The origins of cancer robustness and evolvability
Tianhai
Tian
a,
Sarah
Olson
b,
James M.
Whitacre
*c and
Angus
Harding
*d
aSchool of Mathematics and Statistics, University of Glasgow, Glasgow, G12 8QW, UK
bPrincess Alexandra Hospital, Department of Neurosurgery, Brisbane, Queensland, Australia
cSchool of Computer Science, University of Birmingham, Edgbaston, Birmingham, UK. E-mail: jwhitacre79@yahoo.com
dThe University of Queensland Diamantina Institute, Princess Alexandra Hospital, Brisbane, Queensland 4102, Australia. E-mail: a.harding1@uq.edu.au
First published on 14th October 2010
Abstract
Unless diagnosed early, many adult cancers remain incurable diseases. This is despite an intense global research effort to develop effective anticancer therapies, calling into question the use of rational drug design strategies in targeting complex disease states such as cancer. A fundamental challenge facing researchers and clinicians is that cancers are inherently robust biological systems, able to survive, adapt and proliferate despite the perturbations resulting from anticancer drugs. It is essential that the mechanisms underlying tumor robustness be formally studied and characterized, as without a thorough understanding of the principles of tumor robustness, strategies to overcome therapy resistance are unlikely to be found. Degeneracy describes the ability of structurally distinct system components (e.g. proteins, pathways, cells, organisms) to be conditionally interchangeable in their contribution to system traits and it has been broadly implicated in the robustness and evolvability of complex biological systems. Here we focus on one of the most important mechanisms underpinning tumor robustness and degeneracy, the cellular heterogeneity that is the hallmark of most solid tumors. Based on a combination of computational, experimental and clinical studies we argue that stochastic noise is an underlying cause of tumor heterogeneity and particularly degeneracy. Drawing from a number of recent data sets, we propose an integrative model for the evolution of therapy resistance, and discuss recent computational studies that propose new therapeutic strategies aimed at defeating the adaptable cancer phenotype.
Insight, innovation, integrationDespite an intense global research effort, most adult cancers remain incurable. The challenge facing researchers is that cancer is a complex disease, displaying many trait properties that drive tumor progression. One such trait property is therapy resistance, widely regarded as the greatest obstacle preventing long-term patient survival. Here we integrate findings from mathematical models, experimental systems and clinical studies to provide an updated schema for the evolution of cancer therapy resistance. In this new paradigm, selectively filtered cellular and sub-cellular heterogeneity provides cancer with the crucial property of degeneracy, rendering tumors both robust and evolvable. We then explore the latest generation of conceptual and computational models that, by directly attacking tumor evolvability, have proposed new therapeutic paradigms that may help reduce or overcome therapy resistance in tumors. |
Introduction
Although modern therapies have increased patient lifespan, the majority of adult cancers remain terminal diseases.1 This is because anticancer drugs generally lose efficacy due to the emergence of therapy resistance within tumors, which remains a significant obstacle to long-term patient survival. Some cancers, such as acute myeloid leukemia and ovarian and breast cancers, show an initial response to chemotherapeutics but invariably relapse, with the recurrent cancer often resistant to any further therapeutic intervention.2 Other cancers such as melanoma and pancreatic and colon cancers contain fewer proliferating cells during therapy, but the tumor mass nonetheless remains stable within the patient throughout treatment.2 Tumors utilize many mechanisms to avoid and/or overcome chemotherapeutics. The diversity of drug evasion mechanisms that are observed in tumors, combined with the challenge of effective in vivo drug delivery, renders the identification and targeting of therapy-resistance mechanisms difficult.Trait robustness is a ubiquitous and fundamental property at all organizational scales in biology and is prevalent for instance in gene expression, protein folding, metabolic flux, physiological homeostasis, development, and organismal fitness.3,4 Here we define robustness as ‘a property that allows a system to maintain its function despite internal and external perturbations’.5 Robustness requires the maintenance of system function as opposed to simply maintaining a stable state,4 and biological systems often achieve this robustness through adaptation, a principle dramatically illustrated in the anhydrobiosis of tardigrade, which can suspend their metabolism under conditions of extreme dehydration, surviving for years in a dormant state.6 Adaptive change is not a unique property of extremophiles as cancers, an inherently robust disease system, are able to adapt to and accommodate many different physiological insults, such as low oxygen and metabolic stress.7 In this review we argue that selection occurring over cellular trait heterogeneity is one of the fundamental causes of cancer robustness. In cancer, cell trait heterogeneity originates from under-regulated stochastic processes at the genetic, epigenetic and protein expression levels. Studies of tumor heterogeneity have revealed extensive cellular trait variation within tumors with respect to size, morphology, antigen expression and membrane composition.8 Individual tumor cells also display diverse functional behaviors in terms of proliferation rate, cell–cell interactions, metastatic potential and sensitivity to therapy.8 Sequencing studies have demonstrated surprising levels of genetic diversity between individual patient tumors of the same type.9 Heritable intra-tumoral heterogeneity increases the probability of tumors harboring a therapy-resistant phenotype and has been hypothesized to endow tumors with the necessary adaptability to survive and recur after treatment.7,10,11 More generally, we propose that tumor heterogeneity provides the phenotypic diversity necessary for the rapid evolution that occurs in many cancers.
Evolvability is defined as ‘the capacity to generate heritable, selectable phenotypic variation’.12 Since the seminal paper from Nowell in 1976 describing cancer as an evolutionary system,13 many studies support the idea that tumors are indeed evolving systems.14 In this paradigm individual cancer cells become the reproductive units within the population, and those cells that have acquired a survival advantage through random genetic or heritable epigenetic change are selected through multiple rounds of clonal expansion, during which they acquire further alterations that combine to produce a malignant phenotype.15 For evolution to occur there must be some form of selection pressure combined with sufficient heritable variation within the population. Many such selection pressures exist for tumor cells in vivo, such as limited nutrients, oxidative stress, and competition for space, as well as extrinsic factors such as immuno-surveillance and anticancer therapeutics.14
In melanoma,16 colon cancer17 and esophageal cancer,18 an increasing number of genetic mutations characterize different phases of neoplastic progression, suggesting a model of sequential mutation acquisition during tumor evolution. This has been best characterized in adenocarcinomas of the large intestine, in which the number of oncogeneic mutations correlates with tumor grade and stage.19 Notably, when isolating different stages of neoplasia in the same tumor specimen, Vogelstein and colleagues demonstrated that although identical ras mutations were present in both regions, the more aggressive carcinomatous regions contained at least one mutation that was absent in the less aggressive adenomatous region.17 This sequential model of tumor progression supports the idea that successive mutation enhances the fitness of the tumor cells, followed by positive selection and clonal expansion. However as with any evolutionary process, the sequential progression observed in these studies is likely providing a ‘selection-biased’ perspective on an underlying stochastic process involving genetic drift, hitchhiking and other dynamic population properties.8,20
More direct evidence for tumor evolution was provided recently in breast cancer. By comparing all somatic coding mutations in a metastatic breast cancer relative to the original primary tumor resected nine years previously, Shah et al. revealed that only five of 32 coding mutations found in the metastases were present in the original tumor, demonstrating that evolution had occurred during disease progression.21 A recent genetic analysis of breast tumors has so far provided the clearest picture of tumor evolution in vivo. By studying the cellular composition of breast tumors, Wigler and colleagues identified multiple clonal subpopulations.22 The observation that all subpopulations within a single tumor shared many of the same chromosome breakpoints provides additional supporting evidence for an evolutionary model of tumor growth, with new clones evolving out of pre-existing tumor cells.22
Further support for the idea of evolution driving tumor development comes from studying the emergence of tumor therapy resistance. In lung cancer, mutant clones containing point mutations within the epithelial growth factor (EGF) receptor drive tumor recurrence that is resistant to further EGF receptor inhibition.23 Likewise, resistance to Glivec in chronic myeloid leukemia is often due to mutant clones with point mutations within the BCR-ABL tyrosine kinase.24,25 In the case of Glivec resistance in chronic myeloid leukemia, there is evidence that resistant mutants are present in patient tumors before drug treatment,26 suggesting that cancer therapies select for pre-existent resistant mutants within tumors. This is reminiscent of natural selection acting on standing genetic variation which is believed to occur in many biological contexts27 such as in bacterial populations, where pre-existing resistant mutants drive the evolution of bacterial phage resistance.28
Given these findings, we propose that a better understanding of the principles of tumor evolvability will allow for the design of new therapeutic paradigms that minimize or inhibit tumor evolution, and thus prevent the emergence of therapy resistance. As heritable phenotypic variation is a prerequisite of tumor evolution, we first focus our attention on four mechanisms thought to be responsible for generating tumor heterogeneity in patients: genetic instability, epigenetic instability, stochastic protein dynamics and tumor micro-environments. Next we provide a brief analysis of the crucial relationship between robustness and evolvability. We then present an integrative model of tumor evolution in which we propose how heterogeneity at different biological scales can facilitate evolution and the generation of complex tumor properties. Finally, we explore how a systems biology approach may be used to help overcome robust, evolvable tumors in patients.
