pep2pro: a new tool for comprehensive proteome data analysis to reveal information about organ-specific proteomes in Arabidopsis thaliana†
Katja
Baerenfaller
*a,
Matthias
Hirsch-Hoffmann
a,
Julia
Svozil
a,
Roger
Hull
b,
Doris
Russenberger
a,
Sylvain
Bischof
a,
Qingtao
Lu
c,
Wilhelm
Gruissem
a and
Sacha
Baginsky‡
a
aDepartment of Biology, ETH Zurich, Universitaetstrasse 2, 8092 Zurich, Switzerland. E-mail: kbaerenfaller@ethz.ch; Fax: +41 44 6321044; Tel: +41 44 6323491
bFaculty of Life Sciences, University of Manchester, Brunswick Street, Manchester M13 9PL, UK
cInstitute of Botany, Chinese Academy of Sciences, Haidian Xiangshan Nanxincun 20, Beijing 100093, China
First published on 24th January 2011
Abstract
pep2pro is a comprehensive proteome analysis database specifically suitable for flexible proteome data analysis. The pep2pro database schema offers solutions to the various challenges of developing a proteome data analysis database and because data integrated in pep2pro are in relational format, it enables flexible and detailed data analysis. The information provided here will facilitate building proteome data analysis databases for other organisms or applications. The capacity of the pep2pro database for the integration and analysis of large proteome datasets was demonstrated by creating the pep2pro dataset, which is an organ-specific characterisation of the Arabidopsis thaliana proteome containing 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 522 identified proteins based on 2.6 million peptide spectrum assignments. This dataset provides evidence of protein expression and reveals organ-specific processes. The high coverage and density of the dataset are essential for protein quantification by normalised spectral counting and allowed us to extract information that is usually not accessible in low-coverage datasets. With this quantitative protein information we analysed organ- and organelle-specific sub-proteomes. In addition we matched spectra to regions in the genome that were not predicted to have protein coding capacity and provide PCR validation for selected revised gene models. Furthermore, we analysed the peptide features that distinguish detected from non-detected peptides and found substantial disagreement between predicted and detected proteotypic peptides, suggesting that large-scale proteomics data are essential for efficient selection of proteotypic peptides in targeted proteomics surveys. The pep2pro dataset is available as a resource for plant systems biology at http://www.pep2pro.ethz.ch.
522 identified proteins based on 2.6 million peptide spectrum assignments. This dataset provides evidence of protein expression and reveals organ-specific processes. The high coverage and density of the dataset are essential for protein quantification by normalised spectral counting and allowed us to extract information that is usually not accessible in low-coverage datasets. With this quantitative protein information we analysed organ- and organelle-specific sub-proteomes. In addition we matched spectra to regions in the genome that were not predicted to have protein coding capacity and provide PCR validation for selected revised gene models. Furthermore, we analysed the peptide features that distinguish detected from non-detected peptides and found substantial disagreement between predicted and detected proteotypic peptides, suggesting that large-scale proteomics data are essential for efficient selection of proteotypic peptides in targeted proteomics surveys. The pep2pro dataset is available as a resource for plant systems biology at http://www.pep2pro.ethz.ch.
Insight, innovation, integrationMass spectrometry has become an important tool for obtaining qualitative and quantitative protein information. Proteome data analysis remains a challenge, however, when analysis workflows require integration of results from different search algorithms, and tools allowing for versatile analysis of large integrated datasets are currently not available. We therefore built the pep2pro database. Its capacity for the integration and analysis of large proteome datasets was demonstrated by creating the pep2pro dataset, which represents an organ-specific Arabidopsis thaliana proteome map with 14522 identified proteins available at http://www.pep2pro.ethz.ch. We used this dataset to establish quantitative protein accumulation, to define organ-specific proteomes of mitochondria, peroxisomes and chloroplasts that reflect their specific contributions to plant metabolism, and to revise gene models by proteogenomic mapping.
|
Introduction
The production and integration of genome-scale quantitative transcript, metabolite and proteome data allow investigating the organisational principles and the regulatory mechanisms of living cells and organisms. Systematic information on proteins has been difficult to obtain in the past, yet it is important for systems biology approaches as proteins are the main effectors in cells. Mutations affecting protein abundance, the pattern of post-translational modifications and their ability to interact with other molecular components are the reason for most phenotypic alterations associated with genetic perturbations.1 Because protein expression is regulated at different levels, information about protein presence, abundance and activity cannot be inferred solely from measuring mRNA levels. Mass spectrometry (MS) has now become an important tool for obtaining qualitative and quantitative protein information. However, proteome data integration and analysis of large datasets remain a challenge. This is especially the case when data analysis requires integration of results from different search algorithms, because the different algorithms create different outputs with diverse scores and because the results will be partly overlapping and in some cases contradictory. In addition to the consistent integration of results, the proteome data analysis pipeline should enable flexible data handling and versatile analysis of the results to support efficient data mining and extraction of biologically meaningful information. Existing widely used tools are restricted to a predefined set of search algorithms and detailed analyses of the search results often require downloading of the data and integration into another system like a relational database. For example, in the data analysis workflow for PeptideAtlas2 mass spectrometry data are first loaded into the PeptideAtlas raw data repository, after which they are analysed through the Trans-Proteomic Pipeline (TPP) using either Sequest3 or X!Tandem4 as database-dependent search algorithm followed by statistical assessment by PeptideProphet5 and ProteinProphet.6 The results are then loaded into the PeptideAtlas database from where they can be downloaded or queried using few pre-formed queries. Similarly, for the analysis of mass spectrometry data with the Global Proteome Machine Database (GPMDB)7 a local installation of GPMDB can be created for data analysis using the implemented search algorithm X!Tandem4 and uploading of the result files into the public GPMDB data repository. Other tools, such as Scaffold (Proteome Software) or Phenyx (Genebio), provide some elaborate data analysis and visualisation tools and allow users to validate search results from Mascot (Matrix Science), Sequest3 and X!Tandem.4 While these are doubtlessly powerful tools in many workflows, they are constrained in combining search results from different search algorithms into one integrated dataset by applying user-defined filtering criteria. Thus, they can not be adopted if a workflow requires the use of additional search algorithms.The integration of search results from different search algorithms is challenging, first because results of the different algorithms come in different formats and with different quality scores. Second, the results can contradict each other when the algorithms annotate the same spectrum with different peptide sequences or different post-translational modifications or post-translational modifications at different positions. Third, the definitions of a non-tryptic end or a missed cleavage site can vary between the different algorithms. Sequest/PeptideProphet3,5 for example considers peptides at the C-terminus of a protein as tryptic, whereas for PepSplice8 they are semi-tryptic, and missed cleavage sites where arginine or lysine are followed by proline are not considered as missed cleavage by Sequest/PeptideProphet,3,5 but by PepSplice.8 This will cause problems in the database if not addressed properly when it comes to the definition of true tryptic peptides. In the pep2pro database and analysis tool that we developed these issues were addressed and the database schema allows the integration of search results from Sequest/PeptideProphet,3,5 PepSplice,8 Mascot (Matrix Science) and Inspect9 based on user-defined filtering and cut-off criteria. Following the same building principle additional search algorithms can easily be appended to the workflow.
The handling of post-translational modifications is an additional challenge that has to be dealt with upon setting up a database system for proteome data integration and analysis. This, because a peptide can either be modified or not and when it is modified, it can be modified one to several times at different positions, or it can even be modified at unknown positions as it is often the case for phosphopeptides. Another issue that a proteome analysis database needs to cope with is the management of several databases used for database-dependent spectrum annotation because of new database releases or because one wants to search different databases. Here it is important that the database schema can store datasets searched against the different databases, and that it is known for each peptide and protein identification in which database release it was identified. In order to provide solutions to these and other questions we present the database schema of our pep2pro database. So far the data integrated are exclusively from Arabidopsis, yet in principle the database schema allows for the integration of proteome data from any organism.
