
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interface property–functionality interplay suppresses bimolecular recombination facilitating above 18% efficiency organic solar cells embracing simplistic fabrication†
Top Archie
Dela Peña
 abc,
Ruijie
Ma
*d,
Zengshan
Xing
e,
Qi
Wei
b,
Jafar I.
Khan
f,
Ryan Michael
Young
g,
Yulong
Hai
a,
Sheena Anne
Garcia
h,
Xinhui
Zou
e,
Zijing
Jin
e,
Fai Lun
Ng
i,
King Lun
Yeung
h,
Dayne F.
Swearer
f,
Michael R.
Wasielewski
g,
Jiannong
Wang
e,
Hyojung
Cha
j,
He
Yan
c,
Kam Sing
Wong
e,
Gang
Li
*d,
Mingjie
Li
*bk and
Jiaying
Wu
*al
abc,
Ruijie
Ma
*d,
Zengshan
Xing
e,
Qi
Wei
b,
Jafar I.
Khan
f,
Ryan Michael
Young
g,
Yulong
Hai
a,
Sheena Anne
Garcia
h,
Xinhui
Zou
e,
Zijing
Jin
e,
Fai Lun
Ng
i,
King Lun
Yeung
h,
Dayne F.
Swearer
f,
Michael R.
Wasielewski
g,
Jiannong
Wang
e,
Hyojung
Cha
j,
He
Yan
c,
Kam Sing
Wong
e,
Gang
Li
*d,
Mingjie
Li
*bk and
Jiaying
Wu
*al
aThe Hong Kong University of Science and Technology, Function Hub, Advanced Materials Thrust, Nansha 511400, Guangzhou, P. R. China. E-mail: jiayingwu@ust.hk
bThe Hong Kong Polytechnic University, Faculty of Science, Department of Applied Physics, Kowloon, Hong Kong 000000, P. R. China. E-mail: ming-jie.li@polyu.edu.hk
cThe Hong Kong University of Science and Technology, School of Science, Department of Chemistry, Kowloon, Hong Kong 000000, P. R. China
dDepartment of Electronic and Information Engineering, Research Institute for Smart Energy (RISE), Guangdong-Hong Kong-Macao (GHM) Joint Laboratory for Photonic-Thermal-Electrical Energy Materials and Devices, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 999077, China. E-mail: gang.w.li@polyu.edu.hk; ruijie.ma@polyu.edu.hk
eThe Hong Kong University of Science and Technology, School of Science, Department of Physics, Kowloon, Hong Kong 000000, P. R. China
fNorthwestern University, Department of Chemistry and Department of Chemical and Biological Engineering, Evanston, Illinois 60208, USA
gNorthwestern University, Department of Chemistry and Institute for Sustainability and Energy at Northwestern, Evanston, Illinois 60208, USA
hThe Hong Kong University of Science and Technology, School of Engineering, Department of Chemical and Biomolecular Engineering, Kowloon, Hong Kong 000000, P. R. China
iThe Hong Kong University of Science and Technology, School of Engineering, Department of Electronic and Computer Engineering, Kowloon, Hong Kong 000000, P. R. China
jKyungpook National University, Department of Hydrogen & Renewable Energy, ITA Convergence Graduate School, Daegu 41566, South Korea
kPhotonics Research Institute, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 000000, P. R. China
lThe Hong Kong University of Science and Technology, School of Engineering, Department of Chemical Engineering, Kowloon, Hong Kong 000000, P. R. China
First published on 15th June 2023
Abstract
Efficient exciton-to-charge generation from the introduction of non-fullerene acceptors (NFAs) has been an important breakthrough in organic solar cell (OSC) developments. However, low device fill factors (FFs) following significant free charge carrier recombination loss continue to undermine their marketplace potential. Previous studies have successfully uncovered the importance of donor and acceptor domains in promoting charge transport. However, the functionality of donor/acceptor (D/A) interfaces relevant to free charge recombination remains unclear despite such interfaces being present throughout the material. In this work, the disorder-induced uphill bulk-to-D/A interface transport energy landscape is unveiled to enhance the polaron recombination resistance thereby also mitigating the triplet state formation from back charge transfer. In simple words, increasing the interface disorder while keeping the purer domains highly ordered will impart greater bulk-to-interface differential energy which then serves as a recombination energy barrier. Herein, these are made possible by varying the NFA outer side chains from linear alkyls to bulkier 2D phenylalkyls which influence the donor–acceptor interaction and define the interfacial disorder. Meanwhile, the extent of interface disorder without tradeoff in geminate losses is dependent on electrostatics and nanomorphology driving efficient exciton dissociation. By understanding these interplays, remarkable FFs over 80% and power conversion efficiencies (PCEs) above 18% are revealed to remain accessible even with simple binary component device fabrication without additional components/treatments thereby maintaining the scalability. Consequently, the principles uncovered here will serve as the foundation to reach the optimum potential of binary and simple systems, a prerequisite to ultimately realize the most cost-effective OSC design strategies for practical applications.
Broader contextThe search for renewable energy sources has been one of the major issues to enable continuous and sustainable industrial expansions fueling economic growth. Organic solar cells (OSCs) have been of prime interest for solar energy harvesting owing to their low carbon footprint, mechanical flexibility, lightweight, and solution-processability. Thanks to the extensive efforts from the research community, efficiencies above 18% are already feasible. However, achieving these efficiencies often comes with more complexities undermining the commercial scalability and manufacturing costs. It can be a consequence of the substantial knowledge deficiency concerning the functionality of D/A interfaces, particularly in relation to free charge recombination behavior. Hence, this study uncovered the disorder-induced modulation of the bulk-to-D/A interface energy landscape which is found to regulate the free charge recombination energy barrier. Through this principle, state-of-the-art device performances are revealed to remain accessible even with simple binary component devices (i.e., without the need for additional components/treatments). Consequently, together with the synthetic flexibility of organic molecules and device engineering tailoring such interface properties, the understanding derived in this work will serve as a foundation for reaching the true optimum potential of binary systems thereby fostering the development of efficient yet economical OSCs. |
Introduction
Over the past decades, there have been continuous efforts to improve the power conversion efficiencies (PCEs) of organic solar cells (OSCs).1–8 These developments are motivated by the introduction of non-fullerene acceptors (NFAs) such as BTP-4F (also known as Y6) displaying exciton dissociation efficiencies of near unity and low voltage losses.9,10 Notwithstanding such progress, fill factors (FFs) reaching 80% with concurrent PCEs exceeding 18% are still rarely attainable, particularly for simpler devices such as single-junctions with binary-component bulk heterojunctions (BHJs).3,8,11–16 In trying to overcome these limitations, several different strategies have been explored and are mostly based on the improvement of free charge transport on the basis of acceptor domains and (or) donor network properties. It can be argued as the consequence of the convenience in characterizing molecular domains when compared to the complex nature of D/A interfaces. Hence, understanding the free charge recombination behavior relative to the interaction of acceptor (A) and donor (D) molecules at D/A interfaces remains unclear, more particularly when explicitly considering the latest high-efficiency materials (i.e., defined here with >14% PCEs and >70% FFs). Accordingly, it is of utmost importance to uncover the functionality of D/A interfaces in constructing the complete picture of design principles enabling simple devices toward their maximum efficiency limit. This is essential to definitively recognize the most cost-effective approaches for OSCs to meet the marketplace standards, provided that there could be infinite possibilities in administering additional treatments to enhance the OSC performance.The NFAs BTP-4F-PC6 and BTP-4F-P2EH with molecular structures shown in Fig. 1(a) have been recently introduced by replacing the outer alkyl side chains of Y6 with 2D phenylalkyl side groups.17 It is known that Y6 exhibits excessive self-aggregation and the synthetic modification with bulkier 2D side groups is originally meant to impart steric hindrance reducing the aggregation tendency and benefit exciton dissociation as previously demonstrated when blended with a polymer donor PM6, also known as poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo [1,2b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′c′]dithiophene-4,8-dione)]. Surprisingly, the FF is found to be substantially enhanced, suggesting that there exists a highly efficient charge transport. Yet, the underlying principles and the mechanism of charge carriers explaining the recombination behavior remain unclear.17 It can be observed that previous studies mainly attributed the charge transport to the molecular packing of acceptor molecules in their respective domains while mostly excluding the properties of D/A interfaces in their discussions. However, the concept of domain characteristics cannot justify the reported FFs since highly ordered acceptor domains in PM6:NFA blend films were identified regardless of the Y6 outer side chain. Such observations then enlighten the urgency to delineate the ambiguity in understanding the functionality of D/A interfaces relevant to free charge transport which will be of critical importance in developing simple yet high-efficiency devices. Meanwhile, incorporating 20% thiophene-thiazolothiazole (TTz) units in the PM6 backbone leads to the formation of the terpolymer PM1, which demonstrates excellent batch-to-batch reproducibility and potentially a future work-horse material,18,19 yet it remains less explored.
