can be obtained by X-ray crystallography or NMR. Given that determination of high resolution protein or peptide structures is hard to obtain, an alternate approach is homology modeling. This provides an educated guess about the 3-D structure of a compound (the model compound) by comparing it to the 3-D structure of a related protein (the template compound). Homology modeling determines how well each region of the model compound fits the template compound and thereby the confidence for the proposed 3-D structure. With the increase in the number of proteins with a known 3-D structure, homology modeling has become widely used and has produced satisfactory results for many proteins,22 especially when an optimization procedure with the satisfaction of spatial restraints (e.g. the Needleman-Wunsch algorithm in the MODELLER program) is used.26
The only member of the CHH peptide family with a known 3-D structure is the MIH of the kuruma prawn, Marsupenaeus japonicus.2 We used homology modeling based on the Marja-MIH to predict the shape of the five type I CHH peptides that have been reported in the white shrimp L. vannamei. Bioinformatics approaches were applied to these type I CHH-related sequences from public databases and published reports. Using Modeller software and bioinformatics tools, 3-D structures of these peptides were predicted and potential modification sites were identified.
2. Results
2.1 Homology modeling of CHH
A BLAST and 3DPSSM search of the Litva_CHH sequences disclosed several potential reference structures in the PDB. These included the molt inhibiting hormone from Marsupenaeus japonicus (Marja_MIH; PDB ID# 1J0T) and pentaketide chromone synthase (PCS) from Aloe arborescens (alone, PDB ID# 2D51; in a complex with coenzyme A, PDB ID# 2D3M). Marja_MIH was chosen because it had a greater sequence identity (34%) and similarity (65%) to Litva_CHH, than PCS (25% similarity).The structurally conserved regions (SCRs) of the CHH peptides and Marja_MIH were identified by aligning the individual CHH peptides and the MIH template (see Fig. 1). Atomic coordinates from the template protein (Marja_MIH) for the SCRs, Structurally Variable Regions (SVRs), N-termini and C-termini were assigned to the target sequences based on the satisfaction of spatial restraints. This allowed us to generate an initial 3-D model for each CHH.
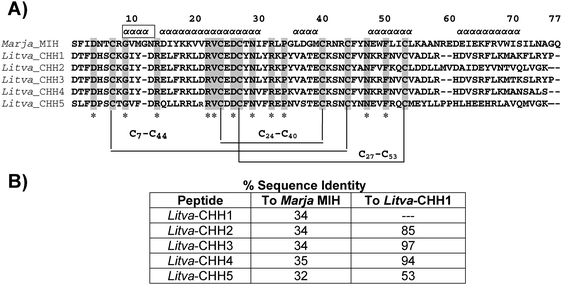 | ||
| Fig. 1 (A) The 3-dimensional conformations of CHH-like peptides and Marja_MIH were stabilized by a number of conserved hydrophobic interactions of fourteen hydrophobic amino acid residues (I/F3, V/I10, I/L16, Y/F17, V/L20, V23, I/V/L30, F/Y31, L/V36, F/Y45, F50, L/V54, I/V63, and F/Y66) and the three disulfide bonds. The fourteen hydrophobic (shown as *) and the 6 cysteine residues were conserved in all of the peptides, suggesting that the CHH-like peptides and MIH have similar folding. The α-helical regions of Marja_CHH are shown as ααα. In addition, seven acidic amino acids (at position 4, 13, 15, 25, 26, 29 and 47) and seven basic amino acids (at position 8, 14, 18, 19, 22, 32 and 41) were strongly conserved in both MIH and CHH-like peptides. (B) The % sequence of identity of the Litva_CHH to Marja_MIH and to Litva_CHH1. | ||
2.2 Validation of the proposed CHH models