The datasets that become integrated into the pep2pro database schema are in a relational format, which gives the user full control over the data and the power of database querying for data analysis. The database schema explained below allows for a detailed analysis of the results and for insights from the data that can not be obtained when the querying capacities are pre-defined and hence limited or when the result files are present in non-relational format. The pep2pro system therefore serves as a proteome data analysis platform. To illustrate its capacity, we report a comprehensive and fully integrated proteome dataset for Arabidopsis that we built by combining different large-scale organ-specific datasets10–12 and adding additional measurements. This large-scale dataset is publicly available on our website http://www.pep2pro.ethz.ch from where all information on peptide and protein identifications can easily be retrieved. In addition, the data in the pep2pro database are available in the PRIDE database.13 The pep2pro dataset extends the efforts to build complete proteome maps for the model organisms Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster and Saccharomyces cerevisiae.10,11,14–16 It also expands other plant proteome databases, such as the Arabidopsis Subcellular Proteomic Database (SUBA),17 plprot18 or the Plant Proteome Database (PPDB),19 which focus mainly on organellar proteomes, to reveal the function of plants at different organisational levels. Several Arabidopsis proteomic databases, including AtProteome,10 SUBA,17 PPDB,19 the phosphorylation databases PhosPhAt20 and RIPP-DB,21 and the spectrum database PROMEX22 have been integrated into the MASCP Gator,23 a proteomics aggregator that summarises information about Arabidopsis gene models and can be accessed at http://gator.masc-proteomics.org/. The pep2pro database is also integrated into this resource and constitutes the entry point for accessing the pep2pro and AtProteome datasets.10
The analyses of the pep2pro dataset that we report here include the identification of organ- and organelle-specific processes based on protein abundance determined by normalised spectral counting.24 This quantification method is comparable to other quantification methods and is reliable, but only in very large datasets.25 Validation of quantitative information requires targeted measurements of a selected set of proteins. One such targeted approach is selected reaction monitoring (SRM) in which quantitative analysis is targeting a predetermined set of peptides and fragment ions while excluding all other peptides in the sample. Setting up SRM experiments requires previous information. First, peptides need to be identified that can be unambiguously assigned to proteins and are easily detectable by mass spectrometry. These peptides are referred to as proteotypic peptides (PTPs).26 Second, for each PTP fragment ions have to be determined that provide optimal signal intensity and stability and that help to discriminate the targeted peptide from other peptides in the sample.27 The pep2pro dataset presented here provides more than 140000 unique peptides that unambiguously identify proteins and 2.6 Mio annotated spectra, vastly expanding PROMEX, a spectral reference database for plant proteins.22 The experimental information provided in pep2pro is valuable for SRM, because experimentally determined PTPs show different physical features than predicted PTPs. In addition, the information provided in the pep2pro website for each peptide assignment will help in choosing the experimental procedure and the plant material that show promise for protein and peptide identification.
Results and discussion
Features of the pep2pro database
The public interface of pep2pro is available at http://www.pep2pro.ethz.ch. The data for all peptides is shown together with the proteogenomic mapping of the peptides onto the genome. In addition to the comprehensive organisation of the AtProteome and pep2pro datasets (see below), we have also successfully used pep2pro for the integration of phosphoproteome data, as demonstrated by the large-scale phosphoproteome analysis of Arabidopsis shoots.28 The phosphoproteome data were analysed with Inspect9 and Mascot (Matrix Science) using the TAIR8 (The Arabidopsis Information Resource)29 protein database. After combining the search results, peptide spectrum assignments to decoy database peptides and spectra for which Mascot and Inspect assigned a different peptide to the same spectrum were not taken into consideration for further analyses. Because manual inspection suggested that Inspect had difficulties with tyrosine phosphorylation site assignments we flagged those tyrosine phosphorylations that were exclusively based on Inspect peptide spectrum assignments. If one search algorithm fulfilled the cut-off criteria for determining the phosphorylation position, but the other search algorithm failed to identify the phosphorylation position, we retained the information that the peptide is phosphorylated but removed the position information. With the fully integrated dataset we investigated phosphoproteome dynamics during a circadian cycle, which resulted in the identification of 1429 phosphoproteins and 3029 phosphopeptides with the exact site of phosphorylation established in 2349 peptides. These data are also available on the pep2pro web site and the phosphoproteins identified in this dataset are interlinked with the Arabidopsis Protein Phosphorylation Site Database (PhosPhAt).20The database schema of pep2pro that we developed for the integration and analysis of the above-described data is shown in Fig. 1 and in more detail in pep2pro.sql in the Supplemental Data.† The database schema consists of several building blocks that have been integrated for different aspects of proteome data analysis. A detailed explanation of the different building blocks can be found in the Supplemental Data,† together with an algorithm for custom proteogenomic peptide mapping (Supplemental Fig. S1†). Supplemental Fig. S2† highlights the fundamental differences between the analysis workflows of pep2pro and PeptideAtlas2 to demonstrate that pep2pro is a novel tool enabling consistent data handling and flexible proteome data analysis.
 | ||
| Fig. 1 Database schema of the pep2pro database. | ||
The pep2pro dataset represents a fully integrated organ-specific proteome map
We recently reported a large-scale organ-specific characterisation of the Arabidopsis proteome and the identification of 86456 unique peptides that matched to 13029 proteins from the TAIR7 Arabidopsis annotated genome database.10 Since then, TAIR8 and TAIR9 database releases have become available and the original AtProteome dataset has been reanalysed (Table 1). We have expanded the original AtProteome dataset by adding two biological replicates of flowers and flower buds, three biological replicates of cotyledons and young leaves (each biological replicate measured in three technical replicates), and shoots at stage 1.06 and two weeks later. Collectively, these data represent the “AtProteome extended” datasets. In order to create a fully integrated organ-specific proteome map we also included the pollen proteome data of Grobei et al.12 and the organ-specific proteome data of Castellana et al.11 (excluding phosphoproteome data) after subjecting the data to our standard protein analysis pipeline, resulting in the dataset termed “pep2pro TAIR9”.