Sources of heterogeneity
1 Genomic instability
Sequencing technologies are now sufficiently advanced to allow researchers to begin assessing intra-tumor genomic heterogeneity. Recent sequencing of a primary breast tumor using next-generation sequencing confirmed that intermediate grade breast tumors do indeed contain clonal subpopulations.21 By developing a new technology termed sector-ploidy-profiling (SPP) Navin et al.22 revealed that primary breast carcinomas consist of either a single major clonal population, or several primary clonal subpopulations.22 Given that this technology currently lacks the sensitivity to detect uncommon clones within populations,22 this first pass almost certainly underestimates the true level of genetic heterogeneity present in solid tumors. Nevertheless these initial studies provide the important proof-of-principle that intra-tumor genetic heterogeneity exists, and begin to shed light on the evolutionary dynamics occurring within tumors.21,22
A firm theoretical foundation supporting the mutator model has been provided by recent modeling studies. The first study, undertaken by Beckman and Loeb, assumed that all potential mechanisms of tumorigenesis are in operation. The major insight of this work was the novel idea that mechanisms that produce malignant lineages most efficiently should be considered the most likely to generate clinical cancers.44 In this model, efficiency is defined as the number of malignant lineages generated in a given time. This schema allowed the first direct comparison of the relative efficiencies of mutator and non-mutator pathways in cancer lineage production,44 and demonstrated that mutator mutations increase the efficiency of tumorigenesis under many realistic simulation conditions.44 Most compellingly, the mutator phenotype became increasingly important as the number of mutations required for cellular transformation rose.44 Thus, for tumors requiring only two mutations, the mutator phenotype offers no advantage.44 However, when the number of mutations required for transformation exceeds six, then the mutator phenotype imparts a significant advantage in the efficiency of tumorgenesis.44
As stated by Jarle Breivik ‘Each random mutation may be regarded as a bet, and the odds are always unfavorable, simply because there are more ways to damage a genome than improve it. As for roulette, you may get a lucky strike, but the more bets you make, the more certain it is that you will lose’.45 Clonal extinction due to reduced fitness is known as negative clonal selection and is one of the most serious criticisms leveled against the mutator model.45 To directly address whether negative clonal selection negates the mutator hypothesis, Beckman recently developed an updated model that incorporated the mutator's reduced fitness within its underlying assumptions.46 This model revealed that even with negative clonal selection, mutator cells still provide the most efficient route to tumorigenesis.46 The model made an important new prediction: that there exists an optimal mutation rate for tumors, above which the deleterious effects of reduced fitness do lead to negative clonal selection and tumor collapse.46 Support for this idea has recently been generated using bacterial competition models, where Loeb and colleagues experimentally confirmed that mutator strains of E. coli displaying a high mutation rate invariably suffer negative clonal selection and die out, whereas those mutator strains that fall within a narrow optimal range of mutation rates consistently prevail and survive in evolutionary competition assays.47 Importantly, these results suggest that mutator cells are not necessarily doomed to extinction due to reduced fitness.47 Beckman's updated model also confirmed two predictions of the earlier model: that mutator mutations are most likely to occur early in tumorigenesis, and that the mutator phenotype becomes increasingly important as more oncogenic mutations are required for transformation.44,46 In an independent study, Zhou et al. explored evolutionary dynamics in silico using a numerical model based on Highly Optimized Tolerance (HOT),48 a mathematical paradigm used to understand how selectively acquired robustness can lead to the evolution of complexity.49 In this model, mutators played a primary role in adaptation, whereas low-mutators preserved well-adapted phenotypes.48 Taken together, these three models support the hypothesis that mutator phenotypes both increase the efficiency of tumorigenesis (and thus increase the probability of tumor initiation) and drive tumor adaptation throughout disease progression.
The correlation between tumor incidence and advanced age has been used to estimate that the minimum number of genetic mutations that drive oncogenesis is five to six.50–52 Pediatric tumors such as retinoblastoma require significantly fewer mutations for transformation,53–55 whereas late-onset adult tumors may require as many as 10–12 events.56 Experimental models initially suggested that 3–4 mutations were sufficient to generate transformed cells.57 However recent experimental data indicates that more mutations are in fact required. Mahale et al. explored the efficiency of transformation using a combination of four oncogenes in a human fibroblast model of transformation.58 Using this established experimental system they made the striking observation that tumorigenicity significantly increased after serially passaging tumor cells either in vitro or in vivo. The observed increase in tumorigenicity correlated with the selection of dominant clones, suggesting that malignant transformation is a stochastic process initiated by the four defined oncogenes, but that full transformation involves clonal selection of tumors harboring further transforming mutations.58 Nicholson and Duesberg then extended these analyses to reveal fundamental differences in the karyotypes and phenotypes of clones derived from a single parent cell with four oncogenes, providing direct evidence that evolution had indeed occurred during expansion both in vitro and in vivo.59 These authors went on to demonstrate that individual clones evolve further during serial passaging in culture, generating either enhanced tumorigenicity or drug resistance in vitro.59 These findings are consistent with recent reports showing a correlation between genomic instability and drug resistance,60,61 supporting the idea that the rate of tumor evolution, including the acquisition of therapy resistance, is significantly enhanced by genomic instability. Taking a complementary approach and working in parallel, the systematic and comprehensive analysis of Ye et al. showed that tumorigenicity is positively associated with genomic diversity in five independent models of tumor progression.62 These combined data sets provide experimental evidence for a direct causal relationship between genomic instability and cancer evolution.
Direct confirmation of this relationship came recently using a mouse model of genomic instability.63 Sotillo et al. induced lung tumor formation in mice by doxycyline-mediated expression of oncogeneic Ras either alone or in combination with Mad2, a known mediator of chromosomal instability.63 The expression of oncogenic Ras alone generated lung tumors, whereas Mad2 expression alone did not. However the addition of chromosome instability through Mad2 co-expression markedly enhanced Ras tumorigenicity, as revealed by a more rapid disease onset and mortality, a more aggressive tumor phenotype, and a two-fold increase in the size of the Ras + Mad2 tumors compared to tumors expressing Ras alone.63 The increased aggressiveness of Ras + Mad2 tumors correlated with increased aneuploidy (a marker of chromosomal instability) and a rise in the diversity of Ras + Mad2 tumor sub-types compared to the Ras-only control.63 When the oncogenic protein expression was ablated by doxycycline withdrawal, both the Ras and the Ras + Mad2 tumors collapsed, consistent with the idea of oncogenic Ras expression driving tumor growth. Ras-only tumors never recurred after doxycycline withdrawal. In striking contrast, approximately half of the Ras + Mad2 tumors returned, with recurrent tumors displaying both increased aneuploidy and new signal transduction pathway activation.63 The most plausible interpretation of these results is that the increased genomic instability of the the Ras + Mad2 tumor allowed the evolution of Ras-independent tumor cells, which then drove tumor recurrence despite the loss of oncogenic Ras expression.
2 Epigenetic instability
It has been cogently argued that as a single genome is capable of generating the diversity of cell phenotypes present in metazoan organisms, the same mechanisms that underpin normal cell diversity may also drive tumor heterogeneity and contribute to tumor evolution.64 Cell diversity in somatic organisms is regulated through epigenetic mechanisms. By epigenetics we mean ‘the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence’.65 This fulfils the biologists definition of ‘a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence’66 while accommodating the existence of pseudo-stable states driven by network dynamics and long-lived stochastic fluctuations.Intriguingly, multiple epigenetic aberrations have been directly linked to genomic instability (reviewed in ref. 70). DNA hypomethylation at repeat sequences increases genomic instability by promoting chromosomal rearrangements.72 Hypomethylation of transposable elements can lead to their activation and translocation, increasing genomic instability.73 Genomic instability can also be caused by epigenetic inactivation of genes encoding DNA repair proteins.74,75 Indeed, the silencing of the DNA repair protein MGMT and global DNA demethylation are thought to be early initiating events in tumor formation.76–78 Thus epigenetic modifications are well placed to contribute directly to genomic instability and tumor heterogeneity, thereby enabling tumor evolution.
One of the primary functions of miRNAs in normal development is the stabilization of cellular phenotypes.79 Regulatory loops play a central role in maintaining robust phenotypic reproducibility of developmental programs.90 This type of system control comprises a collection of feedback loops that monitor and quantitatively regulate the output of signaling networks.91 Negative feedback loops embedded within signaling networks are prevalent and are known to stabilize pathway dynamics.91 Experimental evidence suggests that an important role of miRNAs is to impose stabilizing negative feedback loops during development.92,93 In line with this function, miRNA expression is reduced in late-stage tumors and correlates with tumor aggressiveness, which is thought to be due to an increase in tumor heterogeneity resulting from destabilized pathway dynamics.79
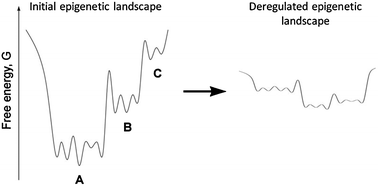 | ||
| Fig. 1 (Left panel) An illustration of hierarchy within an epigenetic landscape for a single transition pathway from stem cell (C) to final cell type (A). (Right panel) Deregulation and modification of the epigenetic landscape as a consequence of genetic instability and the mutator phenotype. The illustration is modified from ref. 177 and was originally created to illustrate the recently discovered existence of hierarchy within the conformational landscape of large proteins. | ||
Recent experimental evidence from malignant melanoma provides support for this model.101 Roesch et al. recently identified a slow-cycling subpopulation of cells within melanoma tumors that functions as the tumor-maintaining cell population.101 These slow-cycling tumor-maintaining cells express JARID1B, a histone 3 K4 (H3K4) demethylase that in healthy organs is highly expressed in regenerative tissues.102–104 In melanoma, JARID1B is associated with negative regulation of the cell cycle.105,106 Knockdown of JARID1B initially stimulated tumor growth, however growth could not be maintained in the absence of JARID1B,101 revealing the JARID1B cell subpopulation as crucial in maintaining continual tumor growth. Intriguingly, JARID1B expression appears to be dynamic, with JARID1B-negative cells able to spontaneously generate JARID1B-positive cells and vice versa.101 Together these results and other findings reviewed in ref. 107 argue against a hierarchical cancer stem cell model, instead suggesting that melanoma tumor-initiating cells are generated spontaneously or induced by environmental cues within the melanoma tumor bulk, consistent with the idea of pseudo-stable attractor states driving tumor growth in aggressive cancers.