Validation of the 3-D model for each CHH peptide was carried out using Ramachandran plots. Table 1 shows the summary of the G factors and the percentages of residues in the various regions of the Ramachandran plots for models of the Litva_CHH peptides based on the Marja_MIH template. When analyzed by PROCHECK, the CHH models had G factors above −0.5, indicating that the secondary structure of each model is realistic. The Φ and Ψ distributions of the residues other than glycine and proline in the Ramachandran plots are summarized in Fig. 2. Altogether, nearly 100% of these residues were in the favored and the allowed regions (Table 1). The final structure was subsequently checked by VERIFY-3D and the results indicate that the location of all residues appears to be reasonable (i.e., a compatibility score >0; Fig. 3). | ||
| Fig. 2 Ramachandran plots showed the residues that were in the most favored regions (red), additional allowed (yellow), generously allowed (light yellow), and disallowed (white) regions. The cysteine residues C7, C23, C27, C40 and C53 in Marja_MIH were in right-handed alpha helixes and C44 was in a left-handed alpha helix. | ||
 | ||
| Fig. 3 The 3D profiles verified results of CHH models. Residues with positive compatibility score are reasonably folded. | ||
| M. japonicus | P. vannamei | |||||
|---|---|---|---|---|---|---|
| MIH | CHH1 | CHH2 | CHH3 | CHH4 | CHH5 | |
| a The core region corresponds to conformations of the polypeptide backbone where there are no steric clashes. b The allowed region corresponds to conformations in which shorter Van der Waals radii are used in the calculation, that is the atoms are allowed steric clashes, permitting consideration of left handed alpha helices. c Disallowed regions involve steric hindrance between the side chain group and main chain atoms and typically occur in turn regions of proteins. d RMSD values for the structures. | ||||||
| Core regionc | 60.0% | 75.7% | 78.6% | 80.9% | 77.7% | 78.8% |
| Allowed regionb | 34.3% | 18.6% | 18.4% | 18.1% | 17.0% | 18.2% |
| Generously Allowed region | 5.7% | 5.7% | 3.0% | 1.1% | 4.3% | 3.0% |
| Disallowed regionc | 00.0% | 00.0% | 00.0% | 00.0% | 1.1% | 00.0% |
| G factor | −00.08 | −0.13 | −0.17 | −0.07 | −0.14 | −0.19 |
| RMSD d | — | 0.85 Å | 0.75 Å | 0.92 Å | 0.97 Å | 0.60 Å |
The conformations of Marja_MIH and the Litva_CHH peptides appear to be stabilized by disulfide bonds and the placement of hydrophobic and hydrophilic amino acids. The positions of the six cysteine residues were highly conserved in all five CHH peptides and appear to form 3 disulfide bonds (7–44, 24–40 and 27–53; see Fig. 1 and Table 2). The characteristics of the disulfide bond length in the homology models of Litva_CHH are similar to Marja_MIH (Table 2). The locations of 14 hydrophobic amino acids (positions 3, 10, 16, 17, 20, 23, 30, 31, 36, 45, 50, 54, 63 and 66) are also conserved in all five peptides, suggesting that hydrophobic interactions between these residues are important in maintaining structure stability. In addition, the positions of seven acidic (4, 13, 15, 25, 26, 29 and 47) and seven basic (8, 14, 18, 19, 22, 32 and 41) residues are conserved in the peptides (Fig. 1). Finally, all of the CHH peptides are predicted to contain four α-helix regions while Marja_MIH has been shown to have five (Fig. 4).