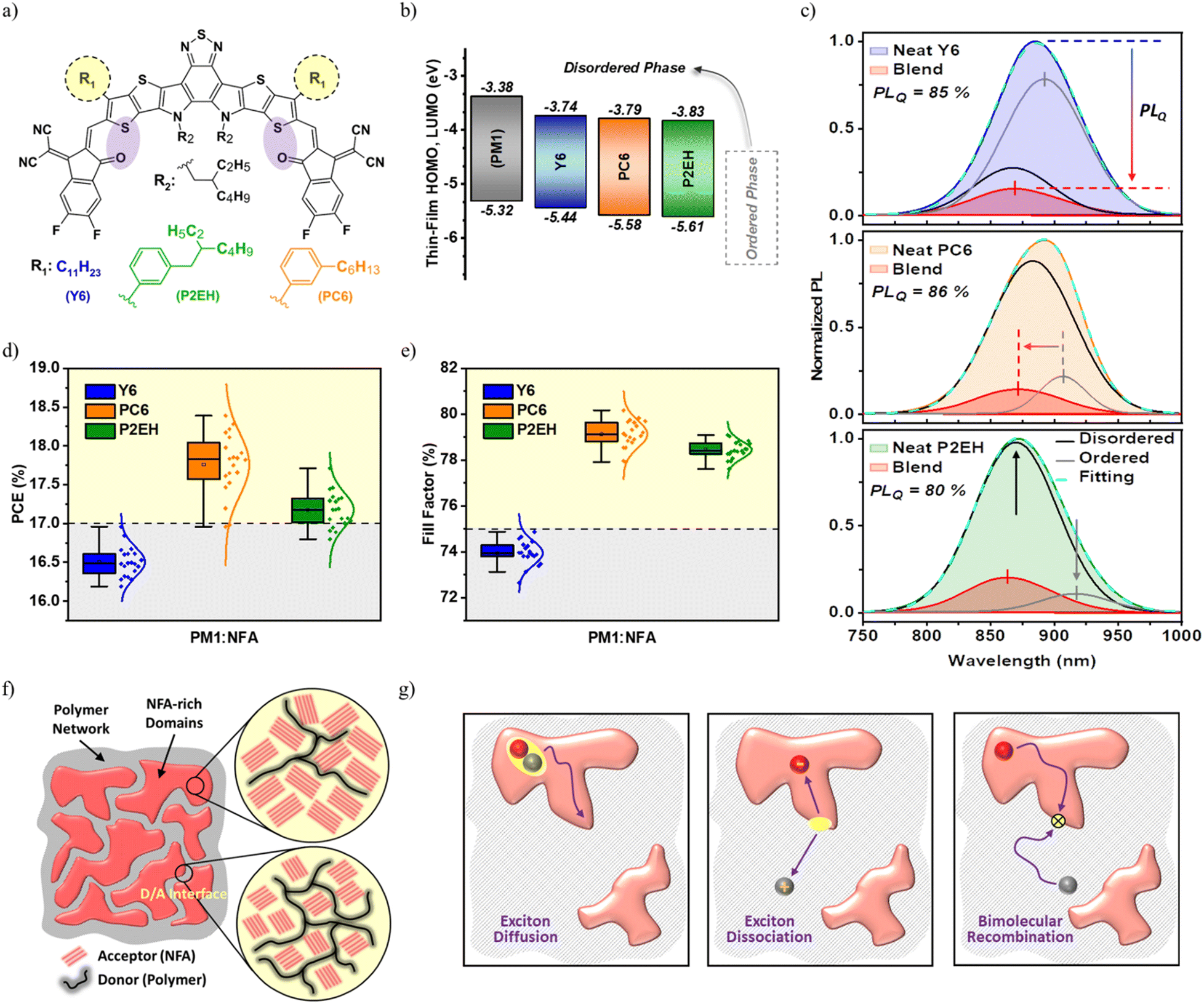 | ||
| Fig. 1 Material and device properties: molecular structures of the NFAs (a), thin-films HOMO and LUMO transport energy levels derived from neat materials and based on photoelectron spectroscopy estimations (b), PL spectra of neat NFAs, their blends with PM1, and NFA exciton PLQ (c), key photovoltaic metrics of PM1:NFA binary devices (d and e), schematic illustration of the D/A interface and NFA-rich domains in the bulk heterojunction (f), and simplified charge carrier dissociation and recombination mechanism in OSCs (g). The PL intensities are normalized to the maximum of the corresponding NFA neat films upon excitation at 633 nm. | ||
Recent studies successfully recognized that energy level cascades, energy offsets, and interpenetrating network morphologies promote efficient charge transfer while domain purity and phase separation are crucial in free charge transport.10,20–24 Although these arguments are essential when rationalizing PCEs and FFs of wide ranges, they remain deficient in explicitly defining the unique framework enabling simple and state-of-the-art systems relative to their recent predecessors, as inferable from earlier works.17,25–27 Accordingly, the energetic disorder of the D/A interface from absorption exponential tail states, measured as Urbach energy (EUrbach), is becoming an attractive figure of merit.28 This is enlightened by former studies recognizing the significance of understanding the D/A material interactions.29–33 It can be observed that the intermolecular charge transfer states (CTSs) have been primarily used as the basis to characterize the properties of D/A interfaces. However, in contrast to fullerene-based systems, the low absorption cross-section of intermolecular CTSs in NFA-based OSCs which substantially overlaps with the acceptor absorption edge leads to indistinguishable features and ambiguities between the D/A interface and purer domains.34,35 Thus, the D/A interface and bulk properties are typically classified indeterminately despite being discrete.36 Recently, Ohkita et al. made a significant contribution to understanding the influence of D/A interface properties in exciton dissociation which refines future device optimizations.21 However, the dependence of free charge recombination in the D/A interface also demands an in-depth understanding to effectively tune the FFs and PCEs while minimizing the overall device fabrication complexities. Likewise, as NFAs generally have more compact nanomorphology while efficiency losses upon degradation remain a serious problem and mechanisms are still to be explored,26,37–41 then understanding the influence of the D/A interface will also be of great importance in the development of stable devices.
In this work, through the blends of PM1 and NFAs with variable outer side groups, the donor–acceptor molecular interaction defining the interface disorder is demonstrated to impose an uphill transport energy landscape from the bulk to the D/A interface. The resulting bulk-to-D/A interface differential energy is demonstrated to serve as a recombination energy barrier that regulates the polaron recombination resistance and therefore can also reduce the maximum triplet state population by 40% herein. Meanwhile, the acceptor–acceptor molecular interaction exhibits highly ordered domains with minimal energy trap states. Consequently, upon suppressing the polaron recombination, a substantial improvement in the free charge transport facilitating over 80% FFs is observed. In addition, the D/A interface disorder is also illustrated to enhance the burn-in device performance stability. However, these merits can be at the expense of geminate losses since the same interface energy landscape will also govern diffusion-limited exciton dissociation. Accordingly, molecular electrostatics and nanomorphology are then unraveled to mitigate such tradeoffs by imparting auxiliary effects for charge transfer and generation. Ultimately, through this framework, even simple binary component BHJ devices are shown to be capable of displaying PCEs above 18%.
Material selection and photovoltaic performance
For simplicity, the NFAs BTP-4F-PC6 and BTP-4F-P2EH will be referred to here as PC6 and P2EH to classify the Y6 outer side chain modification directly. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) transport energies for randomly oriented molecules can be derived from solution-state cyclic voltammetry (CV) while solid-state measurements will be useful to understand the influence of order induced by molecular packing.21 Through photoelectron spectroscopy of pure films (Fig. 1b, Fig. S3, ESI†), it is observed that energy levels for NFAs in the disordered phase (e.g., aggregate–aggregate interfaces) are increased compared to their more ordered phase thereby approaching those reported from solution-state CV, as can also be inferred from previous studies.21 Likewise, the absorption onset of solutions is widely known to display a blue shift (i.e., higher energy) than their more ordered solid-state counterparts.42 The same concept applies to D/A interfaces present in blend films, it can be observed that the NFA photoluminescence (PL) in the blend is blue-shifted relative to that of their self-assemblies in neat films (Fig. 1c). Such spectral transitions can be understood as a consequence of order–disorder phase evolutions influencing the spatial extent of excited states,43 as further discussed in ESI† Notes S1. Simply on the basis of these precursor material property investigations, the more disordered nature of the D/A interfaces relative to purer molecular domains is already suggestive to impose an uphill bulk-to-interface transport energy landscape, as can also be inferred from former studies.21,33 Indeed, this is further verified through more direct measurements presented in succeeding discussions which then enables us to unveil the unique functionality of D/A interfaces tailoring the dynamics of free charge carriers and imparting highly efficient charge transport even without the need for additional components/treatments.Interestingly, the BHJ blends of PC6 and P2EH with PM1 in single-junction devices are demonstrated to reach up to 18.39% and 17.71% PCEs under 1 Sun illumination, respectively, far superior to the Y6-based device and amongst the highest reported to date. Additionally, by considering the PTQ10 donor with the same set of acceptors, the same relative device metrics exist (Table S1, ESI†). As shown in Fig. 1(d and e), these improvements can be attributed to the striking FFs of PC6 and P2EH-based devices (i.e., reaching >80%) since there is only marginal variability in the open circuit voltage (VOC) and short-circuit current density (JSC). The summary of current–voltage (JV) curve characteristics and other device metrics are provided in Table S1 and Fig. S4 (ESI†). In addition, this work also emphasizes that low-cost and synthetically more reproducible random terpolymers (e.g., PM1) can actually compete with other commonly used expensive donor materials. To understand what enables such notable FFs, the charge recombination behavior in relation to both bulk and D/A interface properties, illustrated in Fig. 1(f and g), is investigated.