Unstable epigenetic states add another layer of complexity to tumor biology, the phenomenon of transient therapy resistance. The existence of a transiently resistant drug state within tumors was first proposed based on the observation that some drug-resistant tumors become responsive after a break from treatment.108–110 A recent study has provided experimental confirmation that transient drug resistance does occur in lines derived from multiple cancers, driven by epigenetic modification.111 Transient treatment of tumor cells with various chemotherapeutics has identified a small fraction of quiescent cells that are ∼500 fold less sensitive to anticancer drugs than their parental cells.111 In clonally derived populations, drug tolerance emerges de novo and is reversible, although it can become stabilized over time.111 Transient drug resistance is driven by activation of the insulin growth factor 1 (IGF-1) receptor and an altered chromatin state requiring histone demethylase RBP2.111 Importantly, the transiently drug-resistant subpopulation can be ablated using inhibitors of IGF-1 or chromatin-modeling agents, suggesting an avenue of therapeutic redress for future studies.111 In combination with clinical studies108–110 the study of Sharma et al. provides support for the existence of a transient drug-resistant attractor states that reflect states within a deregulated epigenetic landscape. Moreover, with each cell displaying slightly unique epigenetic landscape properties and different positioning within the landscape, tumors could be endowed with an enormous repertoire of transient cell responses that enhances the tumor's overall robustness in the face of therapy. This idea is well established in bacterial populations where phenotypic outliers contribute to population fitness, one relevant example being the so called ‘persisters’ in bacterial populations that express an increased resistance to penicillin that can then be inherited.112–115
3 Stochastic protein dynamics
Recent work has focused attention on the role of stochastic protein expression fluctuations in generating trait heterogeneity within clonal populations.116 When analyzed by flow-cytometry, protein abundance in clonal mammalian cell populations can vary by as much as three orders of magnitude due to stochastic fluctuation.64,116 This variation imparts several important characteristics on the clonal population. First, the outliers of the population can display very different biological properties,116 showing that purely stochastic effects can generate functionally diverse subpopulations within a clonal group of cells.116 In mammalian cells, such stochastic fluctuations in protein expression can be reasonably long lived, lasting up to 11 days in culture,116 meaning that they can impart phenotypic variety over clinically relevant timeframes. Recent work provides support that these types of stochastic fluctuations do indeed afford tumors protection from anticancer drugs, with Cohen and colleagues discovering significant cell-to-cell variability in the temporal behavior of drug-induced protein expression, which correlated with the ability of cells to resist drug-induced apoptosis.117 This finding, together with the study of Sharma et al. described in the previous section, provide the first evidence that transient non-genetic phenotypic states contribute to therapy resistance in tumors and may explain historical studies showing tumors that repopulate after a drug treatment can sometimes remain sensitive to that drug.118 Even though transient states are, by definition, relatively short lived and therefore invisible to natural selection, transient resistant states can potentially contribute to the evolution of therapy resistance. While several possible pathways to inheritance exist, one plausible mechanism would involve clonal expansions originating from mutations that alter regulation of the epigenetic landscape and stabilize the drug resistant phenotype. This model fits with the recent observation of transient drug resistant states becoming stabilized over time,111 and may help explain the biology of chronic myeloid leukemia (CML), where a rare subpopulation of quiescent CML cells resists conventional therapy and is thought to drive disease relapse.1194 The tumor micro-environment
Genetic, epigenetic and stochastic protein dynamics sources of heterogeneity do not act in isolation, as important cross-scale interactions also take place within tumors. Tumor cells continually interact with the surrounding tumor microenvironment, a relationship that has crucial roles in tumor initiation and progression. One relevant example is regions of low oxygen (hypoxic regions) commonly found within solid tumors that are the result of an imbalance between supply and consumption of oxygen.120 Many cancers including squameous cell carcinoma of the uterine, head and neck cancers, breast cancers and brain tumors have regions of low oxygen in contrast to normal adjacent tissue.121,122 Patients with hypoxic tumors have significantly lower overall survival,121 with hypoxia an independent prognostic factor for poor clinical outcome in many tumors,118 indicating that hypoxic regions play an active role in tumor malignancy.Hypoxic regions within tumors contribute to tumor heterogeneity in at least three ways. First, tumor cells in hypoxic environments display reduced expression of DNA repair genes and corresponding increased levels of genomic instability.120,123 Thus hypoxia can contribute directly to the mutator phenotype and might enhance tumor evolution. Consistent with this idea, hypoxic tumor cells display increased resistance to radiation and drugs124,125 as well as an increased incidence of both apoptosis-resistant126 and invasive clones,127 supporting the hypothesis that hypoxic environments drive tumor cell evolution towards more aggressive phenotypes. Second, hypoxic environments can directly regulate the epigenetic state of tumor cells. Cells in hypoxic regions are dependent on anaerobic glycolytic metabolism,128 which in turn acidifies the hypoxic region through the generation of lactic acid.128 The combination of low oxygen and low pH triggers tumor cell cycle arrest and quiescence,120,128 increasing phenotypic heterogeneity and rendering tumor cells insensitive to many anticancer therapies as described above. A third confounding factor is that the low vascularization responsible for hypoxic regions reduces the concentrations of drugs within hypoxic regions,129 a condition known to favor selection of drug-resistant clones.130
The tumor microenvironment is composed of many non-transformed cell types such as endothelial cells, fibroblasts, and immune cells, all of which interact with tumor cells and modulate the tumor microenvironment.131 There is a large body of research demonstrating that tumorigenesis is strongly influenced by the non-malignant cells within the tumor microenvironment (reviewed in ref. 132). It is likely that tumor cells co-evolve with their micro-environments, and during the course of disease progression changes in micro-environment create local differences in selection pressure, thereby driving some of the heritable differences that are observed across cancer cells within a single tumor. From this perspective, at least some of the phenotypic, genetic and epigenetic diversity observed at the cell population level is likely to be a natural consequence of tumor-microenvironment interactions. These ideas are discussed in detail in recent reviews.14,131,133
Heterogeneity and degeneracy as an enabler of tumor robustness and evolvability
Above we explored how tumor heterogeneity provides the phenotypic variation required for natural selection to act upon, thereby increasing tumor evolvability. Next we briefly examine how tumor heterogeneity may enhance tumor robustness and evolvability by endowing tumors with the system property of degeneracy. Degeneracy is the ability of structurally dissimilar system components to perform the same function or generate the same output.134 Like robustness, degeneracy is also a ubiquitous property of biological systems.134 It is important to note that degeneracy is distinct from the simpler design principle of redundancy. In redundant systems, multiple identical components are present within the system, one important example being the multiplicity of pacemaker cells that robustly regulate heartbeat. Redundancy is common both in engineering and biology, where it provides robustness in response to very specific perturbations, e.g. compensating for the loss or failure of an identical component. In contrast to redundant components, degenerate components perform similar functions within certain contexts but distinct and separate functions in others. For degeneracy to arise, system components must display functional plasticity, i.e. context-sensitivity in the different functional responses generated by each component. Recent analyses indicate that the conditionally overlapping functionality of degenerate components plays a fundamental role in reconciling requirements of robustness and evolvability in nature.135For instance, recent in silico simulation experiments have revealed that networks composed of redundant multi-functional proteins (i.e. proteins having either identical or completely unique functions) are robust but do not provide a system with mutational access to very many distinct heritable phenotypes.135 Allowing multi-functional proteins to partially overlap in functionality (i.e. protein degeneracy) resulted in networks that were both exceptionally robust and exceptionally evolvable.135 This relationship between degeneracy, robustness and evolvability appears to arise at many different scales in biological systems but has yet to be fully understood.134 On the one hand, having diversity amongst functionally similar components will enhance robustness in a manner that is straightforward to understand. If components are somewhat different, they also have somewhat different weaknesses: a perturbation or attack on the system is less likely to present a risk to all components at once.136 Edelman and Gally have documented numerous biological examples of this relationship between degeneracy and robustness.134
One clinically important example of this relationship between degeneracy and tumor cell robustness comes from receptor tyrosine kinase (RTK) coactivation in tumor cells.137 The epidermal growth factor receptor (EGFR) is amplified, mutated or rearranged in over 40% of Glioblastoma multiforme (GBM) tumors,138,139 the most common and aggressive primary brain cancer in adults. Nevertheless most GBM patients whose tumors are driven by oncogenic EGFR fail to respond to specific EGFR inhibitors, despite the fact that oncogenic activation of the EGFR is a crucial transforming event for these tumors.140 It was discovered that multiple RTKs are co-activated in GBM tumors, and as many RTKs share common downstream components, co-activation of multiple RTKs allows GBM tumors to maintain robust signaling simply by switching RTK usage in the presence of specific inhibitors.141,142 Combinations of RTK inhibitors were required to overcome degenerate RTK usage in GBM tumor cells.141,142 The RTK coactivation strategy has subsequently been observed in other tumor types, suggesting that degenerate RTK usage may represent a general pathway by which tumor cells evade targeted therapies (reviewed in ref. 137).
The example above describes how robustness can be achieved through direct functional compensation. While this mechanism is intuitively obvious, degenerate components might also collectively contribute to the stability of many traits simultaneously, distributing robustness throughout a system.143 As reviewed in ref. 136 and 143 there is some evidence to suggest that degenerate components can allow systems to establish networked compensatory pathways whose inherent versatility in resource usage enables buffering against a much greater variety of perturbations than can be accounted for by direct functional compensation alone.135
In some respects, the theoretical relationship between degeneracy and evolvability is also straightforward to understand. Because degenerate components are only conditionally similar, circumstances can arise where the components display unique functions and these can contribute to measurable trait differences.134 At the molecular level, this is observed in the conditional silencing of single nucleotide polymorphisms within protein coding genes. A conditional similarity affords synonymous codons mutational access to amino acids that are the same for some mutations but different for others. For example, in the synonymous arginine codons CGG and CGT, the former can access amino acids {Leu; Pro; Gly; Gln; Trp} through single point mutation, while the latter can reach {Leu; Pro; Gly; His; Ser; Cys}. On the one hand, this provides synonymous codons with higher mutational robustness. On the other hand, by drifting over silent mutations, this also increases mutational access to amino acid residues. It has recently been shown that this conditional silencing can be exploited to enhance the evolvability of bacterial cell lines.144 Degeneracy might also facilitate evolvability in more complex and less direct ways. For instance, it has been proposed that the compensatory actions of degenerate proteins can lead to cryptic differences between cell states that only become realized as measurable trait differences at some later time, e.g. when thresholds for trait stability are crossed.143 In either scenario, it is the conditional similarity amongst degenerate components that is believed to afford robustness while providing the requisite variety of distinct phenotypes that is necessary for evolvability.143 While these theoretical developments and supporting studies appear promising, the complexity of biological systems has so far precluded a thorough experimental assessment of this proposed role of degeneracy in facilitating robustness and evolvability. However the functional divergence of redundant genes in many organisms, combined with large-scale gene deletion studies in yeast, worms and plants provides compelling support for the role of degeneracy as an enabler of robustness and evolution (reviewed in ref. 145).