 | ||
| Fig. 4 Ribbon representations of the constructed α-Helical forms of CHH models generated from the MODELLER program. | ||
| M. japonicus | L. vannamei | |||||
|---|---|---|---|---|---|---|
| MIH | CHH1 | CHH2 | CHH3 | CHH4 | CHH5 | |
| C07 and C44 | 2.26 Å | 2.019 Å | 2.027 Å | 2.007 Å | 2.021 Å | 2.017 Å |
| C24 and C40 | 2.242 Å | 2.029 Å | 2.025 Å | 2.028 Å | 2.026 Å | 2.025 Å |
| C27 and C53 | 1.986 Å | 2.026 Å | 2.027 Å | 2.028 Å | 2.023 Å | 2.021 Å |
2.3 RMS comparisons of the backbone fold
The root-mean-square (RMS) deviation of each model from the Marja_MIH template structure was calculated, based on the carbons (Cα), using the SPDBV (Swiss PDB viewer).27 The backbone RMS deviation of the modeled structures differed only slightly from the NMR structure of Marja_MIH. As shown in Table 1 and Fig. 3, the average RMS deviations are generally within 1 Å. This further indicates that the homology model is reliable and super-imposable on the corresponding template structure.2.4 Identification of potential phosphorylation and myristolylation sites
Prosite analysis predicted possible modification sites in each peptide (Table 3). Marja_MIH contains a protein kinase C (PKC) phosphorylation site (amino acid residues from 06–09), a casein kinase II (CK-2) phosphorylation site (amino acid residues from 01–05), and two N-myristoylation sites (amino acid residues from 09–15 and 38–44). An analysis of the CHH peptides showed putative phosphorylation sites for PKC and TYR (tyrosine kinase phosphorylation site) on all of the CHH peptides except Litva_CHH5, which had an N-glycosylation site and a CK-2 site. The CHH peptideLitva_CHH3 had two potential PKC phosphorylaton sites (Table 3). It was noted that the CHH peptides contained minor variations in primary sequences in most of these putative sites.| Type of modification | Marja_MIH | Litva_CHH | |||
|---|---|---|---|---|---|
| 1 and 2 | 3 | 4 | 5 | ||
| Numbers indicate the amino acids that may be modified. | |||||
| ASN_glycosylation | — | — | — | — | 34 to 38 |
| PKC_phosphorylation | 06 to 09 | 06 to 09 | 06 to 09 | 06 to 09 | — |
| 69 to 72 | |||||
| CK2_phosphorylation | 01 to 05 | — | — | — | 01 to 05 |
| TYR_phosphorylation | — | 21 to 28 | 21 to 28 | 21 to 28 | — |
| N-myristoylation | 09 to 15 | — | — | — | — |
| 38 to 44 | |||||
3. Discussion
CHH peptides were originally identified as regulators of carbohydrate homeostasis. It is now known that members of this CHH family regulate several physiological functions, such as molting, reproduction, osmoregulation, metamorphosis and metabolism.8,15,20,21,28–30 Different functions imply that they may have different 3-D structures, allowing them to bind to different receptor proteins. However, the clarification of a peptide 3-D structure is often difficult for technical reasons (obtaining enough peptide, creating diffracting crystals, etc.). As a result there is no 3-D structure information for many of the peptides registered in GENBANK, including CHH.Homology modeling can be used to assign a tentative 3-D structure to a target protein. This methodology compares the target protein to a template protein with a known 3-D structure that has a sequence similar to the template protein. One approach is to thread the target protein backbone atoms over the backbone atoms of the template protein. However, refinement methods such as PROCHECK are needed to produce realistic models.
The CHH peptide family is divided into two sub-families. The type I subfamily includes peptides with CHH activity and type II subfamily includes peptides with MIH, GIH/VIH and MOIH activity. These divisions are based on the analysis of amino acid sequences and functional divergence.31,32 For example, the translation products for members of the type I subfamily are prepropeptides and contain a hydrophobic signal peptide33 of 30 amino acids, the precursor-related peptide (CPRP) of approximately 36 amino acids (for Litva_CHH CPRPs contain 27 amino acids), a dibasic cleavage site (Lys-Arg) located immediately after the CPRP, and the mature CHH, which usually contains 72–74 amino acids (Fig. 1). Type I CHH peptides have a blocked N-terminus (pyroglutamyl) and an amidated C-terminus. In contrast, the translated product for members of the type II subfamily contains simply a signal peptide of about 28–30 residues and the mature peptide (77–80 residues; Fig. 1).34 Type II CHHs usually have (with a few exceptions) free N- and C-termini.17,32,35
For our analysis, we removed the CPRP residues from the Litva_CHHs because no homologous region occurs in Marja_MIH (Fig. 1). In addition, the mature Litva_CHHs do not contain this element. The Marja_MIH has been shown to have five α-helix regions, while Litva_CHH's are predicted to contain four α-helix regions (Fig. 4). This observation confirms the importance of the glycine residue at position 12 of MIH (Fig. 1)35,36 which is thought to be critical for the structural and functional divergence of type II peptides.37Glycine is absent in this position in type I CHHs, including the Litva_CHHs.