Bulk and D/A interface properties
Recently, it has become a common practice to quantify the energetic disorder by extracting the Urbach energy (EUrbach) from the sensitively-measured EQE.44,45 As shown in Fig. 2b and Table S4 (ESI†), it can be noted that OSCs with PCEs >14% customarily exhibit EUrbach below 30 meV,6,46 indicatively low enough allowing the ambient thermal energy to assist charge transport. Intriguingly, despite having significantly different FFs which reflect the competition between charge transport and recombination (illustrated in Fig. 1g), all the blends considered herein display a low energetic disorder of around 25 meV (Fig. 2a and Table S3, ESI†). This is consistent with several reports observing the superior directional stacking and high molecular rigidity of Y6 molecules.47 Likewise, from grazing-incidence wide-angle X-ray scattering (GIWAXS), both PC6 and P2EH exhibit clear scattering peaks which is a typical characteristic of Y6 derivatives possessing crystalline properties (Fig. 3c). Consequently, these NFA self-assemblies are previously found to have similar ordered packing even in blends.17,48 Hence, it can be roughly estimated that FFs beyond 70% cannot be justified simply through EUrbach. As opposed to the inset of Fig. 2a, it is also interesting to note that their absorption exponential tails at the operational temperature (i.e., 300 K) display no distinguishing features for the D/A interface and NFA-domains up to the extent of our measurement sensitivity. This is commonly observed in NFA-based OSCs with high efficiency as a consequence of smaller optical energy differences (ES1-CTS) between acceptor singlet states and intermolecular CTSs (i.e., intermediate states residing in the D/A interface region), imparting reduced energy losses.35 In fact, there are several reports with CTS energy (ECTS) approaching (or even higher) than the singlet which is consistent with the uphill transport energy levels from NFA domains to D/A interfaces as caused by an increasing disorder (illustrated in Fig. 2c).49 It then follows that the D/A interface remains the least interpreted among former studies, more specifically with regard to efficient free charge transport as there is a very limited number of binary OSCs reaching FFs above 80%.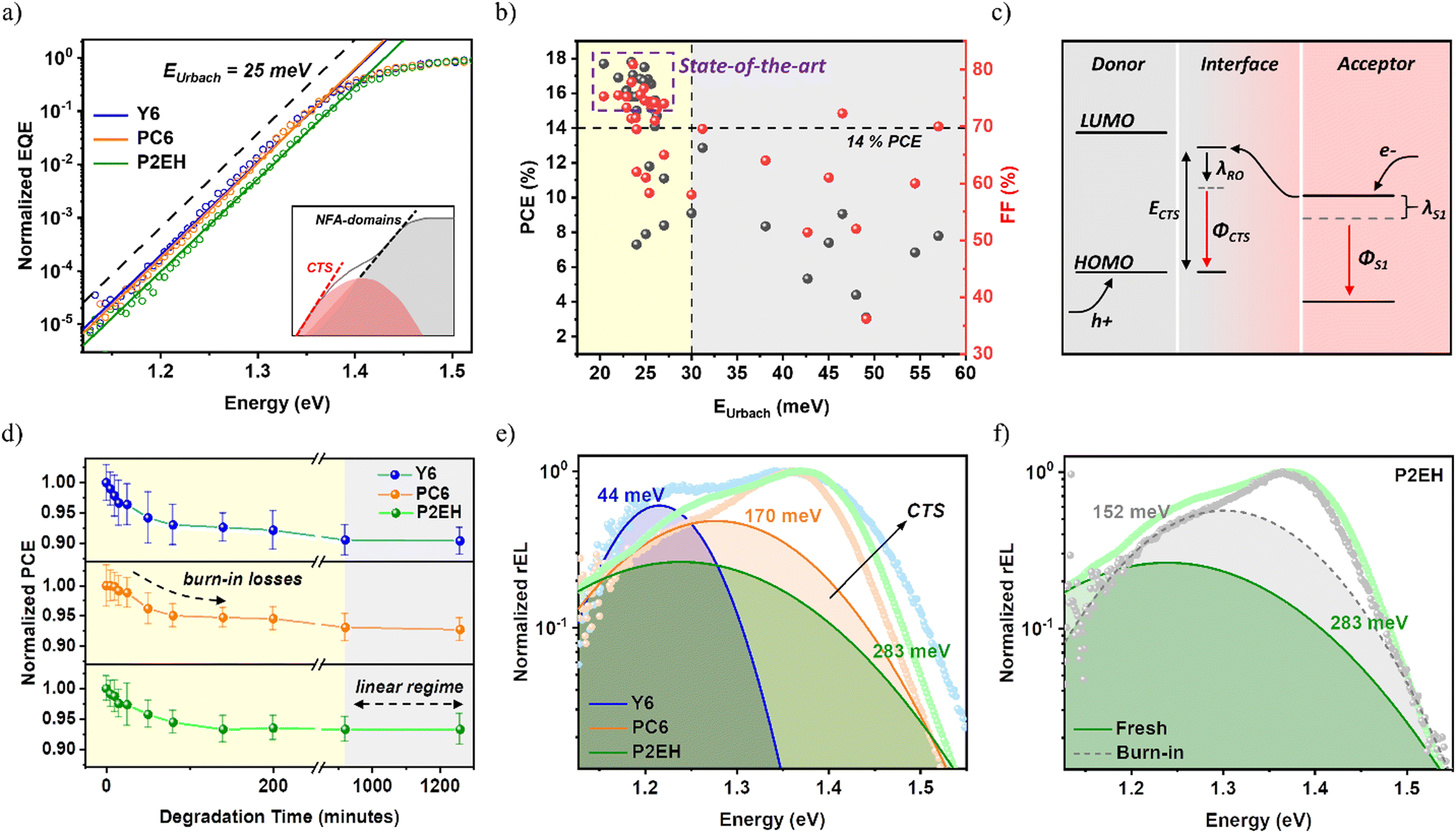 | ||
| Fig. 2 PM1:NFA bulk and D/A interface characteristics: EUrbach from the sensitively measured EQE (a), an overview of the recent literature reports for FFs relative to EUrbach (b), a simplified illustration of electroluminescence (EL) and the CTS energy level on account of the interface disorder (c), normalized PCE as a function of the thermal ageing time (d), and normalized reduced EL with the corresponding CTS fitting curves and λRO based on Marcus theory of electron transfer (e and f). The acceptor and cumulative fittings curves for reduced EL spectra are provided in Fig. S11 (ESI†). | ||
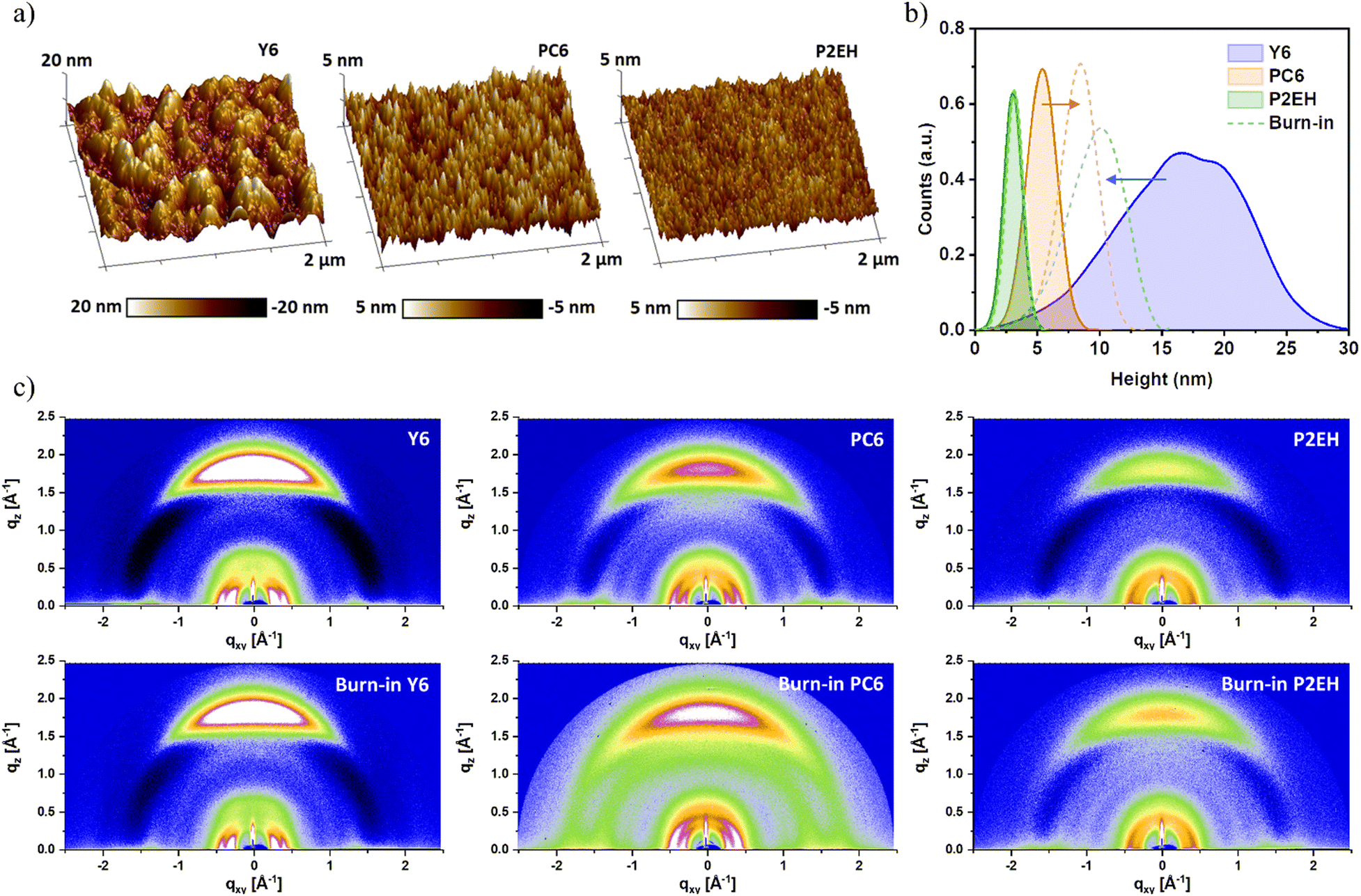 | ||
| Fig. 3 Nanomorphology characterization: scanning probe micrographs of PM1:NFA blends (a), surface height distribution of fresh and burn-in PM1:NFA blends (b), and GIWAXS of fresh and burn-in NFA films (c). | ||
Through the experimental Gaussian width of electroluminescence (EL) spectra comprising contributions from disorder and characterized by reorganization energy (λRO), the disorder of D/A interfaces can then be understood. Interestingly, the Y6-based device displays the lowest λRO indicating relatively more ordered D/A interfaces (Fig. 2e). At a glance, this is somehow confusing given that previous reports are generally suggesting high molecular order to benefit with efficient free charge transport. However, it must be re-emphasized that such a degree of order discussed in former studies generally represents only the acceptor (or donor) domains leaving the D/A interface ambiguously defined, as discussed above. Intuitively, the design optimization principles concerning the functionality of D/A interfaces are uniquely different from those understandings derived from purer domains. Meanwhile, the inductive effects from 2D phenylakyl side groups are found to increase the average permanent dipole moment of the NFA molecules, as observable from the more evident spectral deviation between the 1st and 2nd harmonics electroabsorption (EA) (Fig. S6–1, ESI†). This is also supported by density functional theory (DFT) calculations, as shown in Fig. S6–3 (ESI†). Consequently, the electron-withdrawing ability of PC6 and P2EH is enhanced through electrostatic effects and this is known to contribute to the driving force for exciton dissociation,50,51 as further elaborated in ESI† Notes S2. More importantly, PC6 and P2EH have reduced aggregation behavior enabling smaller domains, as also confirmed by earlier studies.17 This is consistent with their smoother surface obtained from scanning probe microscopy than those of Y6-based blends (Fig. 3a), thereby also promoting exciton dissociation. Likewise, as a consequence of smaller domains, the concomitantly increasing molecular interface area justifies the more pronounced higher energy PL feature (i.e., lower wavelength) in PC6 and P2EH (Fig. 1c). As a consequence, based on electrostatics and nanomorphology, PC6 and P2EH blends offer advantages for efficient free charge generation when compared to the Y6 blend. Although these cannot impart significant improvements to the EQE since the Y6 blend itself is already characterized by a nearly 100% probability of charge dissociation (Pdiss) under short-circuit conditions, as shown in Table S2 (ESI†). On the other hand, these are sought to enhance the resilience of the exciton dissociation efficiency to factors that impact geminate recombination. As will be further explained in later discussions, such greater resilience to geminate losses is fundamental to effectively optimizing the bulk-to-D/A interface transport energy landscape warranting >80% FFs without tradeoffs (i.e., even in simple binary systems).