Degeneracy within a cancer cell appears to play an important role in tumor robustness as seen by the ability of tumor cells to co-activate multiple RTKs.141,142 However, individual cancerous cells within a heterogeneous tumor are also likely to express both distinct and overlapping functional outputs thereby establishing degeneracy at a higher organizational level within the tumor. This idea is supported by the ability of tumor cells to stochastically switch from one cell state to another, such as alternating between tumor-initiating versus proliferative cell states,101 or adopting transient drug-resistant states.111,117 These studies provide the first evidence that individual tumor cells can functionally replace other cell types within the tumor. Cells that switch to a new cell state will not necessarily be identical to those cells being replaced, and therefore could harbor heritable trait differences with survival characteristics, such as therapy resistance. In this way degeneracy within the cell population could directly facilitate tumor evolvability, and may provide a general explanation for the evolution of therapy resistance in aggressive cancers.
The relationship between robustness and evolvability in tumors
A cohesive paradigm characterizing the intimate relationships between robustness and evolvability, which is fundamental to our understanding of biology, has until recently eluded theoreticians.12,143,146–148 While evolvability is repeatedly seen to support the robustness of higher level traits, it is not clear that robustness always supports evolvability. For instance, it is apparent that evolvability increases a cell population's robustness by enabling the population to adapt.5,12 On the other hand, trait robustness within the cell seems to oppose evolvability, as cells that are robust to mutational change would be expected to have difficulty discovering distinct heritable phenotype that allow for adaptation to environmental change.149A series of computational studies have resolved this tension by showing how robustness and evolvability arise at different timescales and furthermore showing phenotypic robustness to be a precondition to evolution.147,150–152 Robust phenotypes allow a population to accumulate neutral mutations, increasing genotypic diversity.147 Because many of these neutral genotypes harbor distinct phenotypically consequential sensitivities to further genetic modification, mutational robustness enhances access to phenotypic diversity over time, facilitating evolution.147 The idea that robustness facilitates evolution has strong experimental support. Bloom et al. found that only robust (thermostable) protein variations could tolerate the destabilizing mutations needed to confer novel activities, whereas non-robust (thermosensitive) proteins could not evolve new activities.153 Measuring evolution of thermotolerance in an RNA virus, McBride and co-workers found that populations derived from robust clones evolved greater resistance to heat shock relative to populations founded by non-robust clones.154 These studies provide direct empirical evidence that robustness can facilitate evolvability. Chaperone proteins such as Hsp90 function to buffer the expression of genetic and epigenetic variation, increasing an organism's robustness to mutation.155 Chaperones have been shown to function as an enabler of evolution in both eukaryotes156 and prokaryotes.157 Tumors make good use of the buffering ability of chaperones; Hsp90 buffers tumor cells against mutations that impinge signaling,158,159 or are lethal.160,161 These combined studies support the hypothesis that molecular and cellular robustness facilitates tumor evolvability.
An integrative model of tumor robustness and evolvability
1 Increasing evolutionary potential through tumor degeneracy
The studies presented in this review emphasize several research trends that we propose could form the basis of a new integrated model of tumor robustness and evolvability. First, a tumor precursor cell that acquires a mutator phenotype within a narrow optimal range has a reasonable probability of acquiring sufficient transforming mutations to become a mature, malignant cancer cell before accumulating deleterious mutations and suffering negative clonal selection. The mutator phenotype is also predicted to generate a destabilized epigenome, allowing cells to transiently adopt multiple discrete cell fates and further increase genomic instability (positive feedback). Stochastic protein dynamics within individual cells can in some cases help to further enhance the diversification of cell states within the tumor, amplifying the phenotypic heterogeneity generated through (epi)genomic instability. The net effect of this three-tiered destabilization is the generation of a high degree of cellular trait heterogeneity within the tumor, which affords the cancer a greater repertoire of responses to the perturbations it encounters during growth, thus rendering it a more adaptable and robust system. For instance, with individual tumor cells able to transiently adopt a variety of cell fates, individual cells of one type can functionally replace other cell types through environment-induced trait plasticity. On the other hand, these compensatory cell transitions do not result in identical cell states (the cells are degenerate) and cells of a similar type will display unique strengths and weaknesses that play out in a competitive environment to the overall benefit of tumor robustness. With many traits having a heritable basis, transient resistance can transform into persistent tumor properties under sustained selective pressure, i.e. genetic assimilation.162,163 In short, we propose that enhanced tumor robustness and evolvability is conferred through the development of degenerate selective repertoires that arise naturally in cell populations presented with genetic instability, epigenetic instability, and stochastic protein dynamics. These proposed relationships between tumor robustness, degeneracy, and evolvability are summarized in Fig. 2.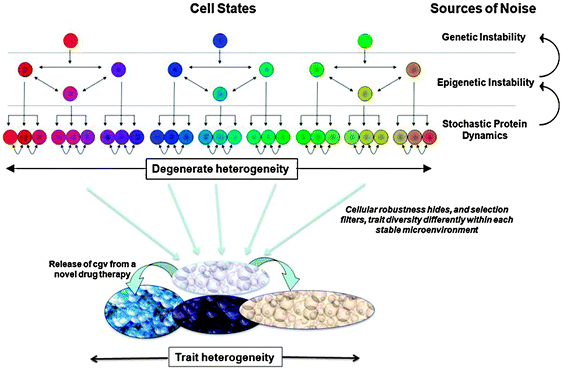 | ||
| Fig. 2 Three tiers of noise-genetic instability, epigenetic instability, stochastic protein dynamics, and the feedback between these tiers-provides a strong source of divergence in the internal and external properties of cancer cells, i.e. the mutator phenotype. Cellular robustness achieved (in part) through degeneracy allows for high amounts of heritable heterogeneity to accumulate in a cell population. While the mutator phenotype introduces new heritable variants at a rapid rate, canalization will hide, and selection will filter, the phenotypic diversity that is actually observed in a microenvironment-dependent fashion. Other factors such as genetic drift and tumor expansion can also influence the speed and extent that heritable variation accumulates. When presented with novel environmental conditions such as the administration of a new drug therapy, directional selection will then act on any of the standing genetic variation that is expressed as selectively relevant phenotypic differences. Some of this phenotypic variation is pre-existing and some is conditionally exposed by the new therapy. The overall extent of heritable phenotypic variation will influence the propensity to evolve a persistent therapy resistance and thus impact the robustness of the cancer. | ||
While the studies reviewed in this article support the model described above, there are aspects of the proposed model that could be modified or elaborated upon and still be supported by the accumulated evidence within these studies. For instance, under the mutation-selection balance that constrains the likelihood of initial disease onset within the mutator hypothesis, a mutator phenotype would not be highly maladaptive if preceded by (selectively passive) mutations that elevate the mutational robustness (i.e. attenuated phenotypic effects from mutations) through so called capacitance or genetic buffering of a pre-cancerous cell. Examples of common tumor mutations that can increase genetic capacitance include p53 loss-of-function (=loss of apoptotic response to DNA damage), constitutive PI3-kinase signaling (pushing cells into a proliferative anti-apoptotic state) and increased chaperone expression (direct increase in genetic buffering). Stochastic fluctuations in protein expression may also serve as a genetic buffering system by compensating for loss-of-function mutations or suppressing deleterious protein expression. With elevated levels of genetic buffering, a clonal population could subsequently acquire a mutator phenotype under nearly neutral conditions, thereby enhancing the overall likelihood of the mutator phenotype pathway. Even in the absence of elevated genetic buffering, mutational robustness is exceptionally high in eukaryotic genomes compared to the more compact genomes of viruses and bacteria,164 and this genetic neutrality should increase the plausibility of a mutator phenotype pathway beyond the conditions suggested from the simulation studies reviewed earlier.
2 Increasing evolutionary potential through cryptic heritable variation in tumors
Counter-intuitively, high levels of mutational robustness163 within cancer cells may also have a direct positive impact on a tumor's ability to evolve therapy resistance. While heritable phenotypic diversity is a precondition for evolutionary adaptation, the competitive environment within a tumor will suppress trait differences that are deleterious to cell survival and fecundity and will thereby impose some constraints on the type and amount of phenotypic heterogeneity that can arise; both within a cell population microenvironment and across the entire tumor. However, due to the cell's inherent genetic and epigenetic capacitance, mutations can readily accumulate in a cell population that appear phenotypically cryptic (or selectively hidden) under stabilizing selection but that become expressed or released under perturbed (e.g. stressful) environmental conditions. This conditional exposure of trait variation is often discussed as a phenomena known as cryptic genetic variation (cgv) or “hidden reaction norms” (for reviews see ref. 165 and 166). cgv describes heritable phenotypic variation that is hidden under “normal conditions” but that is released in the presence of novel alleles or novel environments.Given the high mutational robustness within the human genome, a mutator phenotype should facilitate an accelerated accumulation of high levels of this cryptic genetic variation that can be subsequently (partially) released in ways that depend on the stressful environments encountered. This scenario, inherent mutational robustness combined with elevated (epi)genetic instability, is particularly promising because it would provide the necessary fuel for tumor adaptation under new stressful environments while bypassing the negative clonal selection that limits phenotypic variability under stable conditions.