| Dataset | Spectra | specFDR | Distinct peptides | Loci |
|---|---|---|---|---|
| TAIR7 database | 27029 |
|||
| AtProteome TAIR7 (original)10 | 790181 |
0.9% | 86456 |
13029 |
| AtProteome TAIR7 | 803577 |
1.0% | 86966 |
13154 |
| TAIR8 database | 27235 |
|||
| AtProteome TAIR8 | 799292 |
1.0% | 86763 |
13104 |
| AtProteome TAIR8 extended | 1062435 |
1.0% | 96145 |
14195 |
| TAIR9 database | 27379 |
|||
| AtProteome TAIR9 | 803530 |
1.0% | 87630 |
13290 |
| AtProteome TAIR9 extended | 1066363 |
1.0% | 97010 |
14378 |
| pep2pro TAIR9 | 2664441 |
0.9% | 144824 |
18111 |
| pep2pro TAIR9 wos (without single hits) | 2660852 |
0.7% | 141235 |
14522 |
| Large-scale phosphoproteome analysis of Arabidopsis thaliana shoots TAIR828 | 4905 |
|||
| Pollen proteome map of Arabidopsis thaliana TAIR712 | 4436 |
|||
False positive spectrum peptide assignments randomly hit proteins in the protein database, whereas true positive assignments are mostly consistent with already identified proteins. Consequently, the average number of peptide spectrum assignments per protein increased from 61 in the AtProteome TAIR9 dataset to 147 in pep2pro TAIR9. Adding more spectrum peptide assignments to the dataset therefore has the effect that more false positives accumulate in the group of single protein hits.30 To increase confidence in protein identifications, we therefore created the pep2pro TAIR9 wos dataset, in which 3589 single protein hits were excluded for which the peptide identification was based on the results of one search algorithm only, based on the assumption that good quality peptide spectra would mostly be assigned by both search algorithms. The remaining 1268 single peptide protein identifications show an average of 5 peptide spectrum assignments with a maximum of 124. These proteins have in average 22 theoretical tryptic peptides as compared to 32 for all proteins in the pep2pro TAIR9 wos dataset. The identification of only one peptide per protein can therefore to some extent be attributed to the lower number of detectable peptides. In summary, the pep2pro TAIR9 wos dataset has more than 2.6 million spectra assigned to 141235 peptides and 14522 genome loci. It is available from the pep2pro website and has been exported to the PRIDE database (accessions 15 478-15 540 and 15 564-15 565).13 From the three most prominent centralised and standard compliant databases for proteomics data, PeptideAtlas,2 GPMDB7 and PRIDE,13 PRIDE was chosen as it allows the submission of data that were generated in our data analysis workflow.
Features of identified and non-identified proteins
In order to investigate the depth of analysis and possible biases in the pep2pro TAIR9 wos dataset we first looked at over-represented GO categories of aspect biological process in the proteins from the TAIR9 protein database that we had not identified. The four most over-represented categories were regulation of transcription, DNA-dependent (p < 1e−30), regulation of transcription (p < 1e−30), response to chitin (p = 3.2e−13) and response to auxin stimulus (p = 2.1e−08). Transcription factors and signalling proteins are known to be of low abundance and the bias of proteomics studies for abundant proteins is well documented. For example, we had shown previously for the AtProteome TAIR7 dataset that proteins expressed at higher transcript frequency were preferentially detected.10 The bias for abundant proteins therefore most probably also accounts for the under-representation of proteins from these categories in this large dataset. We next looked at the distribution of INTERPRO domains and SCOP protein folds in identified versus non-identified proteins, for which we downloaded the lists TAIR9_all.domains and SCOP_class.tair9 from TAIR29 (ftp://ftp.arabidopsis.org/home/tair/Proteins/, on www.arabidopsis.org, version 20.6.2009), taking the first splice variant as a representative gene model. The set of non-identified proteins comprised many more proteins containing the following INTERPRO domains than the set of identified proteins: Transcription factor, MADS-box (97 non-identified proteins vs. 11 identified proteins), F-box associated (135 vs. 16) and Pathogenesis-related transcriptional factor and ERF, DNA-binding (130 vs. 16). Likewise, the non-identified proteins contained more proteins with the SCOP protein folds SRF-like (98 vs. 11), NAC domain (100 vs. 16) and DNA-binding domain (139 vs. 27), which are all involved in transcriptional regulation. We next analysed the coverage of plant-specific proteins from the Plant Specific Database (http://genomics.msu.edu/plant_specific/index.html).31 Of the 3831 unique plant-specific loci, only 46% were identified. Because plant-specific proteins include a large proportion (13%) of transcription factors,32 this low coverage might again be explained by the bias against low-abundance proteins. A functional categorisation of the non-identified plant-specific proteins against all plant-specific proteins using the GO ontology with aspect biological process identified regulation of transcription, DNA-dependent (p = 7.3e−20) as the most over-represented category, which is consistent with our interpretation. Taking all these results together, we conclude that protein abundance is a major factor determining if a protein can be identified in high-throughput experiments.In addition to the bias against low-abundance proteins, a bias against low-molecular weight proteins was also reported for the AtProteome TAIR7 dataset, in which the average molecular weight of the identified proteins was 54.7 kDa as compared to the average of 45.9 kDa for all proteins in the TAIR7 protein database.10 The average molecular weight for proteins in the pep2pro TAIR9 wos dataset is 54.2 kDa as compared to the average of 45.0 kDa for all proteins in TAIR9, indicating that we could not reduce the bias against low-molecular mass proteins by combining high-throughput data from different resources into a larger database. This suggests that overcoming this bias would require additional measurements with specific enrichment of under-represented protein categories.
Another bias often attributed to proteomics datasets is that against membrane proteins. We therefore analysed the coverage of proteins containing trans-membrane domains (TMDs) using Membrane_Proteins.tair9 (ftp://ftp.arabidopsis.org/home/tair/Proteins/Properties/, on www.arabidopsis.org, version 20.6.2009), again taking the first splice variant as a representative gene model. Fig. 2 shows that the pep2pro TAIR9 wos dataset does not bias against proteins containing TMDs as TAIR9 proteins predicted to contain more than 14 TMDs were identified at a rate of more than 80%. A likely explanation is that large proteins are identified more easily, and that the average molecular weight of the representative gene models containing at least 14 TMDs is 124 kDa. The reason for the pep2pro TAIR9 wos dataset not showing the bias against trans-membrane proteins might be that the bias observed previously can most probably be attributed to the prevalence of 2D-PAGE analyses in the early days of high-throughput proteomics. Sample preparation for 2D-PAGE analyses has stringent extraction requirements not allowing for buffer compositions with detergents that can solubilise membranes, whereas the protein samples used in pep2pro TAIR9 wos were mostly extracted with buffers containing strong detergents.