Burn-in properties govern the long-term device operational performance critical for commercial viability.52,53 Herein, continuous thermal stress (85 °C) in the dark is applied to the samples to stimulate the burn-in degradation corresponding to the abrupt and substantial loss in most OSCs.52 Considering all the blends, it can be inferred that the burn-in occurs for the first 900 minutes while only gradual changes occur subsequently (Fig. 2d). It can be viewed from Fig. S4(d) (ESI†) that FFs are the principal cause of the burn-in efficiency losses, similar to other high-efficiency OSCs.49 Noticeably, both PC6 and P2EH-based devices exhibit enhanced stability compared to Y6 although both the donor molecule selected and the acceptor molecular backbone are the same which are known to be responsible for their similar ordered packing within purer domains. Consequently, the evolution of the D/A interface could be another important factor influencing their differences in burn-in stability. Meanwhile, scanning probe microscopy indicates that the P2EH blend has negligible domain size changes while those of Y6 and PC6 display significant (and opposite) domain size evolution (Fig. 3b), indicating thermally induced de-mixing and convolution, respectively. To better understand this, the GIWAXS of NFA neat films are also shown in Fig. 3c. Indeed, the burn-in causes Y6 molecules to exhibit reduced crystallinity which is indicative of reduced self-aggregation tendency while the trend is opposite for PC6 and P2EH. It can also be observed that P2EH displays better stability which justifies the marginal enlargement in its domain size features upon burn-in. These are also consistent with their FTIR spectra as a result of vibrational changes and with the evolution of electron mobility (Fig. S10 and S13, ESI†). Interestingly, the λRO of the burn-in P2EH blend decreases (Fig. 2f) which can be understood as molecular ordering limited to around the interface region. Considering that the domain size will influence the charge transport dynamics, the more stable burn-in P2EH blend is explored to further verify the specific impact of D/A interface characteristics. In contrast to drastic changes in FFs, the EUrbach and NFA absorption profiles of all the systems are well maintained (Fig. S7 and S8, ESI†), indicating that there is no formation of deep energetic trap states in the domains upon burn-in.
Charge transfer and exciton dissociation
Turning towards charge carrier properties relevant to the device performance, transient absorption spectroscopy (TAS) is performed to probe the donor hole polaron (PM1+) and NFA electron polaron (NFA−) dynamics (Fig. 4(a–e) and Fig. S16–S18, ESI†). It can be inferred that upon charge generation at D/A interfaces, the resulting polarons begin to dominate at 5 ps while the resulting NFA− photobleached at around 800 nm redshifts at 7 ns following electron migration to acceptor domains. This behavior is previously elaborated to emerge from charge relaxation to a lower energy level.21 Hence, such spectral shifts support the previously mentioned uphill energy landscape from NFA purer domains to D/A interfaces. Conversely, the PM1+ photobleach peak around 640 nm remains for the same time window which is also consistent with studies observing that polymer energy levels are less influenced by the disorder.21 Meanwhile, despite having stronger aggregation and a larger domain size for Y6-based blends, its diffusion-limited component of exciton dissociation generating free charges display a much faster rate than PC6 and P2EH blends at different excitation fluences <10 μJ cm−2 (Fig. 4e and Table S5, ESI†). In this process, singlet excitons will diffuse from purer domains to the D/A interface prior to charge transfer and subsequent charge generation. Because the relative domain size was unable to justify the PC6 and P2EH dissociation rates relative to Y6 blends, it then implies that their substantial differences in D/A interface disorder are playing a critical role. Thermodynamically speaking, the exciton to free charge generation kinetics will reflect the uphill bulk-to-interface transport energy landscape confirming the presence of smaller differential energy present in Y6 blends (i.e., having the least disordered D/A interface) imposing faster rates owing to the reduced dissociation energy barrier. Consistent with the previous discussions, the downshifted NFA transport energy levels at D/A interfaces following interface molecular ordering will minimize the bulk-to-interface energy difference responsible for the kinetics of exciton diffusion to the D/A interface which is prior to free charge generation. Likewise, a downshifted Mott–Schottky (MS) curve implying a lower energy requirement for Y6 dissociation (Fig. S17, ESI†) is observed. This also explains the increasing dissociation rate for the burn-in P2EH blend upon D/A interface ordering as indicated by the decreasing λRO values (Fig. 2f). On the other hand, the relative dissociation rates between PC6 and P2EH blends (i.e., both with higher λRO values) are well reflective of their corresponding nanomorphologies. Nevertheless, the EQEs of Y6 and PC6-based devices are comparably high (>80%) and only slightly reduced for P2EH (Fig. 4f), indicating that exciton dissociation among all the samples remains highly efficient.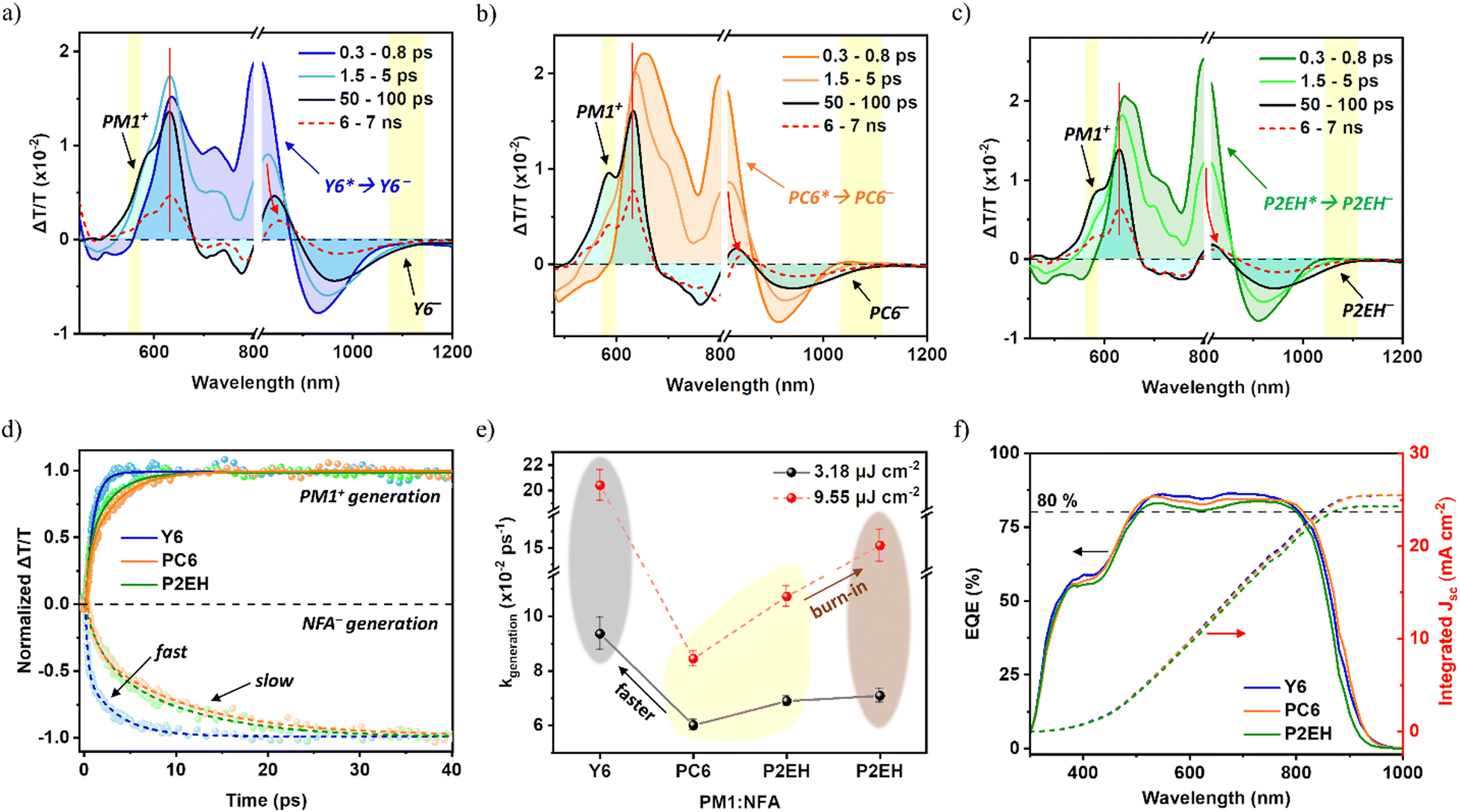 | ||
| Fig. 4 Singlet exciton (NFA*) dissociation and free charge generation in PM1:NFA blends: transient absorption spectral line cuts at various delay times upon selective acceptor excitation with an 800 nm pump laser (a–c), hole and electron polaron generation kinetics by probing PM1+ photobleaching and NFA− PIA (d), summary of the diffusion-limited free generation rate (1/τgeneration) at various fluences <10 μJ cm−2 (e), and device EQE at 1 sun illumination (f). For (a–c), the spectral line cuts shaded with color cyan at 50–100 ps dominantly represent polarons that remain even up to 7 ns while NFA singlets' intrinsic lifetimes are much shorter, <40 ps (Fig. S19, ESI†). The red arrows indicate the redshift acceptor polaron photobleaching upon charge relaxation. Note that 780–820 nm is the spectral overlap region considered to merge the visible probe and near-infrared probe data, and hence the magnitude of the photobleach shifts is not quantified. Furthermore, the regions highlighted with yellow are used to describe the hole and electron polaron dynamics, and these assignments are previously elaborated in great detail by Ohkita et al.21 Also, since the selected NFA- PIA spectral ranges are more exclusive to polarons, they are used to represent the free charge generation rates. Other details are provided in ESI† Notes S4. | ||