All five of the Litva_CHH peptides have 6 cysteine residues at the same relative positions (7, 24, 27, 40, 44 and 53). These allow the formation of three intramolecular disulfide bonds that provide strong stabilization of the 3-D structure (Fig. 1).13,17,33 These bonds, in addition to the similar sequences of the Litva_CHH's, are reflected in their similar structures. In fact, the calculated RMSD of the alpha-carbon (Cα) co-ordinates for protein cores share 50% residue identity at 1 Å, similar to the value that we calculated for the five Litva_CHH's.
The five Litva neuropeptides have similar protein structures and share conserved characteristics in amino acid sequence. Thus, the peptides form part of a unique family of neuropeptides. Previously, Lago-Lestónet al.5 showed that Litva_CHH1 and Litva_CHH2 are more closely related to the type I CHH subgroup than they are to the type II MIH/GIH/MOIH subgroup. Furthermore, they showed that purified recombinant Litva_CHH1 could raise glucose levels in the hemolymph of L. vannamei, but had no effect on molting.38 Tiu et al.23 observed higher levels of Litva_CHH5 messenger in the posterior gills which suggests its potential role in osmo-regulation. They also examined Litva_CHH5 messenger levels in the gills, which decreased when shrimp were exposed to both salinity extremes.
In the present analysis we found several hydrophobic and cysteine residues are conserved in all of the peptides, as shown in Fig. 1, suggesting that the Litva_CHH peptides and Marja_MIH peptides fold in similar ways. Recent reports suggest that the members of the CHH/MIH gene family derived from a common ancestral gene through gene duplication and mutation. These findings also agree with previously published homology and 3-D modeling of CHH family neuropeptides.20,37 Collectively, these conclusions validate further 3-D modeling of crustacean CHHs. On the other hand, photoaffinity-labeling, antibody recognition information and site-directed mutagenesis experiments would be useful for confirming and enhancing the anticipated models. Further, to know whether the MIH or CHH peptides are phosphorylated at any specific site might be explored in vitro using phospho-specific antibodies.
Gade et al.39 found a phosphorylated neuropeptide (Trifa-CC) was released from the corpus cardiacum of the protea beetle. Our sequence analysis indicates that CHH peptides have several putative modification sites (e.g.phosphorylation, myristoylation). Of course, the mere existence of a possible modification site does not mean that it is actually modified. Nevertheless, such modifications, along with variations in amino acid sequences, could explain the multiple functions of these peptides.