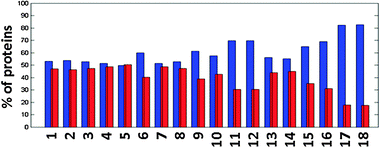 | ||
| Fig. 2 Percent of identified proteins (blue) and unidentified proteins (red) in the pep2pro TAIR9 wos dataset compared to all proteins in the TAIR9 database; 1 all identified proteins, 2 all proteins containing at least one trans-membrane domain (TMD), 3 proteins without a TMD, 4 proteins containing 1 TMD, 5 2 TMDs, 6 3 TMDs, 7 4 TMDs, 8 5 TMDs, 9 6 TMDs, 10 7 TMDs, 11 8 TMDs, 12 9 TMDs, 13 10 TMDs, 14 11 TMDs, 15 12 TMDs, 16 13 TMDs, 17 14 TMDs, 18 ≥15 TMDs. | ||
Organ-specific sub-proteomes and quantification of organ-specific biological processes
The pep2pro database provides information about the proteome of several distinct plant organs and therefore about organ-specific biological processes. Supplemental Table S2† gives an overview over the different plant organs and samples included in the pep2pro TAIR9 wos dataset and the number of assigned spectra, peptides and proteins, and Supplemental Table S3† shows the number of peptide spectrum assignments and the normalised spectral count for all quantified proteins in the different organs. To assess the robustness of organ-specific proteome characterisation, we compared the protein quantification results from different replicate measurements of flowers and leaves. The Spearman rank correlation coefficient for the four juvenile leaf replicates from the AtProteome dataset was between 0.62 and 0.65, and between 0.28 and 0.65 for the four leaf replicates from the Castellana dataset11 that had not been processed with the same method. The AtProteome juvenile leaves correlated with the Castellana leaves with a coefficient of 0.53. The correlation coefficient for the three open flower replicates from the AtProteome dataset was between 0.55 and 0.59, and for the two replicates from the Castellana dataset11 it was 0.76. The AtProteome open flowers correlated with the Castellana flowers with a coefficient of 0.2. Thus, the correlation in intra-laboratory datasets using the same technique is relatively high, whereas it is low for results from different laboratories or between different techniques in the same laboratory. This shows that it is important to apply the same method to different samples, if the results are to be compared, especially when working with relatively small datasets. Because the new pep2pro TAIR9 wos dataset combines poorly correlated organ-specific data from different resources we assessed how the combination of samples created in different laboratories influences the selectivity of protein detection in different organs, i.e. the degree of organ-specificity. For this, we repeated the GO-term classification of the identified proteins with which we had previously reported a number of organ-specific biological processes based on functional classification into TAIR GO categories using GO terms from aspect biological process and topGO.10,33,34 The functional classification of organ-specific sub-proteomes (Supplemental Table S4†) confirmed that in all organ proteomes translation was still over-represented, as well as response to cadmium ion, response to salt stress and response to cold. Other responses and processes associated with protein production were over-represented in the majority of the organ proteomes. Proteins were over-represented in the categories chloroplast organization in leaves, toxin catabolic process in roots, lipid storage in seeds, and RNA processing in cell culture. Together, most of the organ-specific categories previously determined were again identified in the pep2pro TAIR9 wos dataset. New organ-specific over-represented GO categories in this dataset include cellular nitrogen compound, heterocycle and water-soluble vitamin biosynthetic process in roots, as well as pollen germination and pollen tube growth in pollen.In order to refine our analysis of organ-specific processes, we queried pep2pro for organ- and sample-specific biomarkers. We defined as organ-specific biomarkers those proteins, which were identified with at least three spectra in one organ but not at all in all other organs. The proteins that fulfil the criteria were also subjected to a functional classification (Supplemental Table S5†). The most dramatic over-representation was obtained for flower biomarkers with pollen exine formation (Fig. 3A). Fig. 3B shows the quantitative distribution of the proteins in this functional category, which substantiates that most of the proteins in the category pollen exine formation are expressed specifically in flowers and not in other organs. The GO functional categories that were previously found over-represented in organ biomarkers10 and that were identified again with the new biomarker lists are toxin catabolic process in roots, lipid storage, lipid transport, embryonic development ending in seed dormancy and seed dormancy in seeds, and cell wall modification and anthocyanin biosynthetic process in siliques. New categories of organ-specific pathways are microsporogenesis in flowers, zinc ion transport in roots and pollen tube growth in pollen.
 | ||
| Fig. 3 (A) Over-represented functional categories of the biomarkers of each organ (B) Distribution of identified proteins belonging to GO-category pollen exine formation in the different organs in the pep2pro TAIR9 wos dataset as displayed in the quantification section of the pep2pro website. The color is a measure for the protein abundance; the darker red, the higher the normalised spectral count of that protein. | ||
This shows that the large pep2pro TAIR9 wos dataset, in which datasets from different laboratories were integrated, still displays organ-specific processes and that the organ-specific sub-proteomes are functionally significant. The biomarkers belong to processes that are clearly important for establishing organ-specific functions and hence validate the utility of the dataset to reveal insights into the functioning of the different organs of a plant. It also shows that the pep2pro database is instrumental in finding the expression pattern for proteins of interest in the different organs and samples.
Organelle-specific sub-proteomes
Because the depth of the organ-specific sub-proteomes in the pep2pro TAIR9 wos dataset allows the identification of organ-specific processes, we examined if quantitative differences in the organelle-specific sub-proteomes of the different organs could be identified using assembled protein lists for plastids, peroxisomes and mitochondria as examples. Since the contribution of organelles to cellular metabolism in different plant organs is not well understood, this analysis could reveal functional differentiation reflected in the organellar proteomes and thus provide important new information about compartmentalisation principles. While the chloroplast proteome is expected to reflect the ability to perform photosynthesis, the prevalence of other plastid functions in different organs is less well known, as is the contribution of mitochondria or peroxisomes to cell metabolism.To extract plastid proteins from the organ-specific proteome maps we used a previously published list,28 added missing plastid-encoded proteins and removed obvious contaminants (catalases, glycine dehydrogenases, 40S and 60S ribosomal proteins), as well as proteins that no longer exist in TAIR9. For peroxisomes we used the proteins identified by Reumann et al.,35 and for mitochondria we assembled the list by first querying the MitoP2 database36 and then adding all mitochondria-encoded proteins. From the final lists all proteins were removed that were present in more than one list, resulting in 1430 plastid, 127 peroxisomal and 396 mitochondrial proteins (Supplemental Table S6†). For the proteins from those lists that were quantified in the pep2pro TAIR9 wos dataset (Supplemental Table S3†) we assembled organ-specific protein lists. To quantify organ specific functions, we correlated the quantitative organellar proteomes in the different organs (Fig. 4A), which resulted in correlation coefficients ranging from 0.10 for leaves vs. pollen to 0.84 for flowers vs. leaves (Fig. 4A). The values for peroxisomes were similar and ranged from 0.15 for leaves vs. pollen and 0.85 for flowers vs. siliques. The range was much smaller for mitochondria where the lowest correlation coefficient was 0.64 for seeds vs. cell culture. The low correlation coefficients obtained with plastid and peroxisomal proteins indicate the organ-specific functions of these organelles, and illustrate organelle differentiation. In contrast, the correlation coefficients of mitochondrial proteomes are rather high, suggesting that little organ-specific differentiation occurs and that mitochondrial functions are similar in all organs.
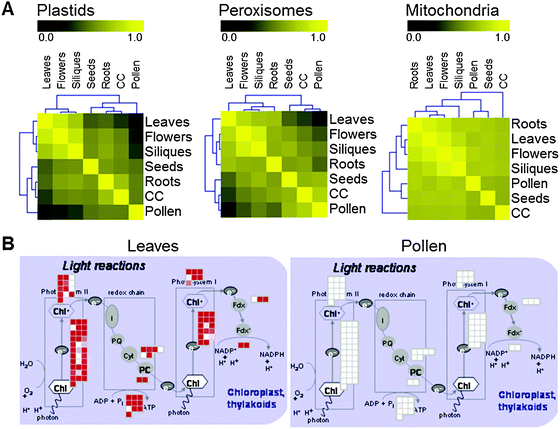 | ||
| Fig. 4 (A) Hierarchical clustering of the correlation matrices that were created by correlating the organ-specific factor lists for plastids, peroxisomes and mitochondria. (B) Abundance of photosynthetic proteins in leaf and pollen datasets. The low correlation of the plastid proteome in these two organs results from differences in photosynthetic capacity. Shown here is a MapMan43 representation of proteins involved in photosynthetic light reactions. | ||
Hierarchical clustering of the different correlation matrices revealed a separation between photosynthetic and non-photosynthetic plant organs for the plastid and peroxisomal proteins, but not for the mitochondrial proteins (Fig. 4A). Cell culture proteins cluster together with non-photosynthetic organs, even though green (illuminated) Arabidopsis cultured cells were used for about half of the MS/MS measurements. Illuminated cultured plant cells also depend on sucrose for growth, however, and therefore do not have a fully functional photosynthesis metabolism for autotrophic growth. This limits the utility of cultured cells for the analysis of quantitative processes at the protein level in plant organelles involved in assembling and regulating photosynthetic function and associated processes.