Polaron recombination and triplet formation
Longer-lived polaron population will start to rise upon singlet exciton dissociation, and as a consequence the TAS features after tens-to-hundreds of ps are similar regardless of whether the acceptor or donor is selectively excited (Fig. 5a). Fig. 5(b and c) and Fig. S16–S18 (ESI†) show that the decay of PM1+ photobleaching and NFA− photo-induced absorption (PIA) is used herein to understand the sub-nanosecond recombination behavior wherein free charges still have sufficient energy to reoccupy the CTSs and undergo bimolecular losses which are previously identified to dominate in NFA systems.30 Indeed, the recombination among all the blends is highly dependent on excitation fluence due to dominantly bimolecular mechanisms such that hole and electron polarons encounter within D/A interfaces prior to recombination. Meanwhile, the relative kinetics remain at different fluence <10 μJ cm−2.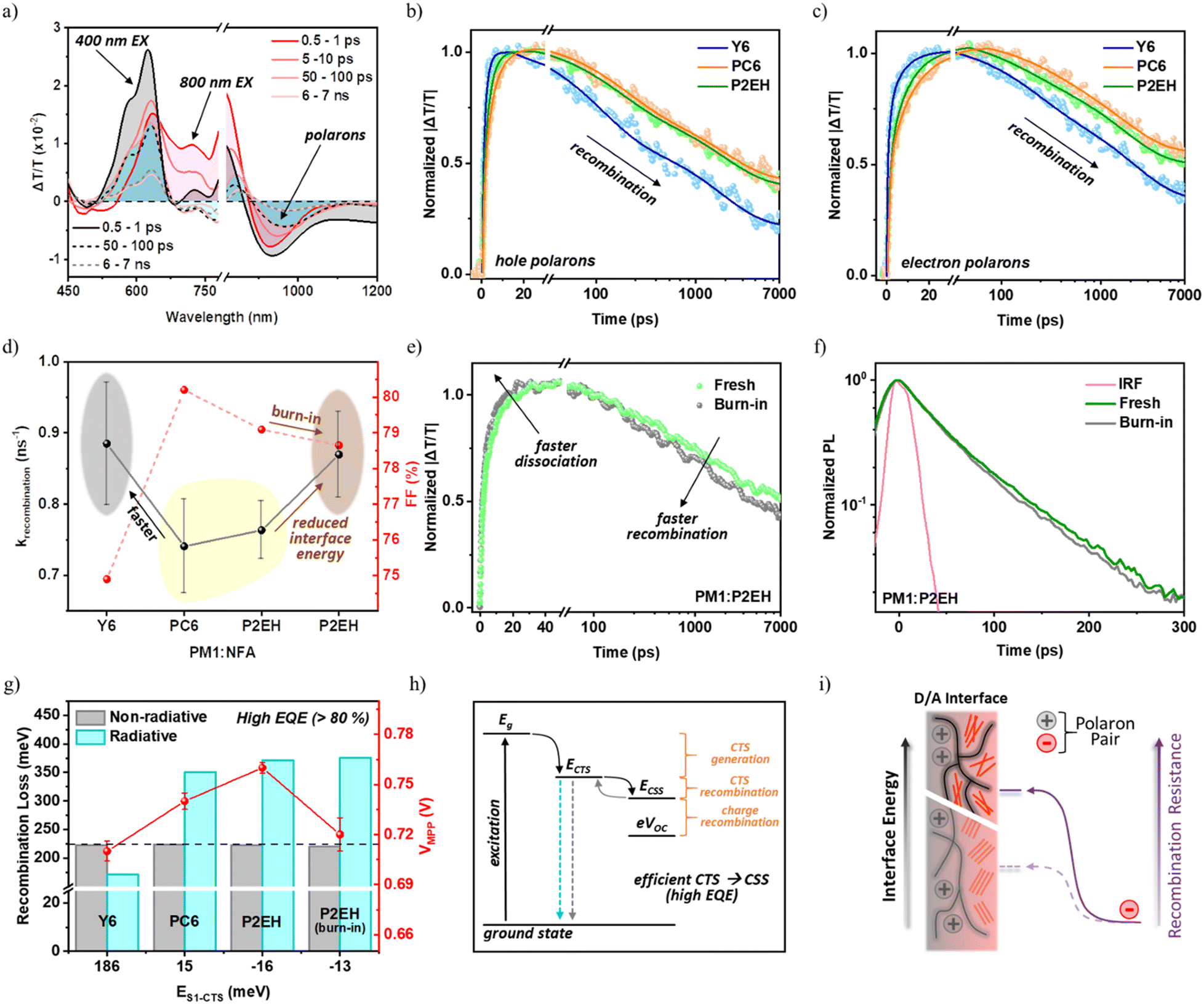 | ||
| Fig. 5 Polaron recombination and energy losses of PM1:NFA devices and films: 400 nm and 800 nm excitation TAS spectral line cuts are achieved at various delay times using PM1:Y6 as the representative system (a), hole polaron generation and recombination dynamics with the corresponding sum of exponential fittings (b), electron polaron generation and recombination dynamics with the corresponding sum of exponential fittings (c), a summary of average hole and electron polaron recombination rates (1/τrecombination) (d), polaron generation and recombination dynamics for fresh and burn-in P2EH blends (e), TRPL decay of fresh and burn-in P2EH blends and the IRF (f), a summary of VMPP and recombination energy losses with respect to singlet and CTS energy difference (g), schematic illustration of energy losses on the basis of the CTS energy level (h), and illustration of the influence of interface energy on recombination resistance (i). The hole and electron polaron dynamics at lower fluence show slower kinetics but similar trends (Fig. S18–2, ESI†). In (h), the cyan dashed arrow represents CTS radiative recombination while the grey dashed arrow represents nonradiative recombination, Eg is the optical gap, CTS is the charge transfer state, and CSS is the charge-separated state. The total radiative energy loss (i.e., CTS generation + recombination) is comparable among all the samples. | ||
It can be observed that both PC6 and P2EH blends display conservatively comparable polaron recombination rates with respect to their domain size (Fig. 5d and Table S6, ESI†). However, despite having the strongest aggregation causing a larger domain size which is known to improve charge transport, a much faster recombination rate from the Y6 blend is observed. This observation attests that understanding the characteristics of purer domains alone while leaving the functionality of D/A interfaces unclear will not be sufficient to completely justify the free charge recombination dynamics which is of paramount importance in advancing the performance of current state-of-the-art OSCs as they remain largely limited by low device FFs. It must be recalled that Y6 blends are previously identified to have the smallest differential energy between the bulk and D/A interface as a consequence of its relatively more ordered D/A interfaces (i.e., lowest λRO). Hence, the free charges in PC6 and P2EH blends are capable of having longer lifetimes owing to the greater thermodynamic energy barrier preventing the interface re-encounter of polarons. In simple words, the functionality of D/A interfaces is critical for achieving efficient charge transport as bimolecular free charge recombination involves the migration of oppositely charged polarons from purer domains to D/A interfaces. Consistently, the interface ordering in P2EH blends as a result of burn-in degradation imparts faster recombination rates (Fig. 5e). It can then be understood that the bulk-to-interface energy landscape regulates the polaron recombination resistance (illustrated in Fig. 5i) that impacts the recombination dynamics similar to diffusion-limited free charge generation. In fact, this energy landscape can be further verified by the excitons generated far away from the interface region thereby becoming highly diffusion-limited with longer dissociation times. Given that P2EH molecules tend to only slightly aggregate upon burn-in, slightly to negligibly slower rates can be expected. Yet, from the time-resolved photoluminescence (TRPL) with the instrument response function (IRF) > 25 ps, the dissociation even becomes sizably accelerated (Fig. 5f). This is justified by the decently lower D/A interface energy (Fig. 2f) reducing the uphill bulk-to-interface energy landscape thereby enabling faster dissociation rates.
Concerning energy losses (illustrated in Fig. 5h), it was previously found that there are trade-offs between radiative (Erad) and nonradiative (Enr) energy losses on account of CTS recombination and that ECTS influences Enr.49 This can be observed in several reports for different devices with various EQEs spanning from 10% up to 80%, thereby defining the preliminary design guidelines. Interestingly, regardless of ECTS, Enr is barely influenced and remains capped at around 200–225 meV under open-circuit conditions (Fig. 5g). This suggests that the reduction of the non-radiative loss on the basis of direct CTS recombination is already at its limit, as also previously inferred.54 This is because all the systems herein exhibit an exclusively high EQE (>80%) such that CTS dissociation is very efficient. Hence, a further reduction of Enr in current state-of-the-art OSC systems can be achieved through factors beyond geminate CTSs. On the other hand, ECTS remains critical for the radiative component on account of the Shockley–Queisser (SQ) limit defining the losses associated with CTS formation and recombination as influenced by the singlet intensity borrowing oscillator strength.49 To understand the influence of stronger polaron recombination resistance, the actual operational condition known as the maximum power point (MPP) is considered wherein charge extraction is present. Indeed, a higher photovoltage at the MPP (VMPP) is obtained from PC6 and P2EH devices, as shown in Fig. 5g.