4. Methodology
The Crustacean Hyperglycemic Hormone (CHH) sequences for L. vannamei (Litva) used in this study were published previously. The sequences for Litva_CHH1, CHH2, and CHH3 (GenBank accession no's. AAN86054, AAN86055, AAN86056, respectively) were described by Lago-Lestónet al.5 The sequence for Litva_CHH4 (GenBank accession no. AAC60516) was described by Sun,6 although it was initially mislabeled as MIH. The sequence for Litva_CHH5 (GenBank accession no. X99731.1) was submitted by Van Wormhoudt to Genbank in 1996. According to Tiu et al.,23Litva_CHH5 can also be considered a putative ion transportCHH peptide (Litva_CHH-ITP), because its C-terminal end is quite different from the other Litva_CHH sequences. This C-terminal region has a high degree of similarity to the insect ion transport peptide.26 The transcript levels of this peptide are most abundant in the posterior gills of L. vannamei.23For homology modeling, we used the solution structure of MIH from the kuruma prawn, Marsupenaeus japonicus PDB (BAA20432 Protein Data Bank) ID# 1J0T2 as the template. These CHH proteins and over 100 homologous sequences from non-crustaceans were found using BLAST (http://www.ncbi.nlm.nih.gov.ilsprod.lib.neu.edu/blast/) and 3D PSSM searches (http://www.sbg.bio.ic.ac.uk/%E2%88%BC3dpssm). It should be noted that the amino acid sequences of the CHHs that we analyzed were obtained by conceptually translating cDNAs. Hence, the initial sequences included the crustacean precursor-related peptide (CPRP; residues 1–27) and cleavage region (28–29) that are not present in the mature peptides. We removed these residues from each of the Litvapeptides before analysis. After trimming, the CHH peptides contained 72–74 residues. In situ, the trimming of type I CHHs leaves a pyroglutamate residue on the N-terminal end and an amidated C-terminus. We did not add these features to the type I CHH sequences analyzed here.
Theoretical 3-D models of Litva_CHH peptides were built using homology modeling. Marja_MIH (PDB ID: 1J0T) was used as a reference (template) structure because its amino acid sequence has a high level of identity to the Litva_CHH (target) peptides (see Fig. 1). The coordinates for the structurally conserved regions (SCRs) of the Litva_CHHs were assigned from the template peptide using multiple sequence alignment, based on the Needleman–Wunsch algorithm.40 The best alignment of the template sequence to each target sequence was used to create a 3-D model for each target peptide of all non-hydrogen atoms using MODELLER software.41 MODELLER consists of a suite of applications that searches for and aligns a template structure to the target sequence before constructing and refining the protein model. One application is the SSR (Satisfaction of Spatial Restraints) program that derives distance and dihedral angle restraints in the form of probability density functions from the template protein. The spatial restraints methodology assumes that geometrical features, such as distances or angles, are conserved when comparing equivalent positions in homologous proteins. Therefore, restraints derived from the template proteins can be used as a guide for the construction of the target model.
The spatial restraints and CHARMM energy terms enforcing proper stereochemistry42 were calculated using routines in the MODELLER software and combined into an objective function. The models with the lowest value were used. Each model was further refined for energy minimization by calculating a molecular mechanics force field (Kollman-All-Atom with Kollman charges) using the SYBYL 6.8 software package (Tripos Inc., St Louis, MO, USA). At the beginning of the energy minimization, the steepest descent technique was used to eliminate steric conflicts between the side-chain atoms until the root mean square (RMS) force was reduced to 50 kcal/(mol Å). After arriving at this threshold, further energy minimization was done by using Powell conjugate gradient optimization method until the maximum force became less than 0.05 kcal/(mol Å).
The final structures were analyzed by Ramachandran's maps using PROCHECK (http://biotech.ebi.ac.uk/).43 The PROCHECK G factor ranks values above −0.5 as positive candidates for homology models. The G-factor provides a measure of how “normal”, or alternatively how “unusual”, a given stereo chemical property is. A low G-factor indicates that the property corresponds to a low-probability conformation. The reliability of the structure for each CHH peptide was determined using VERIFY3D (http://nihserver.mbi.ucla.edu/Verify_3D/).24 Verify3D analyzes the compatibility of an atomic model (3D) with its own amino acid sequence (1D). Each residue is assigned a structural class based on its location and environment (alpha, beta, loop, polar, nonpolar, etc.). A compatibility score of below 1 indicates an acceptable side chain environment.