To further investigate if photosynthetic processes separate the photosynthetic and non-photosynthetic organs into different clusters and constitute the apparent functional proximity between peroxisomal and chloroplast functions, the abundance factors of the proteins from photosynthetic and non-synthetic organs were subjected to separate Spearman rank correlations using pairwise complete observations, followed by hierarchical clustering. Applying the hierarchical threshold clustering method37 with a minimum correlation coefficient of 0.8, the largest cluster from each dendrogram was identified and the proteins of those clusters were subjected to a functional classification. The most significantly over-represented GO biological process in the non-photosynthetic organ cluster was translation (p = 2.3e−18), whereas it was photosynthesis for the photosynthetic organ cluster (p = 8.2e−29). This shows that protein levels of photosynthetic proteins are co-regulated in photosynthetic organs, but less so in non-photosynthetic organs, and that photosynthesis separates the organs into different clusters. The adaptation of plastid and peroxisomal proteomes in different organs is therefore clearly connected to photosynthesis and photorespiration (Fig. 4B).
In summary, plastids and peroxisomes display high correlation coefficients between photosynthetic organs and photosynthetic organs clustered together for plastids and peroxisomes but not for mitochondria. In plants, peroxisomes differentiate into several variants. Two major variants are glyoxysomes that are induced upon seed germination and contain enzymes that are required to convert storage oil into carbohydrates, and leaf peroxisomes that are present in photosynthetic mesophyll cells where they play an important role in photorespiration.38 Photorespiration is of major importance for photosynthesis and therefore leaf peroxisomes are in close functional interaction with chloroplasts in photosynthetic organs. This is well reflected in the cluster analysis because we find a close coordination of plastid and peroxisomal functions, which is clearly driven by the photosynthetic capacity of the organs. This demonstrates that the protein abundance factors in the pep2pro TAIR9 wos dataset contain valuable information on organelle-specific differences in the different organs and hence can be used to obtain further insights into the processes taking place in different parts of a plant.
Expression evidence for alternative gene models
The high-density proteome map reported here is well suited to provide expression evidence for genome regions in which no protein coding capacity is currently predicted. We originally reported 57 new gene models based on the criteria that at least two peptides were assigned to the whole genome database to a region less than 3 kb apart.10 Of the 57 gene models in the AtProteome TAIR7 dataset, 10 were again identified in TAIR9 and the other 47 had been updated by the database curators (Table 2 and Supplemental Table S7†). The TAIR9 genome release information (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR9_genome_release/readme_TAIR9.txt on www.arabidopsis.org) reports that the proteome data provided by AtProteome10 and Castellana et al.11 and helped to reclassify 99 pseudogenes as protein coding genes while an additional 9 pseudogenes were merged with existing protein coding genes. Indeed, of the 15 pseudogenes reported in the AtProteome TAIR7 dataset, 10 have now been reclassified as protein coding genes in TAIR9, and the remaining 5 are now annotated as transposable elements. In addition, 158 peptides from AtProteome were used to update gene structures. Castellana et al.11 had published the predicted protein sequences of 2308 revised gene models from 1473 loci in the TAIR7 protein database. These were based on whole genome peptide identifications searching a 6-frame translation of the whole genome database and a spliced-exon database with Inspect, followed by gene model building using a gene finding program. Of these gene models, 591 were examined by TAIR and 339 were incorporated into TAIR9 gene models. Of the 261 peptides from the originally reported 57 different loci in the AtProteome TAIR7 dataset, 171 peptides (66%) from 35 loci (61%) match to the predicted protein sequences of the revised gene models published by Castellana et al.11 Of the 219 peptides from 49 gene models in the AtProteome TAIR7 dataset that had been curated in the TAIR9 database, 133 peptides (61%) from 28 loci (57%) match to the Castellana et al.11 gene models. Together, this demonstrates that the different methods for the identification of whole genome hits lead to orthogonal datasets that both contribute to the improvement of the accuracy of genome annotation.| AtProteome TAIR7 | AtProteome extended TAIR8 | pep2pro TAIR9 wos | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Loci | Peptides | Spectra | Loci | Peptides | Spectra | Loci | Peptides | Spectra | |
| TAIR7 whole genome hits | 57 | 261 | 2611 | 49 | 216 | 2978 | 10 | 56 | 1342 |
| TAIR8 whole genome hits | 31 | 31 | 225 | 31 | 31 | 314 | 16 | 19 | 162 |
| TAIR9 whole genome hits | 12 | 12 | 26 | 14 | 17 | 39 | 28 | 40 | 213 |
| Total | 100 | 304 | 2862 | 94 | 264 | 3331 | 54 | 115 | 1717 |
With the AtProteome extended dataset searched against the TAIR8 database we included those whole genome hits that were identified with at least three spectra and analysed in greater detail the 31 newly identified gene models (AT00058 to AT00090). In order to validate a selected set of the new gene models, we performed PCR reactions for 6 intron and 2 intergenic regions. Primers for the intron regions were designed based on flanking exon sequences in the TAIR8 protein database and for the intergenic regions based on gene predictions by Fgenesh39 and GenScan.40 The PCR products were verified by sequencing. Fig. 5 shows that PCR evidence could be found for 7 of these 8 new gene models. The PCR product for AT00080 was 450 bp shorter than expected and its sequence shows that the exon/intron structure of the gene model is quite different from the prediction in TAIR8. The PCR product for AT00067 was 100 bp longer than expected and sequencing showed that the genome sequence in TAIR8 seems to lack 100 bp (Supplemental Fig. S3†). Of the 31 new gene models identified in the AtProteome extended TAIR8 dataset, 16 were still identified in TAIR9 and 15 had been revised by the curators. Searching the pep2pro TAIR9 wos dataset for whole genome hits applying the same criteria as above revealed 28 additional gene models (AT00091 to AT00119), resulting in expression evidence based on 115 peptides for a total of 54 gene models not annotated as protein coding genes in the TAIR9 protein database (Table 2 and Supplemental Table S7†). The expression evidence for these 54 gene models both came from the Castellana et al. dataset,11 which contributed 907 spectra assigned to 64 whole genome peptides from 30 gene models, as well as the extended AtProteome and pollen data, which gave 810 spectra assigned to 90 peptides from 45 gene models.
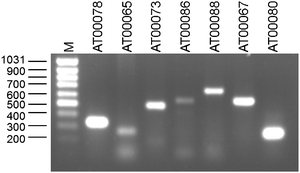 | ||
| Fig. 5 PCR products for the different new gene models were separated on a 1% agarose gel stained with ethidium bromide and visualised on a UV transilluminator. The first two gene models are from category intergenic and the last five from category intronic. The size of the gene products corresponds with what was expected, except for AT00067, which is around 100 bp too long and AT00080, which is around 450 bp too short. | ||
The release of the TAIR9 database has shown that the accuracy of genome annotation can be improved in cycles of genome annotation, identification of peptides from genome regions without annotated protein-coding capacity, and refinement of the annotation. With the data provided here we are continuing our effort in providing experimental evidence for expression from genome regions that are not annotated to be protein coding in the TAIR protein database, and hence contribute to a better annotation of the Arabidopsis genome.