Consistent observations can be also realized upon further exploration of the recombination behavior in view of triplet states. As shown in Fig. 6(a–c and g) and their corresponding kinetics in Fig. 6(d and f), the NFA triplet PIA starts to increase only after hundreds of ps wherein the PIA of most singlet excitons already decayed, indicating that they are bimolecularly formed through back transfer from the CSS. Indeed, more pronounced and accelerated triplet formation can be observed from Y6 and burn-in P2EH blends (Fig. 6h). This is consistent with the discussed stronger polaron recombination resistance of PC6 and P2EH blends induced by their higher D/A interface energy. As illustrated in Fig. 6i, it restricts the bimolecular encounter of free charges which dominantly generate spin-triplet CTSs that can ultimately be converted into typically non-extractable triplets. In fact, former studies have already provided in-depth mechanisms whereby the triplets account for about 90% of charge losses in other structurally similar and high-performing NFA OSCs.30 Thus, achieving remarkable FFs can be delegated to the suppression of triplet formation via back charge transfer bimolecular recombination. Meanwhile, the faster charge generation kinetics of Y6 and burn-in P2EH blends, indicative of the singlet PIA lifetime, directly reflect those previously discussed from the hole and electron polaron dynamics.
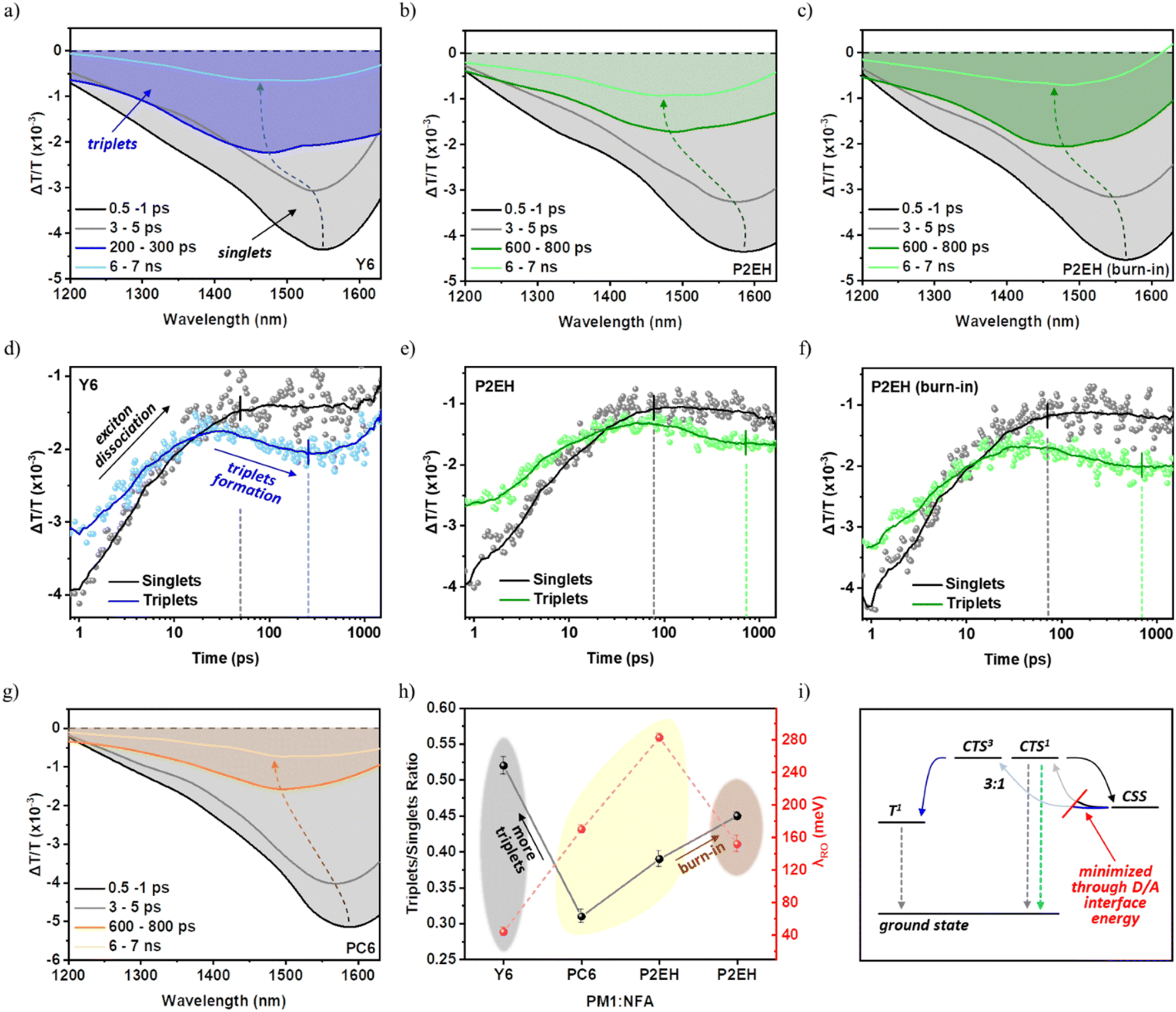 | ||
| Fig. 6 NFA bimolecular triplet formation in PM1:NFA blends: TAS spectral line cuts at 1200–1630 nm range (a–c and g), the kinetics of NFA singlet excitons (black) and triplets (colored) (d–f), a summary of the maximum triplet state population to the initial singlet exciton ratio based on their PIA intensities (h), and schematic illustration of free charge recombination with triplet formation (i). Herein, CTS1 is the spin-singlet CTS and CTS3 is the spin-triplet CTS. The kinetics of NFA singlet excitons are extracted from 1550–1630 nm while 1400–1500 nm for NFA triplets (T1). These assignments have already been previously elaborated in great detail.21,30 For (a–c), the delay time chosen to represent the triplets (i.e., 200–300 ps for the Y6 blend while 600–800 ps for PC6 and P2EH blends) corresponds to the maximum triplet population as indicated by colored vertical lines in (d–f) whereby the corresponding intensity and the initial NFA singlet PIA intensity are used to define the ratio in (h). The black vertical lines indicate the lifetime of singlet exciton PIA. For (i), the gray dashed arrows represent non-radiative recombination while the cyan dashed arrow represents radiative recombination. | ||
Discussion
The above results reveal that despite the substantially sizeable FF variability, a single absorption exponential tail with a low EUrbach value (i.e., comparably within 25 meV) and highly ordered NFA self-assemblies are common among the high-efficiency OSC systems considered herein. Similar to most measurement cases reported, the D/A interface absorption features cannot be distinguished from the bulk. Interestingly, even though the D/A interface emissions show decent evolution upon burn-in, each device sustains such an EUrbach value. Hence, these findings ascertain that the identified EUrbach value can only represent the energetic order of NFA-rich domains, leaving the D/A interface property undefined. This can be due to significant efforts done in the past few years to optimize the characteristics of purer domains through synthetic molecular designs and/or device engineering and therefore state-of-the-art donor and acceptor molecules tend to benefit from the low density of energetic trap states and the highly crystalline packing in their self-assemblies. As a consequence, the properties of purer domains may no longer be able to specifically define the unique characteristics influencing the differences in the device metrics among those recent and explicitly high-efficiency materials. On the other hand, despite the D/A interfaces being known to exist throughout the photoactive layer, there remain no clear strategies for D/A interface optimization more specifically when considering its simultaneous impact in free charge generation and recombination which practically defines the PCE. The co-existing dependence of different charge carrier events together with the difficulties of characterizing such D/A interfaces warrants the highly complex mechanism interpreting the property–functionality interplay of D/A interfaces. Intuitively, there is an urgent need to provide an in-depth understanding of D/A interfaces to continue advancing the development of OSCs.Herein, the broadened CTS emission linewidth causing larger λRO values for PC6 and P2EH-based blends indicates more disordered D/A interfaces when compared to Y6 blends (Fig. 2d). This can be understood as a consequence of the outer side chain variability between the NFA molecules. From the molecular perspective, the fused core and intramolecular attractions (e.g., S–O) in the considered NFAs will induce high backbone planarity and π–π staking in their self-assemblies thereby associating them with their tight-packing reducing the system energy.3,55 As a consequence, the side chains that protrude out from the molecular plane will dominantly govern their intermolecular assembly.3 Hence, it is not surprising that side chain substitution with bulkier side groups will give rise to more random molecular configurations, particularly at D/A interfaces due to dissimilar donor and acceptor molecular geometries.
Given that the NFA transport energy levels tend to increase from the ordered to a more disordered phase, the uphill energy landscape from NFA purer domains to D/A interfaces can be established. This is well evident from the consistent correlations between the different charge carrier mechanisms investigated. More specifically, the photobleached electron polarons red-shift upon relaxation from D/A interfaces to acceptor domains and the slower kinetics in the hole polaron generation, electron polaron generation, and singlet dissociation in PC6 and P2EH-based blends (i.e., with more disordered D/A interfaces) as limited by the exciton diffusion process. Kinetically speaking, the disorder-induced larger differential energy between the bulk and the D/A interface will impose slower diffusion of photogenerated singlet excitons from purer domains to the interfaces, thereby imparting slower diffusion-limited rates. It is also suggestive from the PL blueshift of blend films owing to the presence of disordered D/A interfaces and from the relative MS curves representing the energy barrier for charge generation. Indeed, these disorder-induced modulations of the NFA transport energy levels are also recently noted,21 although there is previously no clear understanding of how it can impact the free charge recombination.
It must be noted that the bimolecular losses are governed by the Coulombic attraction of oppositely charge states (i.e., electrons and holes) in close proximity and require migration to D/A interfaces prior to recombination which reduces the device photocurrent output and FFs. Considering the uphill bulk-to-D/A interface energy landscape, the bimolecular recombination rates are demonstrated here to be substantially reduced for PC6 and P2EH blends owing to their more disordered nature of D/A interfaces. In simple words, the interface energy landscape can prompt stronger polaron recombination resistance, thereby substantially enhancing the charge transport and reflecting FFs > 80% and higher attainable VMPP. Such behaviors are evident considering both the recombination dynamics of free charges and the generation of nongeminate triplet states following the formation of spin-triplet CTSs from back charge transfer at D/A interfaces.