While cryptic genetic variation has been observed in a variety of species and cell populations,165,166 the origins of cgv are not fully understood, making it unclear when or how genetic buffering mechanisms will permit cgv to arise. Very recent simulation studies have found that each of the hallmark features of cgv are readily observed in biological simulations when mutational robustness is achieved through biomolecular degeneracy.167 Because degeneracy is ubiquitous at protein, complex assembly, and molecular pathway levels within the cell, it would thus seem plausible that cgv can accumulate in tumor cell populations. Moreover, under the mutator phenotype scenario, cgv should accumulate rapidly and strongly influence the evolvability of cancer.
Using a systems biology approach to attack robust and evolvable tumors
Keeping firmly in mind Horrobin's fears that biomedical research is becoming a ‘glass bead game’ with little contact with reality,168 we now focus on the design of new therapeutic strategies that combat tumor evolvability in an effort to mitigate therapy resistance. Our overarching hypothesis is that an understanding of the principles of tumor evolvability will allow the design of general therapeutic paradigms that minimize tumor evolution, in the hope of preventing or delaying the emergence of therapy resistance. There has been a significant body of research devoted to developing mathematical models of the evolution of therapy resistance, with the aim of developing general dosing strategies to inhibit tumor evolution.169–173 Below we focus on three recently published modeling approaches that illustrate how combining simulations, theory, and empirical evidence could help in the development of therapeutic strategies that can overcome the evolution of therapy resistance.As even a small number of resistant cells at the start of the therapy can prevent a cure, Foo and Michor recently modeled the worst-case scenario of the inevitable emergence of therapy resistance due to a single (epi)genetic mechanism.174 Importantly, they have taken into account the effects of drug toxicity and side effects174 in an approach designed to give the best outcome for the patient by comparing continuous or pulsed therapy regimes, determined by the maximal time before tumor recurrence occurs.174 The assumptions used in this analysis are consistent with the high probability of resistance experienced in clinical trials. Foo and Michor found that strategies involving drugs delivered in high dose pulses, effectively slowing the net growth of resistant cells, provided the best outcome for patients in silico with respect to delay of tumor recurrence and drug toxicity.174 This high-dose-pulse approach may be useful in identifying optimum therapy schedules to avoid or delay resistance driven by a single (epi)genetic event.174
In their recent manuscript, Silva and Gatenby have taken an ecological approach in their war on cancer.175 Inspired by successful biological control of pest species in ecosystems, they seek to stabilize rather than cure patient tumors, thereby avoiding the introduction of strong selective forces that drive the evolution of therapy resistance. Fundamental to their model is the assumption, based on experimental data sets and historic modeling results, that resistant cells are present within the tumor at low numbers due to their reduced fitness compared to sensitive tumor cells.175 The aim of their therapeutic approach is to maintain a sufficient number of the rapidly proliferating, sensitive cells throughout therapy to compete with and suppress the emergence of the slower-cycling resistant clones. An additional insight in the work of Silva and Gatenby is the incorporation of the role of the tumor microenvironment, specifically hypoxic regions, in modulating drug accessibility to resistant tumor cells within the hypoxic zone. To overcome the hypoxic barrier to therapy, Silva and Gatenby took advantage of tumor cell dependence on glycolytic metabolism by using the glucose competitor 2-deoxy-glucose to target resistant cells within the hypoxic tumor core. This was combined with a standard chemotherapy that targeted sensitive, proliferating cells on the tumor edge. How well do patients do on this ‘adaptive therapy’ strategy compared to traditional therapy regimes? In silico simulations suggest that the patients would survive significantly longer when the two therapies were administered as separate doses, with the best results obtained when the resistant cells were first targeted with 2-deoxy-glucose then sensitive cells attacked with the chemotherapy.175 This approach managed to eradicate the resistant subpopulation, heralding the possibility of tumor elimination and patient remission.175 A potential criticism of this work is the untested biological assumptions that underpin the model. However, previous work by the same group has shown adaptive therapy maintains a significantly lower tumor burden than conventional therapy approaches in an established animal tumor model,176 providing promising experimental support for the efficacy of this approach.
Our model of tumor evolution introduced in the previous section highlights important relationships arising in natural evolution that could inform the development of new therapeutic paradigms. For instance, the cgv pathway outlines a process by which tumor adaptation arises due to drug therapy induced traits that are otherwise selectively hidden within the extant genetic and epigenetic diversity. Even with the high (epi)genetic instability that is associated with a mutator phenotype, any accumulation of cgv will take time and this imposes important restrictions on the adaptive response capabilities of a tumor. For instance, if drug therapies cause the release of cgv under directional selection, this would also act to momentarily reduce cgv and transiently lower the tumor's evolvability to additional stresses. Assuming the cgv pathway significantly contributes to tumor evolution, we propose that a drug regimen that cycles through drug therapy sequences with a timing that maximizes the rate of cgv release could drive tumors to a more fragile state and help lead to their ultimate demise.
While the perspectives on tumor evolution proposed in our model are all reliant upon the onset of degenerate heritable phenotypes through genetic and epigenetic destabilization, there are differences in the timing and conditions for trait heterogeneity expression that could have significant implications to therapeutic strategies. As robustness arises from the presence of multiple partially overlapping pathways for the establishment and maintenance of traits, we predict that this would confer a predisposition towards single target resistance because suppressed pathways are compensated for by degenerate pathways. For polygenic traits that have a large and distributed mutational target, directed selection (under new stress conditions) is more likely to evolve cells with enhanced degenerate pathways,134,135 which according to one study could potentially lead to multiplicative effects on cellular robustness over time.143 While degeneracy at the cell population level (cgv) can be theoretically eliminated using the sequential drug strategy suggested above, this would be less effective against late stage cancers if a mutually supporting network of new degenerate pathways were to become fixated within the cancer genome. In these circumstances of newly adapted cellular robustness, multi-target therapies acting on complementary pathways might provide the only promising avenue for complete eradication.
Conclusion
Cancer is a complex disease, displaying emergent properties that are driven by an evolvable (epi)genome that is fueled by stochastic noise and the contextual, dynamic interactions that occur within tumor environments. One such emergent property is therapy resistance, widely regarded as the greatest obstacle to long-term patient survival. Recent studies using mathematics, cell biology, animal models and clinical data have started to unravel mechanisms underpinning evolvability in tumors. These ideas have in turn inspired the development of mathematical models that, by integrating an understanding of the mechanisms of tumor robustness, therapy resistance and tumor evolvability, are providing a new tool in the identification of novel dosing strategies that may help to delay or prevent the emergence of therapy resistance in human cancer patients.By viewing cancer as a robust, evolvable system, a number of researchers are now coming to the conclusion that single therapeutic targets might be fundamentally unsuitable as a general treatment strategy because the inherent heritable variation in cancer makes it a moving and elusive target. As emphasized in ref. 107, targeted therapy approaches are likely to fail if the molecular targets are present in only a subset of proliferating cancer cells. Instead, we propose that directly attacking the origins of cancer evolvability using therapeutic strategies that reduce heritable variation could provide a rational alternative approach.
Acknowledgements
TT and AH are supported by the Australian Research Council. SO and AH are supported by the Australian National Health and Medical Research Council (NHMRC). AH is an NHMRC CJ Martin Research Fellow. JW is supported in part by the Australian Defence Science and Technology Organization (DSTO). AH thanks Prudence Donovan and Rohan Tweedale for their insightful comments and revisions.References
- B. Tran and M. A. Rosenthal, J. Clin. Neurosci., 2010, 17, 417–421 CrossRef CAS.
- S. Rottenberg and J. Jonkers, Drug Resist. Updates, 2008, 11, 51–60 CrossRef CAS.
- J. A. de Visser, J. Hermisson, G. P. Wagner, L. Ancel Meyers, H. Bagheri-Chaichian, J. L. Blanchard, L. Chao, J. M. Cheverud, S. F. Elena, W. Fontana, G. Gibson, T. F. Hansen, D. Krakauer, R. C. Lewontin, C. Ofria, S. H. Rice, G. von Dassow, A. Wagner and M. C. Whitlock, Evolution, 2003, 57, 1959–1972 CrossRef.
- H. Kitano, Mol. Syst. Biol., 2007, 3, 137, DOI:10.1038/msb4100179.
- H. Kitano, Nat. Rev. Genet., 2004, 5, 826–837 CrossRef CAS.
- J. H. Crowe and L. M. Crowe, Nat. Biotechnol., 2000, 18, 145–146 CrossRef CAS.
- H. Kitano, Nat. Rev. Cancer, 2004, 4, 227–235 CrossRef CAS.
- A. Marusyk and K. Polyak, Biochim. Biophys. Acta, 2010, 1805, 105–117 CAS.
- L. L. Campbell and K. Polyak, Cell Cycle, 2007, 6, 2332–2338 Search PubMed.
- J. A. Tischfield and C. Shao, Nat. Genet., 2003, 33, 5–6 CrossRef CAS.
- B. Baisse, H. Bouzourene, E. P. Saraga, F. T. Bosman and J. Benhattar, Int. J. Cancer, 2001, 93, 346–352 CrossRef CAS.
- M. Kirschner and J. Gerhart, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8420–8427 CrossRef CAS.
- P. C. Nowell, Science, 1976, 194, 23–28 CrossRef CAS.
- L. M. Merlo, J. W. Pepper, B. J. Reid and C. C. Maley, Nat. Rev. Cancer, 2006, 6, 924–935 CrossRef CAS.
- J. J. Salk, E. J. Fox and L. A. Loeb, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 51–75 Search PubMed.