Features of unidentified theoretical tryptic peptides
The identified unique peptides in the AtProteome extended TAIR9 dataset represent a substantial reference dataset for the extraction of features that influence the identification of peptides in MS/MS and that distinguish the 71618 detected true tryptic peptides (TTP: no missed cleavage, full tryptic ends, unambiguously identifying a protein) from the 589276 predicted but undetected TTP. This information is important when selecting optimal peptides for targeted analyses using SRM methods. The histograms in Fig. 6 show that the identified peptides have a lower average positive charge than the unidentified peptides (Fig. 6A1 and A2), which is consistent with the longer average peptide length (Fig. 6C1 and C2) and the corresponding higher molecular mass of the identified peptides (Fig. 6D1 and D2). Furthermore, the identified peptides have a lower average pI (Fig. 6E1 and E2). This analysis suggests that the most important feature that distinguishes detectable from undetected peptides is peptide length, which has been observed previously16 and can be explained by the higher information content of MS/MS spectra from larger peptides.
 | ||
| Fig. 6 Histograms of different peptide features. Row A Average positive charge, row B Positive charge, row C Peptide length, row D Peptide molecular weight, row E Peptide pI; Column 1 Theoretical tryptic peptides, which have been identified in the AtProteome extended TAIR9 dataset in blue and those not identified in red. Column 2 Theoretical tryptic peptides of proteins, which have been identified in the AtProteome extended TAIR9 dataset with more than 100 spectra in blue, and those not identified in red. Column 3 Experimental proteotypic peptides (ePTP, blue); PeptideSieve predicted and identified proteotypic peptides (psPTP, magenta); peptides that are both ePTP and psPTP (green). | ||
Computational prediction of proteotypic peptides with PeptideSieve26 produced 39741 predicted proteotypic TTP with a minimum probability of 0.9, of which 7802 (20%) had been identified in the AtProteome extended TAIR9 dataset. Comparing the features of the predicted and identified peptides with those that were predicted but not identified did not show significant differences (data not shown). We therefore analysed the abundance of the proteins containing these predicted proteotypic peptides by counting the spectra that identified them in the AtProteome extended TAIR9 dataset. The average number of spectra for proteins containing identified and PeptideSieve-predicted proteotypic peptides (psPTP) was 158, whereas it was only 31 for proteins containing PeptideSieve-predicted but not identified peptides. We then defined peptides that were identified by PeptideProphet5 with at least three spectra in at least one fraction in the AtProteome extended TAIR9 dataset to be experimental proteotypic peptides (ePTP) and compared the features of the 17928 ePTP with those of the 7802 psPTP, also including those 1535 peptides that are both ePTP and psPTP. All psPTP have a charge of 2 and display a quite narrow distribution of the peptide length with an average of only 10.9, whereas the ePTP show a broader distribution with an average peptide length of 17.1, which is even higher than the average peptide length of all identified peptides. The pI values of the ePTP also show a broader distribution than those of the psPTP and the average pI of the ePTP is again close to the average pI of all identified peptides. The peptides that are both ePTP and psPTP display similar patterns as the psPTP (Fig. 6, column 3), which shows that the prediction does not cover the range of physical features with which peptides can be identified. The deviation between prediction and experimental validation emphasises that the experimental proteome data are important for identifying proteotypic peptides that are useful as protein markers in quantitative proteome surveys.
This conclusion is further substantiated by a comparison of the AtProteome extended TAIR9 dataset with another experimental dataset of Arabidopsis proteotypic peptides that was downloaded from the GPMDB7 website (ftp://ftp.thegpm.org/projects/xhunter/libs/eukaryotes/peptide/ath1_cmp_20.fasta, version 06/07/2010). The FASTA file contains 62573 peptides and for each peptide a GPM identifier is given as description. A closer look at these peptides revealed that 1505 of them do not occur in the TAIR9 protein database but originate from other organisms or were identified by searching earlier versions of the Arabidopsis protein database, and some of the peptides no longer seem to be present in the GPMDB. For 15504 peptides it was found that they either contain up to eight missed cleavage sites or don't have tryptic ends, and 3487 peptides are ambiguous and fit to several different proteins with up to 101 loci per peptide. A comparison with the remaining 42077 TTPs gave that 35402 (84%) were also identified in the AtProteome extended TAIR9 dataset and 15987 (38%) were ePTPs. This high overlap is most probably a consequence of data exchange between GPMDB and PRIDE into which part of the data in the AtProteome extended dataset had already been integrated. A deeper analysis of the different sets of experimental proteotypic peptides would be interesting, yet this would require information on the peptide identifications also for the GPMDB dataset, as for example the data source, the number of identifying spectra, the protein database searched, the sample, the experimental procedure and the mass spectrometry method applied.
In summary, the more than 2.6 million MS/MS spectra in the pep2pro database together with the pep2pro interface from which all information about the peptide spectrum assignments can easily be retrieved will be instrumental in generating valuable quantitative proteome data for systems biology approaches.
Experimental
Interpretation of MS/MS spectra, data filtering and export to PRIDE
Peptide spectrum assignment and data filtering was done following earlier described methods.10 In brief, these consisted of searching the MS/MS spectra (1) with TurboSEQUEST and PeptideProphet5 using the Trans-Proteomic Pipeline (TPP) against the corresponding database with concatenated decoy database supplemented with contaminants, and (2) with PepSplice8 both against the protein database with an extended search space and the genome database. Post-translational modifications included in the searches with both algorithms were C iodoacetamide derivative as static and M oxidation as variable modification. In addition, PepSplice searches in the extended search space included the variable modifications N-term acetyl, C without iodoacetamide, C S-Methyl, D iodoacetamide derivative, D Phospho, H iodoacetamide derivative, H Phospho, K Acetyl/Carboxy/Trimethyl, K Hypusine, S Phospho, T Phospho, W Oxidation and Y Phospho/Sulfo, for which the modified amino acids are not isobaric to un-modified amino acids in the specified mass tolerance window. For PeptideProphet search results, the cut-off was set to a minimum probability of 0.9, for PepSplice the false discovery rate was adjusted to <0.01, assessed for each search space separately. All peptide spectrum assignments above the determined threshold, except those of known contaminants, were filtered for ambiguity. Peptides matching to several proteins were excluded from further analyses. This does not apply to different splice variants of the same protein or to different loci sharing exactly the same sequence. All remaining spectrum assignments were entered into the pep2pro database. After database upload, the following spectrum assignments were flagged and not taken into consideration for further analyses: (1) spectrum assignments to decoy database peptides, and (2) spectra for which PeptideProphet and PepSplice assign a different peptide to the same spectrum (including different posttranslational modifications and differently charged peptides). PRIDE 2.1 XML files were created from the final fully integrated dataset and exported to the PRIDE database.13Quantification of proteins
In pep2pro proteins are quantified by calculating the expected contribution of each individual protein to the samples total peptide pool10 based on a modification of the APEX-indexing method.24 For this we are assuming an equimolar distribution of proteins and correct the assumption with a correction factor that balances between detected and expected number of spectra for a given protein based on the depth of analysis and the number of theoretical tryptic peptides a protein can contribute. This is done according to the formula: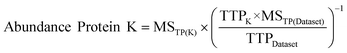
MSTP(K) = Measured spectra of trypic peptides from protein K
TTPK = Theoretical tryptic peptides of protein K
MSTP(Dataset) = Measured spectra of tryptic peptides in the dataset
TTPDataset = Theoretical tryptic peptides of the identified proteins in the dataset
Supplemental Table S3† provides for each organ the normalised spectral count and the number of spectra of true tryptic peptides for the quantified proteins in the pep2pro TAIR9 wos dataset.