However, since the D/A interface is also of prime importance for the singlet exciton dissociation, then diffusion-limited exciton dissociation efficiency can be compensated beyond certain thresholds for the differential energy between the D/A interface and NFA domains. This can impart higher geminate losses leading to reduced EQE and PLQ, as reflected in P2EH-based devices having the largest λRO values. This one of the reasons why most fullerene-based OSCs (i.e., typically having disordered interfaces) generally result in poorer exciton dissociation efficiency than NFAs. Different donor–acceptor combinations may have their characteristic tolerance, which is illustrated here to depend on factors facilitating the exciton dissociation, including but not limited to the molecular electrostatics and nanomorphology. Thanks to the highly efficient exciton dissociation characteristics of many Y-series NFAs,56 such tolerance is demonstrated herein to be high enough wherein substantial PCE improvements are realizable. More specifically, the enhanced electrostatics of PC6 endows it with a more suitable molecular aggregation behavior than that of Y6 permitting >80% FFs without EQE tradeoff. Further discussions can be found in ESI† Notes S5.
Meanwhile, the improved morphological stability of the P2EH-based device also agrees with its relative D/A interface disorder. It is known that the low-frequency vibrational modes increase with more entangled assembly which restricts molecular motions and renders enhanced morphological durability.57 Similarly, owing to the compensating effects of PC6 blend thermal aggregation and slight interface energy reduction, its performance stability is also somehow improved than Y6. In this regard, the D/A interface also plays a vital role in determining the device's long-term operational performance while simply understanding the stability of individual blend components may not be sufficient. By taking advantage of the more stable nanomorphology of P2EH blends, the impact of interface disorder in free charge generation and recombination on the basis of the transport energy landscape is further verified. Hence, it is evident that the interplay between the D/A interface property and functionality is a characteristic of molecular interfaces rather than the selected materials. As a new design principle with direct physical meanings, this will foster developments to achieve the full performance potential and stability of binary systems, a critical foundation in advancing the fabrication of simpler and cost-effective devices.
Conclusion
In summary, this work unveils the functionality of D/A interfaces in simultaneously regulating both free charge generation and recombination, which determines the device efficiency. The device FFs following free charge recombination have been the current primary bottleneck in improving the OSC performance, and hence, previous studies mainly focused on optimizing the charge transport based on the characteristics of purer domains. Yet, understanding the influence of D/A interfaces relevant to free charge recombination is lagging, thereby preventing the complete realization of the optimum performance potential in simple binary systems. This study illustrated that the energy of the D/A interface relative to NFA-rich domains will function as the basis of polaron recombination resistance administering the efficiency of free charge transport. This is in view of the disorder-induced uphill energy landscape for free electrons at NFA domains to encounter free holes at D/A interfaces, as illustrated in Fig. 7. Hence, by thermodynamically restricting the back charge transfer which upon spin-statistics will dominantly create non-geminate spin-triplet CTSs, the uphill bulk-to-D/A interface energy landscape also serves as a feasible approach to substantially suppress the formation of triplet states which are typically non-extractable reducing the photocurrent output. Consequently, state-of-the-art PCEs above 18% on account of remarkable FFs of over 80% are revealed to remain accessible even with simple binary component BHJ devices. Also, through enhanced electrostatic effects and molecular interactions defining the solid-state nanomorphology as imposed by bulky 2D phenyl outer side groups, the diffusion-limited geminate recombination of singlet excitons regulated by the same interface energy landscape is demonstrated to be resilient with increasing interfacial energy, thereby diminishing potential tradeoffs. In simple words, increasing the D/A interface disorder while maintaining high molecular order in purer domains will impose a larger energy barrier mitigating the bimolecular recombination of free charges, thereby enhancing charge transport and FFs. However, the extent of how disordered the D/A interface can be without tradeoffs in singlet exciton dissociation is identified to be dependent on the NFA permanent dipole moment and domain size which are driving the charge dissociation. Herein, the outer side chain modifications of Y6 with bulkier 2D phenylalkyl side groups are taken advantage of to improve the NFA molecular electrostatics and reduce the excessive self-aggregation tendency. Meanwhile, the D/A interface is also uncovered to substantially impact the burn-in losses owing to its strong influence on FFs, enlightening future stability enhancement studies. Overall, the direct physical meanings of the D/A interface's property–functionality interplay will serve as a fundamental basis to realize the true maximum potential of material systems while ensuring the simplicity of device fabrications. Together with the synthetic flexibility of organic molecules, this will revolutionize future device optimizations and direct more cost-effective strategies from the countless number of approaches, thereby fostering marketplace standard realization (Fig. S20, ESI†).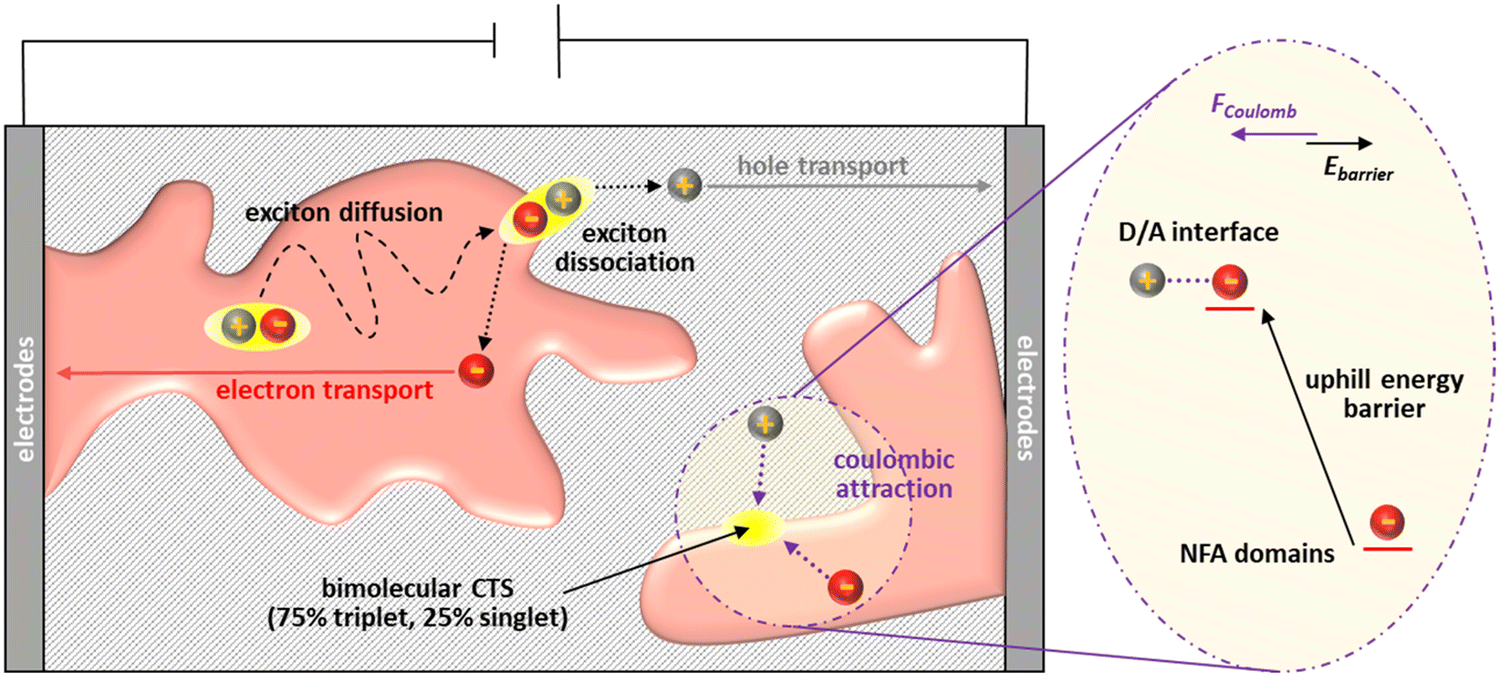 | ||
| Fig. 7 Schematic illustration of free charge generation to the collection with the influence of the uphill bulk-to-D/A interface transport energy landscape as imposed by interface disorder. | ||
Methods
Details are provided in the ESI.†Data availability
Available upon reasonable request to the corresponding authors.Author contributions
Conceptualization: T. A. D. P., R. M., and J. W.; methodology: T. A. D. P. and R. M.; investigation: T. A. D. P., R. M., Z. X., Q. W., J. I. K., R. M. Y., Y. H., S. A. G., X. Z., and F. L. N.; resources: R. M., Z. J., D. F. S., M. R. W., J. W., H. C., H. Y., K. S. W., G. L., M. L., and J. W.; writing – original draft: T. A. D. P.; writing – review & editing: R. M., J. I. K., G. L., M. L., and J. W.; supervision: G. L., M. L., and J. W.; and funding acquisition: G. L., M. L., and J. W.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. Wu thanks the Guangdong government and the Guangzhou government for funding (2021QN02C110) and the Guangzhou Municipal Science and Technology Project (No. 2023A03J0097 and 2023A03J0003). M. Li acknowledges the financial support from the Shenzhen Science, Technology and Innovation Commission (JCYJ20210324131806018) and the Research Grant Council of Hong Kong (25301522). G. Li acknowledges the support from the Research Grants Council of Hong Kong (Project No. 15320216, 15221320, C5037-18G), the RGC Senior Research Fellowship Scheme (SRFS2122-5S04), the National Natural Science Foundation of China (51961165102), the Shenzhen Science and Technology Innovation Commission (JCYJ20200109105003940, SGDX2019081623220944), the Hong Kong Polytechnic University Internal Research Funds: Sir Sze-yuen Chung Endowed Professorship Fund (8-8480), RISE (1-CDA5), 1-W15V, and the Guangdong-Hong Kong-Macao Joint Laboratory for Photonic-Thermal-Electrical Energy Materials and Devices (GDSTC No. 2019B121205001). This work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under award no. DE-FG02-99ER14999 (M. R. W.). R. Ma is thankful for the support through the PolyU Distinguished Postdoc Fellowship. T. A. Dela Peña is also thankful for the support from HKUST Materials Characterization and Preparation Facility (MCPF) Clear Water Bay (CWB) and Guangzhou (GZ).References
- F. Liu, et al., Organic Solar Cells with 18% Efficiency Enabled by an Alloy Acceptor: A Two-in-One Strategy, Adv. Mater., 2021, 33, 2100830 CrossRef CAS PubMed.
- Q. Liu, et al., 18% Efficiency organic solar cells, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- C. Li, et al., Non-fullerene acceptors with branched side chains and improved molecular packing to exceed 18% efficiency in organic solar cells, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- A. Shang, et al., Over 18% binary organic solar cells enabled by isomerization of non-fullerene acceptors with alkylthiophene side chains, Sci. China: Chem., 2022, 1–9 Search PubMed.