- G. B. Balaban, M. Herlyn, W. H. Clark, Jr. and P. C. Nowell, Cancer Genet. Cytogenet., 1986, 19, 113–122 CrossRef CAS.
- B. Vogelstein, E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, Y. Nakamura, R. White, A. M. Smits and J. L. Bos, N. Engl. J. Med., 1988, 319, 525–532 CAS.
- M. T. Barrett, C. A. Sanchez, L. J. Prevo, D. J. Wong, P. C. Galipeau, T. G. Paulson, P. S. Rabinovitch and B. J. Reid, Nat. Genet., 1999, 22, 106–109 CrossRef CAS.
- E. R. Fearon and B. Vogelstein, Cell, 1990, 61, 759–767 CrossRef CAS.
- M. F. Greaves, J. Rao, G. Hariri, W. Verbi, D. Catovsky, P. Kung and G. Goldstein, Leuk. Res., 1981, 5, 281–299 CrossRef CAS.
- S. P. Shah, R. D. Morin, J. Khattra, L. Prentice, T. Pugh, A. Burleigh, A. Delaney, K. Gelmon, R. Guliany, J. Senz, C. Steidl, R. A. Holt, S. Jones, M. Sun, G. Leung, R. Moore, T. Severson, G. A. Taylor, A. E. Teschendorff, K. Tse, G. Turashvili, R. Varhol, R. L. Warren, P. Watson, Y. Zhao, C. Caldas, D. Huntsman, M. Hirst, M. A. Marra and S. Aparicio, Nature, 2009, 461, 809–813 CrossRef CAS.
- N. Navin, A. Krasnitz, L. Rodgers, K. Cook, J. Meth, J. Kendall, M. Riggs, Y. Eberling, J. Troge, V. Grubor, D. Levy, P. Lundin, S. Maner, A. Zetterberg, J. Hicks and M. Wigler, Genome Res., 2010, 20, 68–80 CrossRef CAS.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Janne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, N. Engl. J. Med., 2005, 352, 786–792 CrossRef CAS.
- M. E. Gorre, M. Mohammed, K. Ellwood, N. Hsu, R. Paquette, P. N. Rao and C. L. Sawyers, Science, 2001, 293, 876–880 CrossRef CAS.
- N. P. Shah, J. M. Nicoll, B. Nagar, M. E. Gorre, R. L. Paquette, J. Kuriyan and C. L. Sawyers, Cancer Cell, 2002, 2, 117–125 CrossRef CAS.
- C. Roche-Lestienne and C. Preudhomme, Semin. Hematol., 2003, 40, 80–82 CAS.
- R. D. Barrett and D. Schluter, Trends Ecol. Evol., 2008, 23, 38–44 CrossRef.
- S. E. Luria and M. Delbruck, Genetics, 1943, 28, 491–511 CAS.
- S. Jones, X. Zhang, D. W. Parsons, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, H. Kamiyama, A. Jimeno, S. M. Hong, B. Fu, M. T. Lin, E. S. Calhoun, M. Kamiyama, K. Walter, T. Nikolskaya, Y. Nikolsky, J. Hartigan, D. R. Smith, M. Hidalgo, S. D. Leach, A. P. Klein, E. M. Jaffee, M. Goggins, A. Maitra, C. Iacobuzio-Donahue, J. R. Eshleman, S. E. Kern, R. H. Hruban, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, Science, 2008, 321, 1801–1806 CrossRef CAS.
- C. Greenman, P. Stephens, R. Smith, G. L. Dalgliesh, C. Hunter, G. Bignell, H. Davies, J. Teague, A. Butler, C. Stevens, S. Edkins, S. O'Meara, I. Vastrik, E. E. Schmidt, T. Avis, S. Barthorpe, G. Bhamra, G. Buck, B. Choudhury, J. Clements, J. Cole, E. Dicks, S. Forbes, K. Gray, K. Halliday, R. Harrison, K. Hills, J. Hinton, A. Jenkinson, D. Jones, A. Menzies, T. Mironenko, J. Perry, K. Raine, D. Richardson, R. Shepherd, A. Small, C. Tofts, J. Varian, T. Webb, S. West, S. Widaa, A. Yates, D. P. Cahill, D. N. Louis, P. Goldstraw, A. G. Nicholson, F. Brasseur, L. Looijenga, B. L. Weber, Y. E. Chiew, A. DeFazio, M. F. Greaves, A. R. Green, P. Campbell, E. Birney, D. F. Easton, G. Chenevix-Trench, M. H. Tan, S. K. Khoo, B. T. Teh, S. T. Yuen, S. Y. Leung, R. Wooster, P. A. Futreal and M. R. Stratton, Nature, 2007, 446, 153–158 CrossRef CAS.
- D. W. Parsons, S. Jones, X. Zhang, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, I. M. Siu, G. L. Gallia, A. Olivi, R. McLendon, B. A. Rasheed, S. Keir, T. Nikolskaya, Y. Nikolsky, D. A. Busam, H. Tekleab, L. A. Diaz, Jr., J. Hartigan, D. R. Smith, R. L. Strausberg, S. K. Marie, S. M. Shinjo, H. Yan, G. J. Riggins, D. D. Bigner, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, Science, 2008, 321, 1807–1812 CrossRef CAS.
- T. C. G. A. R. Network, Nature, 2008, 455, 1061–1068 CrossRef.
- T. Sjoblom, S. Jones, L. D. Wood, D. W. Parsons, J. Lin, T. D. Barber, D. Mandelker, R. J. Leary, J. Ptak, N. Silliman, S. Szabo, P. Buckhaults, C. Farrell, P. Meeh, S. D. Markowitz, J. Willis, D. Dawson, J. K. Willson, A. F. Gazdar, J. Hartigan, L. Wu, C. Liu, G. Parmigiani, B. H. Park, K. E. Bachman, N. Papadopoulos, B. Vogelstein, K. W. Kinzler and V. E. Velculescu, Science, 2006, 314, 268–274 CrossRef.
- L. D. Wood, D. W. Parsons, S. Jones, J. Lin, T. Sjoblom, R. J. Leary, D. Shen, S. M. Boca, T. Barber, J. Ptak, N. Silliman, S. Szabo, Z. Dezso, V. Ustyanksky, T. Nikolskaya, Y. Nikolsky, R. Karchin, P. A. Wilson, J. S. Kaminker, Z. Zhang, R. Croshaw, J. Willis, D. Dawson, M. Shipitsin, J. K. Willson, S. Sukumar, K. Polyak, B. H. Park, C. L. Pethiyagoda, P. V. Pant, D. G. Ballinger, A. B. Sparks, J. Hartigan, D. R. Smith, E. Suh, N. Papadopoulos, P. Buckhaults, S. D. Markowitz, G. Parmigiani, K. W. Kinzler, V. E. Velculescu and B. Vogelstein, Science, 2007, 318, 1108–1113 CrossRef CAS.
- R. T. Prehn and J. M. Main, J. Natl. Cancer Inst., 1957, 18, 769–778 CAS.
- N. H. Segal, D. W. Parsons, K. S. Peggs, V. Velculescu, K. W. Kinzler, B. Vogelstein and J. P. Allison, Cancer Res., 2008, 68, 889–892 CrossRef CAS.
- R. J. Leary, J. C. Lin, J. Cummins, S. Boca, L. D. Wood, D. W. Parsons, S. Jones, T. Sjoblom, B. H. Park, R. Parsons, J. Willis, D. Dawson, J. K. Willson, T. Nikolskaya, Y. Nikolsky, L. Kopelovich, N. Papadopoulos, L. A. Pennacchio, T. L. Wang, S. D. Markowitz, G. Parmigiani, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16224–16229 CrossRef CAS.
- L. A. Loeb, C. F. Springgate and N. Battula, Cancer Res., 1974, 34, 2311–2321 CAS.
- Y. Ionov, M. A. Peinado, S. Malkhosyan, D. Shibata and M. Perucho, Nature, 1993, 363, 558–561 CrossRef CAS.
- R. Fishel, M. K. Lescoe, M. R. Rao, N. G. Copeland, N. A. Jenkins, J. Garber, M. Kane and R. Kolodner, Cell, 1993, 75, 1027–1038 CrossRef CAS.
- C. Lengauer, K. W. Kinzler and B. Vogelstein, Nature, 1998, 396, 643–649 CrossRef CAS.
- J. M. Schvartzman, R. Sotillo and R. Benezra, Nat. Rev. Cancer, 2010, 10, 102–115 CrossRef CAS.
- S. L. Thompson, S. F. Bakhoum and D. A. Compton, Curr. Biol., 2010, 20, R285–295 CrossRef CAS.
- R. A. Beckman and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14140–14145 CrossRef CAS.
- J. Breivik, Semin. Cancer Biol., 2005, 15, 51–60 CrossRef CAS.
- R. A. Beckman, PLoS One, 2009, 4, e5860 CrossRef.
- E. Loh, J. J. Salk and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1154–1159 CrossRef CAS.
- T. Zhou, J. M. Carlson and J. Doyle, J. Theor. Biol., 2005, 236, 438–447 CrossRef.
- J. M. Carlson and J. Doyle, Phys. Rev. Lett., 2000, 84, 2529–2532 CrossRef CAS.
- P. Armitage and R. Doll, Br. J. Cancer, 1954, 8, 1–12 CAS.
- P. Armitage and R. Doll, Br. J. Cancer, 2004, 91, 1983–1989 CrossRef CAS.
- P. Armitage and R. Doll, Int. J. Epidemiol., 2004, 33, 1174–1179 Search PubMed.
- D. Duke, J. Castresana, L. Lucchina, T. H. Lee, A. J. Sober, W. P. Carey, D. E. Elder and R. L. Barnhill, Cancer, 1993, 72, 3239–3243 CrossRef CAS.
- A. G. Knudson, J. Cancer Res. Clin. Oncol., 1996, 122, 135–140 CrossRef CAS.