GO functional classification
Assignment of protein functions was based on the TAIR GO categories from aspect biological process (download ATH_GO_GOSLIM_20090722.txt).34 The assignment was performed in R (version 2.8.1, http://www.r-project.org) using the elim method from the topGO package33 that is part of the Bioconductor project.41 Fisher's exact test was used for assessing the GO term significance.RNA isolation and RT-PCR
RNA was extracted from flower buds using Trizol (Invitrogen) according to manufacturer's instructions with subsequent DNase I treatment. For RT-PCR 1 μg total RNA was reverse-transcribed using an oligo(dT) primer and reverse transcriptase (SuperScript II, Invitrogen). Aliquots of the generated cDNA corresponding to 100 ng total RNA were used as template for PCR with the primers listed in Supplemental Table S8.†Proteotypic peptides
Experimental peptides are considered to be proteotypic when they unambiguously identify a protein and in case they were detected with at least 3 spectra by PeptideProphet in at least one fraction. Prediction of proteotypic peptides was performed using the PeptideSieve algorithm26 with parameters -M 6000 -p 0.9, -L 50, taking the resulting peptides for PAGE_ESI.Conclusion
The proteomics field is rapidly expanding and the amount of reported proteome data has increased exponentially over the last few years. The generated raw data are typically interpreted using different search algorithms, the search results integrated and the final data analysed and made publicly available. In order to make sure that the data can be fully exploited by the scientific community, meta-data (data about the data) are provided together with the proteome data, which describe the samples and the analysis methods in a standardised format following the Minimum Information about a Proteomics Experiment (MIAPE) checklist developed by the Proteomics Standards Initiative (PSI).42 For coherent proteome data integration and analysis of large datasets a relational database is best suited, but the setup of such a database is not trivial and often poses unforeseen challenges that must be dealt with. These issues were addressed in the pep2pro database schema. Together with the pep2pro database we present the pep2pro TAIR9 wos dataset, which integrates the two largest high-throughput proteomics experiments in Arabidopsis thaliana published so far10,11 and adds additional organ-specific measurements. The large integrated dataset contains 14522 identified proteins based on more than 2.6 Mio annotated spectra, corresponding to 53% of all proteins in the TAIR9 protein database. All the information concerning these protein identifications and the peptide spectrum assignments can easily be retrieved from the http://www.pep2pro.ethz.ch interface where we also provide a proteogenomic mapping of the peptides. We therefore expect that the pep2pro system will be a useful tool for comprehensive proteome data analysis and that the organ-specific proteome maps will constitute an important source of information for the plant community.
Acknowledgements
We thank the Functional Genomics Center Zurich for providing infrastructure and technical support. We also thank Tanja Kosterman from the Multimedia & E-learning Services at the University of Zurich for help with graphics and design of the pep2pro website. This work was supported by the SystemsX initiative in the framework of the C-MOP project, and by the European Framework Programme 6 AGRON-OMICS project to W.G. (LSHG-CT-2006-037704). Q.L. was supported by the Chinese State Scholarship Program (grant number [2006]3036#).References
- M. Gstaiger and R. Aebersold, Applying mass spectrometry-based proteomics to genetics, genomics and network biology, Nat. Rev. Genet., 2009, 10(9), 617–27 CrossRef CAS.
- E. W. Deutsch, The PeptideAtlas Project, Methods Mol. Biol., 2010, 604, 285–96 CrossRef CAS.
- J. K. Eng, A. L. Mccormack and J. R. Yates, An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database, J. Am. Soc. Mass Spectrom., 1994, 5(11), 976–989 CrossRef.
- R. Craig and R. C. Beavis, TANDEM: matching proteins with tandem mass spectra, Bioinformatics, 2004, 20(9), 1466–7 CrossRef CAS.
- A. Keller, A. I. Nesvizhskii, E. Kolker and R. Aebersold, Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search, Anal. Chem., 2002, 74(20), 5383–92 CrossRef CAS.
- A. I. Nesvizhskii, A. Keller, E. Kolker and R. Aebersold, A statistical model for identifying proteins by tandem mass spectrometry, Anal. Chem., 2003, 75(17), 4646–58 CrossRef CAS.
- R. Craig, J. P. Cortens and R. C. Beavis, Open source system for analyzing, validating, and storing protein identification data, J. Proteome Res., 2004, 3(6), 1234–42 CrossRef CAS.
- F. F. Roos, R. Jacob, J. Grossmann, B. Fischer, J. M. Buhmann, W. Gruissem, S. Baginsky and P. Widmayer, PepSplice: cache-efficient search algorithms for comprehensive identification of tandem mass spectra, Bioinformatics, 2007, 23(22), 3016–23 CrossRef CAS.
- S. H. Payne, M. Yau, M. B. Smolka, S. Tanner, H. Zhou and V. Bafna, Phosphorylation-specific MS/MS scoring for rapid and accurate phosphoproteome analysis, J. Proteome Res., 2008, 7(8), 3373–81 CrossRef CAS.
- K. Baerenfaller, J. Grossmann, M. A. Grobei, R. Hull, M. Hirsch-Hoffmann, S. Yalovsky, P. Zimmermann, U. Grossniklaus, W. Gruissem and S. Baginsky, Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics, Science, 2008, 320(5878), 938–41 CrossRef CAS.
- N. E. Castellana, S. H. Payne, Z. Shen, M. Stanke, V. Bafna and S. P. Briggs, Discovery and revision of Arabidopsis genes by proteogenomics, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(52), 21034–8 CrossRef CAS.
- M. A. Grobei, E. Qeli, E. Brunner, H. Rehrauer, R. Zhang, B. Roschitzki, K. Basler, C. H. Ahrens and U. Grossniklaus, Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function, Genome Res., 2009, 19(10), 1786–800 CrossRef CAS.
- J. A. Vizcaino, R. Cote, F. Reisinger, H. Barsnes, J. M. Foster, J. Rameseder, H. Hermjakob and L. Martens, The Proteomics Identifications database: 2010 update, Nucleic Acids Res., 2010, 38(Database), D736–42 CrossRef CAS.
- E. Brunner, C. H. Ahrens, S. Mohanty, H. Baetschmann, S. Loevenich, F. Potthast, E. W. Deutsch, C. Panse, U. de Lichtenberg, O. Rinner, H. Lee, P. G. Pedrioli, J. Malmstrom, K. Koehler, S. Schrimpf, J. Krijgsveld, F. Kregenow, A. J. Heck, E. Hafen, R. Schlapbach and R. Aebersold, A high-quality catalog of the Drosophila melanogaster proteome, Nat. Biotechnol., 2007, 25(5), 576–83 CrossRef CAS.
- L. M. de Godoy, J. V. Olsen, J. Cox, M. L. Nielsen, N. C. Hubner, F. Frohlich, T. C. Walther and M. Mann, Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast, Nature, 2008, 455(7217), 1251–4 CrossRef CAS.
- S. P. Schrimpf, M. Weiss, L. Reiter, C. H. Ahrens, M. Jovanovic, J. Malmstrom, E. Brunner, S. Mohanty, M. J. Lercher, P. E. Hunziker, R. Aebersold, C. von Mering and M. O. Hengartner, Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes, PLoS Biol., 2009, 7(3), e48 CrossRef.
- J. L. Heazlewood, R. E. Verboom, J. Tonti-Filippini, I. Small and A. H. Millar, SUBA: the Arabidopsis Subcellular Database, Nucleic Acids Res., 2007, 35(Database), D213–8 CrossRef CAS.
- T. Kleffmann, M. Hirsch-Hoffmann, W. Gruissem and S. Baginsky, plprot: a comprehensive proteome database for different plastid types, Plant Cell Physiol., 2006, 47(3), 432–6 CrossRef CAS.
- Q. Sun, B. Zybailov, W. Majeran, G. Friso, P. D. Olinares and K. J. van Wijk, PPDB, the Plant Proteomics Database at Cornell, Nucleic Acids Res., 2009, 37(Database), D969–74 CrossRef CAS.
- P. Durek, R. Schmidt, J. L. Heazlewood, A. Jones, D. MacLean, A. Nagel, B. Kersten and W. X. Schulze, PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update, Nucleic Acids Res., 2010, 38(Database), D828–34 CrossRef CAS.
- H. Nakagami, N. Sugiyama, K. Mochida, A. Daudi, Y. Yoshida, T. Toyoda, M. Tomita, Y. Ishihama and K. Shirasu, Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants, Plant Physiol., 2010, 153(3), 1161–74 CrossRef CAS.