A structure-based sequence alignment of the template structure was obtained using the program JOY.44 JOY is freely available for academic users and can be run on a UNIX-based platform (http://tardis.nibio.go.jp/joy/). The amino acid sequences of the Marja_MIH template and the CHH peptides (models generated by the MODELLER program) were submitted to the JOY program (protein sequence structure representation and analysis).44 This program converts 3D coordinates from PDB files to calculate secondary structural and local environmental features and displays the same in a color-coded format in the sequence alignment.
The identification of possible modification sites (e.g.phosphorylation and myristylation) was determined using SDSC Biologyworkbench3.2 (http://workbench.sdsc.edu/). In view of the importance of the disulfide bond for proper protein folding, disulfide bonds were created manually in each CHH modeled peptide using the ‘Create Disulfide’ functionality in SYBYL 6.8 software package (Tripos Inc., St Louis, MO, USA).
5. Conclusions
We have presented a detailed structural analysis of type I Litva_CHH neuropeptides. Computational analyses were used to determine structural information about these type I Litva_CHH neuropeptide hormones and aid the identification of potential phosphorylation sites. The structural recognition of these Litva_CHH peptides offers the basis for future functional studies of these signaling molecules. The comparative study of Litva_CHH neuropeptides will offer new insights into the functional consequences of peptide multiplicity and peptide evolution.Acknowledgements
This research work was supported by a grant (IBN-0611447) from National Science Foundation, USA. We are thankful to the Bioinformatics Division, Osmania University, for providing the software facility and allowing us to pursue our data analysis there.References
- D. W. Borst, in Encyclopedia of Hormones, ed. H. L. Henry and A. W. Norman, Elsevier, Amsterdam, 2003, vol. 1, pp. 340–351 Search PubMed.
- Huberman, Aquaculture, 2000, 191, 191–208 CrossRef.
- D. Soyez, Ann. N. Y. Acad. Sci., 1997, 814, 319–323 CrossRef CAS.
- Udomkit, S. Chooluck, B. Sonthayanon and S. Panyim, J. Exp. Mar. Biol. Ecol., 2000, 244, 145–56 CrossRef.
- A. Lago-Lestón, E. Ponce and M. E. Muñoz, Aquaculture, 2007, 270, 343–357 CrossRef CAS.
- P. S. Sun, Mol. Mar. Biotechnol., 1994, 3, 1–6 Search PubMed.
- H. Y. Chen, R. D. Watson, J. C. Chen, H. F. Liu and C. Y. Lee, Gen. Comp. Endocrinol., 2007, 151, 72–81 CrossRef CAS.
- E. S. Chang, G. D. Prestwich and M. J. Bruce, Biochem. Biophys. Res. Commun., 1990, 171, 818–826 CrossRef CAS.
- H. Dircksen, D. Bocking, U. Heyn, C. Mandel, J. S. Chung, G. Baggerman, P. Verhaert, S. Daufeldt, T. Plosch, P. P. Jaros, E. Waelkens, R. Keller and S. G. Webster, Biochem. J., 2001, 356, 159–170 CrossRef CAS.
- P. Edomi, E. Azzoni, R. Mettulio, N. Pandolfelli, E. A. Ferrero and P. G. Giulianini, Gene, 2000, 284, 93–102.
- P. L. Gu and S. M. Chan, FEBS Lett., 1998, 441, 397–403 CrossRef CAS.
- D. Soyez, F. Van Herp, J. P. Le Caer, C. P. Tensen and R. Lafont, J. Exp. Biol. Chem., 1994, 269, 18295–18298 Search PubMed.
- G. Wainwright, S. G. Webster, M. C. Wilkinson, J. S. Chung and H. H. Rees, J. Biol. Chem., 1999, 271, 12749–12754.
- M. Nithya and N. Munuswami, Hydrobiologia, 2002, 486, 325–333 CrossRef CAS.
- D. Sedlmeier, Amer. Zool., 1985, 25, 223–232 Search PubMed.
- L. Serrano, G. Balanvillain, D. Soyez, G. Charmantier, E. Grousset, F. Aujoulat and C. Spanings-Pierrot, J. Exp. Biol., 2003, 206, 979–978 CrossRef CAS.
- A. Yasuda, Y. Yasuda, T. Fujita and Y. Naya, Gen. Comp. Endocrinol., 1994, 95, 387–398 CrossRef CAS.
- E. S. Chang, S. A. Chang, B. S. Beltz and E. A. Kravitz, J. Comp. Neurol., 1999, 414, 50–56 CrossRef CAS.
- E. S. Chang, R. Keller and S. A. Chang, Gen. Comp. Endocrinol., 1998, 111, 359–366 CrossRef CAS.
- S. H. Chen, C. Y. Lin and C. M. Kuo, Mar. Biotechnol., 2004, 6, 83–94 CrossRef CAS.
- J. S. Chung, H. Dircksen and S. G. Webster, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13103–13107 CrossRef CAS.
- H. Nielsen, B. Soren and G. Von Heijen, Protein Eng., Des. Sel., 1999, 12, 3–9 CrossRef CAS.
- S. H. K. Tiu, J. G. He and S. M. Chan, Gene, 2007, 396, 226–235 CrossRef CAS.
- R. Lüthy, J. U. Bowie and D. Eisenberg, Nature, 1992, 356, 83–85 CrossRef CAS.
- M. A. Marti-Renom, V. A. Ilyin and A. Sali, Bioinformatics, 2001, 17, 746–747 CrossRef CAS.
- J. E. Phillips, J. Meredith, N. Audsley, N. Richardson, A. Macins and M. Ring, Amer. Zool., 1998, 38, 461–470 Search PubMed.
- N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714–2723.
- P. L. Gu, K. L. Yu and S. M. Chan, FEBS Lett., 2000, 472, 122–128 CrossRef CAS.
- G. P. C. Nagaraju, Aquaculture, 2007, 272, 39–54 CrossRef CAS.
- G. P. C. Nagaraju, G. L. V. Prasad and P. S. Reddy, Int. J. Appl. Sci. Eng., 2005, 3, 61–68 Search PubMed.
- D. Böcking, H. Dircksen and R. Keller, in The Crustacean Nervous System, ed. K. Wiese, Springer, Berlin, 2002, pp. 84–97 Search PubMed.
- Lacombe, P. Grève and G. Martin, Neuropeptides, 1999, 33, 71–80 CrossRef.
- R. Newcomb, E. Stuenkel and I. Cooke, Amer. Zool., 1985, 25, 157–172 Search PubMed.
- M. Khayat, W. J. Yang, K. Aida, H. Nagasawa, A. Tietz, B. Funkenstein and E. Lubzens, Gen. Comp. Endocrinol., 1998, 110, 307–318 CrossRef CAS.
- H. Katayama and H. Nagasawa, Zool. Sci., 2004, 21, 1121–1124 CrossRef CAS.
- T. Ohira, H. Katayama, K. Aida and H. Nagasawa, Fish. Sci., 2003, 69, 95–100 CrossRef CAS.
- H. Katayama, K. Nagata, T. Ohira, F. Yumoto, M. Tanokura and H. Nagasawa, J. Biol. Chem., 2003, 278, 9620–9623 CrossRef CAS.
- E. Sánchez-Castrejón, E. R. Ponce, M. B. Aguilar and F. Díaz, Elect. J. Biotechnol., 2008, 4, 1–9 Search PubMed.
- G. Gade, P. Simek, K. D. Clark and L. Auerswald, Biochem. J., 2006, 393, 705–13 CrossRef.
- S. B. Needleman and C. D. Wunsch, J. Mol. Biol., 1970, 48, 443–453 CAS.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- A. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- R. A. Laskoswki, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef.
- K. Mizuguchi, C. M. Deane, T. L. Blundell, M. S. Johnson and J. P. Overington, Bioinformatics, 1998, 14, 617–623 CrossRef CAS.
Footnote |
| † All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2011 |