- A. G. Knudson, Nat. Rev. Cancer, 2001, 1, 157–162 CrossRef CAS.
- M. J. Renan, Mol. Carcinog., 1993, 7, 139–146 CrossRef CAS.
- A. Rangarajan, S. J. Hong, A. Gifford and R. A. Weinberg, Cancer Cell, 2004, 6, 171–183 CrossRef CAS.
- A. M. Mahale, Z. A. Khan, M. Igarashi, G. J. Nanjangud, R. F. Qiao, S. Yao, S. W. Lee and S. A. Aaronson, Cancer Res., 2008, 68, 1417–1426 CrossRef CAS.
- J. M. Nicholson and P. Duesberg, Cancer Genet. Cytogenet., 2009, 194, 96–110 CrossRef CAS.
- C. Swanton, B. Nicke, M. Schuett, A. C. Eklund, C. Ng, Q. Li, T. Hardcastle, A. Lee, R. Roy, P. East, M. Kschischo, D. Endesfelder, P. Wylie, S. N. Kim, J. G. Chen, M. Howell, T. Ried, J. K. Habermann, G. Auer, J. D. Brenton, Z. Szallasi and J. Downward, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8671–8676 CrossRef CAS.
- S. E. McClelland, R. A. Burrell and C. Swanton, Cell Cycle, 2009, 8, 3262–3266 Search PubMed.
- C. J. Ye, J. B. Stevens, G. Liu, S. W. Bremer, A. S. Jaiswal, K. J. Ye, M. F. Lin, L. Lawrenson, W. D. Lancaster, M. Kurkinen, J. D. Liao, C. G. Gairola, M. P. Shekhar, S. Narayan, F. R. Miller and H. H. Heng, J. Cell. Physiol., 2009, 219, 288–300 CrossRef CAS.
- R. Sotillo, J. M. Schvartzman, N. D. Socci and R. Benezra, Nature, 2010, 464, 436–440 CrossRef CAS.
- A. Brock, H. Chang and S. Huang, Nat. Rev. Genet., 2009, 10, 336–342 CrossRef CAS.
- V. E. A. Russo, R. A. Martienssen and A. D. Riggs, Cold Spring Harbor, Lanoratory Press, Woodbury, 1996 Search PubMed.
- S. L. Berger, T. Kouzarides, R. Shiekhattar and A. Shilatifard, Genes Dev., 2009, 23, 781–783 CrossRef CAS.
- P. A. Jones and S. B. Baylin, Cell, 2007, 128, 683–692 CrossRef CAS.
- K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS.
- S. Sharma, T. K. Kelly and P. A. Jones, Carcinogenesis, 2010, 31, 27–36 CrossRef CAS.
- E. S. McKenna and C. W. Roberts, Cell Cycle, 2009, 8, 23–26 Search PubMed.
- M. F. Fraga, E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero, K. Petrie, N. G. Iyer, A. Perez-Rosado, E. Calvo, J. A. Lopez, A. Cano, M. J. Calasanz, D. Colomer, M. A. Piris, N. Ahn, A. Imhof, C. Caldas, T. Jenuwein and M. Esteller, Nat. Genet., 2005, 37, 391–400 CrossRef CAS.
- A. Eden, F. Gaudet, A. Waghmare and R. Jaenisch, Science, 2003, 300, 455 CrossRef CAS.
- G. Howard, R. Eiges, F. Gaudet, R. Jaenisch and A. Eden, Oncogene, 2008, 27, 404–408 CrossRef CAS.
- F. V. Jacinto and M. Esteller, Mutagenesis, 2007, 22, 247–253 CrossRef CAS.
- C. Sawan, T. Vaissiere, R. Murr and Z. Herceg, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2008, 642, 1–13 CrossRef CAS.
- M. Esteller, S. R. Hamilton, P. C. Burger, S. B. Baylin and J. G. Herman, Cancer Res., 1999, 59, 793–797 CAS.
- K. Suzuki, I. Suzuki, A. Leodolter, S. Alonso, S. Horiuchi, K. Yamashita and M. Perucho, Cancer Cell, 2006, 9, 199–207 CrossRef CAS.
- A. P. Feinberg, R. Ohlsson and S. Henikoff, Nat. Rev. Genet., 2006, 7, 21–33 CrossRef CAS.
- P. M. Voorhoeve, Biochim. Biophys. Acta, 2010, 1805, 72–86 CAS.
- G. A. Calin, C. D. Dumitru, M. Shimizu, R. Bichi, S. Zupo, E. Noch, H. Aldler, S. Rattan, M. Keating, K. Rai, L. Rassenti, T. Kipps, M. Negrini, F. Bullrich and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15524–15529 CrossRef CAS.
- G. A. Calin, C. Sevignani, C. D. Dumitru, T. Hyslop, E. Noch, S. Yendamuri, M. Shimizu, S. Rattan, F. Bullrich, M. Negrini and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2999–3004 CrossRef CAS.
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebert, R. H. Mak, A. A. Ferrando, J. R. Downing, T. Jacks, H. R. Horvitz and T. R. Golub, Nature, 2005, 435, 834–838 CrossRef.
- N. Yanaihara, N. Caplen, E. Bowman, M. Seike, K. Kumamoto, M. Yi, R. M. Stephens, A. Okamoto, J. Yokota, T. Tanaka, G. A. Calin, C. G. Liu, C. M. Croce and C. C. Harris, Cancer Cell, 2006, 9, 189–198 CrossRef CAS.
- S. Volinia, G. A. Calin, C. G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R. L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C. C. Harris and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2257–2261 CrossRef CAS.
- S. Ambs, R. L. Prueitt, M. Yi, R. S. Hudson, T. M. Howe, F. Petrocca, T. A. Wallace, C. G. Liu, S. Volinia, G. A. Calin, H. G. Yfantis, R. M. Stephens and C. M. Croce, Cancer Res., 2008, 68, 6162–6170 CrossRef CAS.
- L. He, J. M. Thomson, M. T. Hemann, E. Hernando-Monge, D. Mu, S. Goodson, S. Powers, C. Cordon-Cardo, S. W. Lowe, G. J. Hannon and S. M. Hammond, Nature, 2005, 435, 828–833 CrossRef CAS.
- S. Costinean, N. Zanesi, Y. Pekarsky, E. Tili, S. Volinia, N. Heerema and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7024–7029 CrossRef CAS.
- H. Hermeking, Cancer Cell, 2007, 12, 414–418 CrossRef CAS.
- P. P. Medina and F. J. Slack, Cell Cycle, 2008, 7, 2485–2492 Search PubMed.
- J. Stelling, U. Sauer, Z. Szallasi, F. J. Doyle and J. Doyle, Cell, 2004, 118, 675–685 CrossRef CAS.
- M. Freeman, Nature, 2000, 408, 313–319 CrossRef CAS.
- N. J. Martinez, M. C. Ow, M. I. Barrasa, M. Hammell, R. Sequerra, L. Doucette-Stamm, F. P. Roth, V. R. Ambros and A. J. Walhout, Genes Dev., 2008, 22, 2535–2549 CrossRef CAS.
- J. Tsang, J. Zhu and A. van Oudenaarden, Mol. Cell, 2007, 26, 753–767 CrossRef CAS.
- S. Kauffman, Nature, 1969, 224, 177–178 CrossRef CAS.
- S. A. Kauffman, J. Theor. Biol., 1969, 22, 437–467 CrossRef CAS.
- S. Huang, I. Ernberg and S. Kauffman, Semin. Cell Dev. Biol., 2009, 20, 869–876 CrossRef CAS.
- M. H. Tomasson, J. Cell. Biochem., 2009, 106, 745–749 CrossRef CAS.
- A. Trumpp and O. D. Wiestler, Nature Clinical Practice, 2008, 5, 337–347 Search PubMed.
- P. N. Kelly, A. Dakic, J. M. Adams, S. L. Nutt and A. Strasser, Science, 2007, 317, 337 CrossRef CAS.
- E. Quintana, M. Shackleton, M. S. Sabel, D. R. Fullen, T. M. Johnson and S. J. Morrison, Nature, 2008, 456, 593–598 CrossRef CAS.
- A. Roesch, M. Fukunaga-Kalabis, E. C. Schmidt, S. E. Zabierowski, P. A. Brafford, A. Vultur, D. Basu, T. Vogt and M. Herlyn, Cell, 2010, 141, 583–594 CrossRef CAS.
- T. Vogt, M. Kroiss, M. McClelland, C. Gruss, B. Becker, A. K. Bosserhoff, G. Rumpler, T. Bogenrieder, M. Landthaler and W. Stolz, Lab. Invest., 1999, 79, 1615–1627 Search PubMed.
- A. Roesch, B. Becker, S. Meyer, C. Hafner, P. J. Wild, M. Landthaler and T. Vogt, Mod. Pathol., 2005, 18, 565–572 Search PubMed.
- A. Roesch, B. Becker, S. Meyer, P. Wild, C. Hafner, M. Landthaler and T. Vogt, Mod. Pathol., 2005, 18, 1249–1257 Search PubMed.
- A. Roesch, B. Becker, W. Schneider-Brachert, I. Hagen, M. Landthaler and T. Vogt, J. Invest. Dermatol., 2006, 126, 1850–1859 CrossRef CAS.
- A. Roesch, A. M. Mueller, T. Stempfl, C. Moehle, M. Landthaler and T. Vogt, Int. J. Cancer, 2008, 122, 1047–1057 CAS.
- M. Greaves, Semin. Cancer Biol., 2010, 20, 65–70 CrossRef.
- S. Cara and I. F. Tannock, Ann. Oncol., 2001, 12, 23–27 CrossRef CAS.
- T. Kurata, K. Tamura, H. Kaneda, T. Nogami, H. Uejima, G. Asai Go, K. Nakagawa and M. Fukuoka, Ann. Oncol., 2004, 15, 173–174 CrossRef CAS.