- J. Hummel, M. Niemann, S. Wienkoop, W. Schulze, D. Steinhauser, J. Selbig, D. Walther and W. Weckwerth, ProMEX: a mass spectral reference database for proteins and protein phosphorylation sites, BMC Bioinformatics, 2007, 8, 216 CrossRef.
- H. J. Joshi, M. Hirsch-Hoffmann, K. Baerenfaller, W. Gruissem, S. Baginsky, R. Schmidt, W. X. Schulze, Q. Sun, K. J. van Wijk, V. Egelhofer, S. Wienkoop, W. Weckwerth, C. Bruley, N. Rolland, T. Toyoda, H. Nakagami, A. M. Jones, S. P. Briggs, I. Castleden, S. K. Tanz, A. H. Millar and J. L. Heazlewood, MASCP Gator: An aggregation portal for the visualization of Arabidopsis proteomics data, Plant Physiol., 2010 Search PubMed.
- P. Lu, C. Vogel, R. Wang, X. Yao and E. M. Marcotte, Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation, Nat. Biotechnol., 2007, 25(1), 117–24 CrossRef CAS.
- S. Baginsky, L. Hennig, P. Zimmermann and W. Gruissem, Gene expression analysis, proteomics, and network discovery, Plant Physiol., 2010, 152(2), 402–10 CrossRef CAS.
- P. Mallick, M. Schirle, S. S. Chen, M. R. Flory, H. Lee, D. Martin, J. Ranish, B. Raught, R. Schmitt, T. Werner, B. Kuster and R. Aebersold, Computational prediction of proteotypic peptides for quantitative proteomics, Nat. Biotechnol., 2007, 25(1), 125–31 CrossRef CAS.
- V. Lange, P. Picotti, B. Domon and R. Aebersold, Selected reaction monitoring for quantitative proteomics: a tutorial, Mol. Syst. Biol., 2008, 4, 222.
- S. Reiland, G. Messerli, K. Baerenfaller, B. Gerrits, A. Endler, J. Grossmann, W. Gruissem and S. Baginsky, Large-scale Arabidopsis phosphoproteome profiling reveals novel chloroplast kinase substrates and phosphorylation networks, Plant Physiol., 2009, 150(2), 889–903 CrossRef CAS.
- D. Swarbreck, C. Wilks, P. Lamesch, T. Z. Berardini, M. Garcia-Hernandez, H. Foerster, D. Li, T. Meyer, R. Muller, L. Ploetz, A. Radenbaugh, S. Singh, V. Swing, C. Tissier, P. Zhang and E. Huala, The Arabidopsis Information Resource (TAIR): gene structure and function annotation, Nucleic Acids Res., 2008, 36(Database), D1009–14 CAS.
- L. Reiter, M. Claassen, S. P. Schrimpf, M. Jovanovic, A. Schmidt, J. M. Buhmann, M. O. Hengartner and R. Aebersold, Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry, Mol. Cell. Proteomics, 2009, 8(11), 2405–17 CrossRef CAS.
- R. A. Gutierrez, M. D. Larson and C. Wilkerson, The plant-specific database. Classification of Arabidopsis proteins based on their phylogenetic profile, Plant Physiol., 2004, 135(4), 1888–92 CrossRef CAS.
- R. A. Gutierrez, P. J. Green, K. Keegstra and J. B. Ohlrogge, Phylogenetic profiling of the Arabidopsis thaliana proteome: what proteins distinguish plants from other organisms?, GenomeBiology, 2004, 5(8), R53 CrossRef.
- A. Alexa, J. Rahnenfuhrer and T. Lengauer, Improved scoring of functional groups from gene expression data by decorrelating GO graph structure, Bioinformatics, 2006, 22(13), 1600–7 CrossRef CAS.
- T. Z. Berardini, S. Mundodi, L. Reiser, E. Huala, M. Garcia-Hernandez, P. Zhang, L. A. Mueller, J. Yoon, A. Doyle, G. Lander, N. Moseyko, D. Yoo, I. Xu, B. Zoeckler, M. Montoya, N. Miller, D. Weems and S. Y. Rhee, Functional annotation of the Arabidopsis genome using controlled vocabularies, Plant Physiol., 2004, 135(2), 745–55 CrossRef CAS.
- S. Reumann, S. Quan, K. Aung, P. Yang, K. Manandhar-Shrestha, D. Holbrook, N. Linka, R. Switzenberg, C. G. Wilkerson, A. P. Weber, L. J. Olsen and J. Hu, In-depth proteome analysis of Arabidopsis leaf peroxisomes combined with in vivo subcellular targeting verification indicates novel metabolic and regulatory functions of peroxisomes, Plant Physiol., 2009, 150(1), 125–43 CrossRef CAS.
- M. Elstner, C. Andreoli, T. Klopstock, T. Meitinger and H. Prokisch, The mitochondrial proteome database: MitoP2, Methods Enzymol., 2009, 457, 3–20 CAS.
- K. Horan, C. Jang, J. Bailey-Serres, R. Mittler, C. Shelton, J. F. Harper, J. K. Zhu, J. C. Cushman, M. Gollery and T. Girke, Annotating genes of known and unknown function by large-scale coexpression analysis, Plant Physiol., 2008, 147(1), 41–57 CrossRef CAS.
- S. Reumann and A. P. Weber, Plant peroxisomes respire in the light: some gaps of the photorespiratory C2 cycle have become filled—others remain, Biochim. Biophys. Acta, Mol. Cell Res., 2006, 1763(12), 1496–510 CrossRef CAS.
- V. Solovyev, P. Kosarev, I. Seledsov and D. Vorobyev, Automatic annotation of eukaryotic genes, pseudogenes and promoters, GenomeBiology, 2006, 7(Suppl 1), S10 CrossRef.
- C. B. Burge and S. Karlin, Finding the genes in genomic DNA, Curr. Opin. Struct. Biol., 1998, 8(3), 346–54 CrossRef CAS.
- R. C. Gentleman, V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis, L. Gautier, Y. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R. Irizarry, F. Leisch, C. Li, M. Maechler, A. J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney, J. Y. Yang and J. Zhang, Bioconductor: open software development for computational biology and bioinformatics, GenomeBiology, 2004, 5(10), R80 CrossRef.
- C. F. Taylor, N. W. Paton, K. S. Lilley, P. A. Binz, R. K. Julian, Jr., A. R. Jones, W. Zhu, R. Apweiler, R. Aebersold, E. W. Deutsch, M. J. Dunn, A. J. Heck, A. Leitner, M. Macht, M. Mann, L. Martens, T. A. Neubert, S. D. Patterson, P. Ping, S. L. Seymour, P. Souda, A. Tsugita, J. Vandekerckhove, T. M. Vondriska, J. P. Whitelegge, M. R. Wilkins, I. Xenarios, J. R. Yates, 3rd and H. Hermjakob, The minimum information about a proteomics experiment (MIAPE), Nat. Biotechnol., 2007, 25(8), 887–93 CrossRef CAS.
- O. Thimm, O. Blasing, Y. Gibon, A. Nagel, S. Meyer, P. Kruger, J. Selbig, L. A. Muller, S. Y. Rhee and M. Stitt, MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes, Plant J., 2004, 37(6), 914–39 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0ib00078g |
| ‡ Present address: Martin-Luther University Halle-Wittenberg, Institute of Biochemistry and Biotechnology, Weinbergweg 22, 06120 Halle (Saale), Germany. |
| This journal is © The Royal Society of Chemistry 2011 |