- X. Wang, et al., Hammer throw-liked hybrid cyclic and alkyl chains: A new side chain engineering for over 18% efficiency organic solar cells, Nano Energy, 2022, 101, 107538 CrossRef CAS.
- Y. Cui, et al., Single-junction organic photovoltaic cells with approaching 18% efficiency, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- J. Huang, et al., Tandem Self-Powered Flexible Electrochromic Energy Supplier for Sustainable All-Day Operations, Adv. Energy Mater., 2022, 12, 2201042, DOI:10.1002/aenm.202201042.
- R. Ma, et al., In situ and ex situ investigations on ternary strategy and co-solvent effects towards high-efficiency organic solar cells, Energy Environ. Sci., 2022 Search PubMed.
- J. Yuan, et al., Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- J. Liu, et al., Fast charge separation in a non-fullerene organic solar cell with a small driving force, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- S. Bao, et al., Volatilizable solid additive-assisted treatment enables organic solar cells with efficiency over 18.8% and fill factor exceeding 80%, Adv. Mater., 2021, 33, 2105301 CrossRef CAS PubMed.
- Z. Zheng, et al., A highly efficient non-fullerene organic solar cell with a fill factor over 0.80 enabled by a fine-tuned hole-transporting layer, Adv. Mater., 2018, 30, 1801801 CrossRef PubMed.
- F. Bai, et al., A highly crystalline non-fullerene acceptor enabling efficient indoor organic photovoltaics with high EQE and fill factor, Joule, 2021, 5, 1231–1245 CrossRef CAS.
- L. Zhu, et al., Single-junction organic solar cells with over 19% efficiency enabled by a refined double-fibril network morphology, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- N. Gasparini, et al., Designing ternary blend bulk heterojunction solar cells with reduced carrier recombination and a fill factor of 77%, Nat. Energy, 2016, 1, 1–9 Search PubMed.
- X. Zhang, et al., High fill factor organic solar cells with increased dielectric constant and molecular packing density, Joule, 2022, 6, 444–457, DOI:10.1016/j.Joule.2022.01.006.
- J. Zhang, et al., Alkyl-Chain Branching of Non-Fullerene Acceptors Flanking Conjugated Side Groups toward Highly Efficient Organic Solar Cells, Adv. Energy Mater., 2021, 11, 2102596 CrossRef CAS.
- J. Wu, et al., Random terpolymer based on thiophene-thiazolothiazole unit enabling efficient non-fullerene organic solar cells, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- G. Zhang, et al., Naphthalenothiophene imide-based polymer exhibiting over 17% efficiency, Joule, 2021, 5, 931–944 CrossRef CAS.
- S. Karuthedath, et al., Intrinsic efficiency limits in low-bandgap non-fullerene acceptor organic solar cells, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- S.-i Natsuda, et al., Cascaded energy landscape as a key driver for slow yet efficient charge separation with small energy offset in organic solar cells, Energy Environ. Sci., 2022, 15, 1545–1555 RSC.
- F. Zhao, C. Wang and X. Zhan, Morphology control in organic solar cells, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- L. Perdigón-Toro, et al., Barrierless free charge generation in the high-performance PM6: Y6 bulk heterojunction non-fullerene solar cell, Adv. Mater., 2020, 32, 1906763 CrossRef PubMed.
- L. Zuo, et al., Dilution effect for highly efficient multiple-component organic solar cells, Nat. Nanotechnol., 2022, 17, 53–60 CrossRef CAS PubMed.
- Y. Zeng, et al., Exploring the charge dynamics and energy loss in ternary organic solar cells with a fill factor exceeding 80%, Adv. Energy Mater., 2021, 11, 2101338 CrossRef CAS.
- Y. Wang, et al., Recent progress and challenges toward highly stable nonfullerene acceptor-based organic solar cells, Adv. Energy Mater., 2021, 11, 2003002 CrossRef CAS.
- V. Coropceanu, X.-K. Chen, T. Wang, Z. Zheng and J.-L. Brédas, Charge-transfer electronic states in organic solar cells, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- S. M. Menke, et al., Order enables efficient electron-hole separation at an organic heterojunction with a small energy loss, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- L. Perdigon-Toro, et al., Excitons dominate the emission from PM6: Y6 solar cells, but this does not help the open-circuit voltage of the device, ACS Energy Lett., 2021, 6, 557–564 CrossRef CAS.
- A. J. Gillett, et al., The role of charge recombination to triplet excitons in organic solar cells, Nature, 2021, 597, 666–671 CrossRef CAS PubMed.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- H. Chen, et al., A guest-assisted molecular-organization approach for >17% efficiency organic solar cells using environmentally friendly solvents, Nat. Energy, 2021, 6, 1045–1053 CrossRef CAS.
- S. Sweetnam, et al., Characterization of the Polymer Energy Landscape in Polymer:Fullerene Bulk Heterojunctions with Pure and Mixed Phases, J. Am. Chem. Soc., 2014, 136, 14078–14088, DOI:10.1021/ja505463r.
- A. Armin, et al., Limitations of charge transfer state parameterization using photovoltaic external quantum efficiency, Adv. Energy Mater., 2020, 10, 2001828 CrossRef CAS.
- F. D. Eisner, et al., Hybridization of local exciton and charge-transfer states reduces nonradiative voltage losses in organic solar cells, J. Am. Chem. Soc., 2019, 141, 6362–6374 CrossRef CAS PubMed.
- N. Jain, et al., Interfacial disorder in efficient polymer solar cells: the impact of donor molecular structure and solvent additives, J. Mater. Chem. A, 2017, 5, 24749–24757 RSC.
- A. T. Kleinschmidt and D. J. Lipomi, Unfavourable interactions enable stability, Nat. Mater., 2021, 20, 447–448 CrossRef CAS PubMed.
- M. Ghasemi, et al., A molecular interaction–diffusion framework for predicting organic solar cell stability, Nat. Mater., 2021, 20, 525–532 CrossRef CAS PubMed.
- M. Ghasemi, et al., Delineation of thermodynamic and kinetic factors that control stability in non-fullerene organic solar cells, Joule, 2019, 3, 1328–1348 CrossRef CAS.
- L. Hong, et al., Simultaneous Improvement of Efficiency and Stability of Organic Photovoltaic Cells by using a Cross-Linkable Fullerene Derivative, Small, 2021, 17, 2101133 CrossRef CAS PubMed.
- C. Yan, J. Qin, Y. Wang, G. Li and P. Cheng, Emerging Strategies toward Mechanically Robust Organic Photovoltaics: Focus on Active Layer, Adv. Energy Mater., 2022, 12, 2201087, DOI:10.1002/aenm.202201087.
- H. Fu, et al., High efficiency (15.8%) all-polymer solar cells enabled by a regioregular narrow bandgap polymer acceptor, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- D. Kroh, et al., Identifying the Signatures of Intermolecular Interactions in Blends of PM6 with Y6 and N4 Using Absorption Spectroscopy, Adv. Funct. Mater., 2022, 32, 2205711 CrossRef CAS.
- S. Liu, et al., High-efficiency organic solar cells with low non-radiative recombination loss and low energetic disorder, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- C. Zhang, et al., Unraveling Urbach Tail Effects in High-Performance Organic Photovoltaics: Dynamic vs Static Disorder, ACS Energy Lett., 2022, 7, 1971–1979 CrossRef CAS.
- Y. Zhang, et al., An electron acceptor analogue for lowering trap density in organic solar cells, Adv. Mater., 2021, 33, 2008134 CrossRef CAS PubMed.
- G. Kupgan, X.-K. Chen and J.-L. Bredas, Molecular packing of non-fullerene acceptors for organic solar cells: distinctive local morphology in Y6 vs. ITIC derivatives, Mater. Today Adv., 2021, 11, 100154 CrossRef CAS.
- Z. Luo, et al., Asymmetric side-chain substitution enables a 3D network acceptor with hydrogen bond assisted crystal packing and enhanced electronic coupling for efficient organic solar cells, Energy Environ. Sci., 2022, 15, 4601–4611 RSC.
- T. A. Dela Peña, et al., Understanding the Charge Transfer State and Energy Loss Trade-offs in Non-fullerene-Based Organic Solar Cells, ACS Energy Lett., 2021, 6, 3408–3416 CrossRef.
- J. Li, et al., Influence of Large Steric Hinderance Substituent Position on Conformation and Charge Transfer Process for Non-Fused Ring Acceptors, Small Methods, 2022, 6, 2200007 CrossRef CAS PubMed.
- Y. Xu, H. Yao, L. Ma, J. Wang and J. Hou, Efficient charge generation at low energy losses in organic solar cells: a key issues review, Rep. Prog. Phys., 2020, 83, 082601 CrossRef CAS PubMed.
- J. Kong, et al., Long-term stable polymer solar cells with significantly reduced burn-in loss, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- Q. Burlingame, M. Ball and Y.-L. Loo, It's time to focus on organic solar cell stability, Nat. Energy, 2020, 5, 947–949 CrossRef.
- X.-K. Chen, et al., A unified description of non-radiative voltage losses in organic solar cells, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- J. Xu, et al., The Molecular Ordering and Double-Channel Carrier Generation of Nonfullerene Photovoltaics within Multi-Length-Scale Morphology, Adv. Mater., 2022, 34, 2108317 CrossRef CAS PubMed.
- F. Eisner and J. Nelson, Barrierless charge generation at non-fullerene organic heterojunctions comes at a cost, Joule, 2021, 5, 1319–1322 CrossRef.
- J. W. Lee, et al., Efficient, Thermally Stable, and Mechanically Robust All-Polymer Solar Cells Consisting of the Same Benzodithiophene Unit-Based Polymer Acceptor and Donor with High Molecular Compatibility, Adv. Energy Mater., 2021, 11, 2003367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, supplementary figures, supplementary tables, supplementary discussions, and supplementary notes. See DOI: https://doi.org/10.1039/d3ee01427d |
| This journal is © The Royal Society of Chemistry 2023 |