- S. Yano, E. Nakataki, S. Ohtsuka, M. Inayama, H. Tomimoto, N. Edakuni, S. Kakiuchi, N. Nishikubo, H. Muguruma and S. Sone, Oncol. Res., 2005, 15, 107–111.
- S. V. Sharma, D. Y. Lee, B. Li, M. P. Quinlan, F. Takahashi, S. Maheswaran, U. McDermott, N. Azizian, L. Zou, M. A. Fischbach, K. K. Wong, K. Brandstetter, B. Wittner, S. Ramaswamy, M. Classon and J. Settleman, Cell, 2010, 141, 69–80 CrossRef CAS.
- A. C. Dean and C. Hinshelwood, Proc. R. Soc. London, Ser. B, 1954, 142, 45–60 CrossRef CAS.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS.
- W. J. Blake, G. Balazsi, M. A. Kohanski, F. J. Isaacs, K. F. Murphy, Y. Kuang, C. R. Cantor, D. R. Walt and J. J. Collins, Mol. Cell, 2006, 24, 853–865 CrossRef CAS.
- M. C. Smith, E. R. Sumner and S. V. Avery, Mol. Microbiol., 2007, 66, 699–712 CrossRef CAS.
- H. H. Chang, M. Hemberg, M. Barahona, D. E. Ingber and S. Huang, Nature, 2008, 453, 544–547 CrossRef CAS.
- A. A. Cohen, N. Geva-Zatorsky, E. Eden, M. Frenkel-Morgenstern, I. Issaeva, A. Sigal, R. Milo, C. Cohen-Saidon, Y. Liron, Z. Kam, L. Cohen, T. Danon, N. Perzov and U. Alon, Science, 2008, 322, 1511–1516 CrossRef CAS.
- J. J. Kim and I. F. Tannock, Nat. Rev., 2005, 5, 516–525 CAS.
- D. J. Barnes and J. V. Melo, Cell Cycle, 2006, 5, 2862–2866 Search PubMed.
- P. Vaupel, A. Mayer and M. Hockel, Methods Enzymol., 2004, 381, 335–354 CAS.
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS.
- Y. Kim, Q. Lin, P. M. Glazer and Z. Yun, Curr. Mol. Med., 2009, 9, 425–434 Search PubMed.
- R. S. Bindra, M. E. Crosby and P. M. Glazer, Cancer Metastasis Rev., 2007, 26, 249–260 CrossRef CAS.
- E. K. Rofstad, K. Sundfor, H. Lyng and C. G. Trope, Br. J. Cancer, 2000, 83, 354–359 CrossRef CAS.
- M. Wartenberg, F. C. Ling, M. Muschen, F. Klein, H. Acker, M. Gassmann, K. Petrat, V. Putz, J. Hescheler and H. Sauer, FASEB J., 2003, 17, 503–505 CAS.
- T. G. Graeber, C. Osmanian, T. Jacks, D. E. Housman, C. J. Koch, S. W. Lowe and A. J. Giaccia, Nature, 1996, 379, 88–91 CrossRef CAS.
- S. J. Lunt, N. Chaudary and R. P. Hill, Clin. Exp. Metastasis, 2009, 26, 19–34 CrossRef.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891–899 CrossRef CAS.
- J. Lankelma, H. Dekker, F. R. Luque, S. Luykx, K. Hoekman, P. van der Valk, P. J. van Diest and H. M. Pinedo, Clin. Cancer Res., 1999, 5, 1703–1707 CAS.
- W. S. Dalton, B. G. Durie, D. S. Alberts, J. H. Gerlach and A. E. Cress, Cancer Res., 1986, 46, 5125–5130 CAS.
- K. A. Rejniak and L. J. McCawley, Exp. Biol. Med., 2010, 235, 411–423 Search PubMed.
- M. Hu and K. Polyak, Curr. Opin. Genet. Dev., 2008, 18, 27–34 CrossRef CAS.
- K. Polyak, I. Haviv and I. G. Campbell, Trends Genet., 2009, 25, 30–38 CrossRef CAS.
- G. M. Edelman and J. A. Gally, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13763–13768 CrossRef CAS.
- J. Whitacre and A. Bender, J. Theor. Biol., 2010, 263, 143–153 CrossRef.
- J. M. Whitacre and A. Bender, Theor. Biol. Med. Modell., 2010, 7, 20 Search PubMed.
- A. M. Xu and P. H. Huang, Cancer Res., 2010, 70, 3857–3860 CrossRef CAS.
- M. Nagane, F. Coufal, H. Lin, O. Bogler, W. K. Cavenee and H. J. Huang, Cancer Res., 1996, 56, 5079–5086 CAS.
- A. El-Obeid, E. Bongcam-Rudloff, M. Sorby, A. Ostman, M. Nister and B. Westermark, Cancer Res., 1997, 57, 5598–5604 CAS.
- J. N. Rich, D. A. Reardon, T. Peery, J. M. Dowell, J. A. Quinn, K. L. Penne, C. J. Wikstrand, L. B. Van Duyn, J. E. Dancey, R. E. McLendon, J. C. Kao, T. T. Stenzel, B. K. Ahmed Rasheed, S. E. Tourt-Uhlig, J. E. Herndon, 2nd, J. J. Vredenburgh, J. H. Sampson, A. H. Friedman, D. D. Bigner and H. S. Friedman, J. Clin. Oncol., 2004, 22, 133–142 CAS.
- P. H. Huang, A. Mukasa, R. Bonavia, R. A. Flynn, Z. E. Brewer, W. K. Cavenee, F. B. Furnari and F. M. White, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12867–12872 CrossRef CAS.
- J. M. Stommel, A. C. Kimmelman, H. Ying, R. Nabioullin, A. H. Ponugoti, R. Wiedemeyer, A. H. Stegh, J. E. Bradner, K. L. Ligon, C. Brennan, L. Chin and R. A. DePinho, Science, 2007, 318, 287–290 CrossRef CAS.
- J. M. Whitacre, Theor. Biol. Med. Modell., 2010, 7, 6 Search PubMed.
- G. Cambray and D. Mazel, PLoS Genet., 2008, 4, e1000256 Search PubMed.
- A. Wagner, BioEssays, 2005, 27, 176–188 CrossRef CAS.
- J. M. Carlson and J. Doyle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(Suppl 1), 2538–2545 CrossRef.
- A. Wagner, Proc. Biol. Sci., 2008, 275, 91–100 Search PubMed.
- L. S. Yaeger, HFSP J., 2009, 3, 328–339 CrossRef.
- J. A. Draghi, T. L. Parsons, G. P. Wagner and J. B. Plotkin, Nature, 2010, 463, 353–355 CrossRef CAS.
- M. A. Huynen, P. F. Stadler and W. Fontana, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 397–401 CrossRef CAS.
- S. Ciliberti, O. C. Martin and A. Wagner, PLoS Comput. Biol., 2007, 3, e15 Search PubMed.
- B. C. Daniels, Y. J. Chen, J. P. Sethna, R. N. Gutenkunst and C. R. Myers, Curr. Opin. Biotechnol., 2008, 19, 389–395 CrossRef CAS.
- J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5869–5874 CrossRef CAS.
- R. C. McBride, C. B. Ogbunugafor and P. E. Turner, BMC Evol. Biol., 2008, 8, 231 CrossRef.
- S. L. Rutherford and S. Lindquist, Nature, 1998, 396, 336–342 CrossRef CAS.
- L. E. Cowen and S. Lindquist, Science, 2005, 309, 2185–2189 CrossRef CAS.
- N. Tokuriki and D. S. Tawfik, Nature, 2009, 459, 668–673 CrossRef CAS.
- Y. Xu and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7074–7078 CrossRef CAS.
- S. F. Falsone, S. Leptihn, A. Osterauer, M. Haslbeck and J. Buchner, J. Mol. Biol., 2004, 344, 281–291 CrossRef.
- S. Takayama, J. C. Reed and S. Homma, Oncogene, 2003, 22, 9041–9047 CrossRef CAS.
- D. D. Mosser and R. I. Morimoto, Oncogene, 2004, 23, 2907–2918 CrossRef CAS.
- M. J. West-Eberhard, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(suppl_1), 6543–6549 CrossRef CAS.
- M. Pigliucci, C. J. Murren and C. D. Schlichting, J. Exp. Biol., 2006, 209, 2362–2367 CrossRef.
- R. Sanjuan and S. F. Elena, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14402–14405 CrossRef CAS.
- G. Gibson and I. Dworkin, Nat. Rev. Genet., 2004, 5, 681–690 CrossRef CAS.
- C. D. Schlichting, Ann. N. Y. Acad. Sci., 2008, 1133, 187–203 CrossRef.
- J. Whitacre, M., ALIFE XII, 2010 Search PubMed , in press.
- D. F. Horrobin, Nat. Rev. Drug Discovery, 2003, 2, 151–154 CrossRef CAS.
- A. J. Coldman and J. H. Goldie, Bull. Math. Biol., 1986, 48, 279–292 CAS.
- A. J. Coldman and J. M. Murray, Math. Biosci., 2000, 168, 187–200 CrossRef CAS.
- R. S. Day, Cancer Res., 1986, 46, 3876–3885 CAS.
- Y. Iwasa, F. Michor and M. A. Nowak, Proc. Biol. Sci., 2003, 270, 2573–2578 Search PubMed.
- M. P. Little, Biol. Direct, 2010, 5, 19 Search PubMed ; discussion 19.
- J. Foo and F. Michor, PLoS Comput. Biol., 2009, 5, e1000557 Search PubMed.
- A. S. Silva and R. A. Gatenby, Biol. Direct, 2010, 5, 25 Search PubMed.
- R. A. Gatenby, A. S. Silva, R. J. Gillies and B. R. Frieden, Cancer Res., 2009, 69, 4894–4903 CrossRef CAS.
- A. Kurakin, Theor. Biol. Med. Modell., 2009, 6, 6 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |