Waste materials – catalytic opportunities: an overview of the application of large scale waste materials as resources for catalytic applications
M.
Balakrishnan
a,
V. S.
Batra
a,
J. S. J.
Hargreaves
*b and
I. D.
Pulford
b
aCentre for Energy and Environment, TERI University, Darbari Seth Block, Habitat Place, Lodhi Road, New Delhi, 110003, India
bWestCHEM, School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: justinh@chem.gla.ac.uk
First published on 10th December 2010
Abstract
In this overview, we present examples of the use of high volume waste materials in catalysis or for catalyst synthesis. Waste materials derived from both industrial and biological sources have attracted interest and this is briefly summarized. The materials described include red mud, aluminium dross, fly ash, blast furnace slag, rice husk and various kinds of shell.
Introduction
Large-scale waste products afford opportunities in terms of catalysis. Whilst the use of such wastes in this context, by definition, does not alleviate disposal problems, which are significant and also costly in some cases, high volume waste can provide an inexpensive route to active systems. A survey of the literature demonstrates that this area is starting to attract more attention. This interest is primarily driven by a combination of economic and environmental considerations. A number of different approaches to the utilization of waste materials in this context has been reported: (i) their direct application as active materials, (ii) their direct use as pre-catalysts (i.e. materials which undergo transformation to active phases under reaction conditions), (iii) their modification to yield catalytically active phases and (iv) their use as precursors for the synthesis of active catalysts. In the sections which follow, we will present selected examples of large scale waste materials being used in these ways to provide an overview of some of the current activity in this area. The waste materials we describe can be broadly divided into those generated by large scale industrial processes (e.g. red mud, aluminium dross) and those which originate from biological sources (e.g. chicken egg shells and rice husks) and so we have sub-divided them in this manner. In terms of the latter class of materials, we have concentrated specifically on wastes rather than the development of renewable resources for catalytic applications. The reported production rates for a number of the industrial and biological wastes described are reproduced in Table 1.| Waste material | Reported production rate | Reference |
|---|---|---|
| Red mud | >70 million tonnes globally per annum | 2 |
| Aluminium dross | 5 million tonnes globally per annum | 22 |
| Fly ash | 430 million tonnes globally in 2003 | 26 |
| Slag from iron manufacture | 240–290 million tons globally in 2008 | 51 |
| Chicken egg shells | 4 million tonnes annually in China alone | 53 |
| Rice husk ash | About 70 million tonnes globally per annum | 75 |
Large-scale industrial wastes
(i) Red mud
Red mud is the large scale waste product of the Bayer process for alumina production. In this process, bauxite ore is digested in hot caustic soda to extract most of the aluminium content which is subsequently processed. The insoluble residues of the digestion procedure, which are filtered off, constitute red mud, which is a finely divided highly alkaline heterogeneous mixture of different oxidic phases. The exact composition of red mud reflects that of the bauxite ore from which it is generated and there can be significant geographical and temporal variations. An example of the composition of red mud from an Indian source is presented in Table 2.1 The current global production of red mud is around 70 million tonnes per annum2 and it is mostly disposed of by land dumping. Since it is such an abundant resource, there has been much interest in its valorization.3–5 In terms of catalysis, the primary interest has centred upon the relatively large iron content of red muds, which is expected to impart a wide range of interesting catalytic behaviour, including oxidation and hydrocarbon activation activity. Whilst red mud has been applied directly as received to catalytic reactions both as an active component and a catalyst support phase, activation procedures which are designed to increase its activity by enhancement of surface area and/or minimization of the effects of the variability of its composition, are possible. One of the most commonly applied activation procedures is that reported by Pratt and Christoverson in which initial acid dissolution is followed by base precipitation, filtering and calcination yielding a material with significantly enhanced surface area.6 Red mud has been applied to a very wide range of catalytic reactions including hydrogenation,6,7 coal liquefaction,8 hydrodechlorination,9 exhaust gas treatment10 and the conversion of polymers into fuel oil11 as has been recently reviewed elsewhere.5 Recent developments have detailed its direct application as a pre-catalyst to various processes. It has been demonstrated to be active for the cracking of methane to yield hydrogen and graphitic carbon.1,12 In the course of this process, the oxidic iron components of the red mud are reduced generating iron metal and iron carbide which imparts magnetic properties that may facilitate application of the resultant carbon in, for example, water treatment through ease of separation. In this way, it is possible to valorize two waste products (methane from land fill gas as well as red mud) to generate two valuable products (hydrogen and magnetically separable carbon). Fig. 1 presents the hydrogen production profiles for three Indian red muds of different origins which illustrate the degree of variation in reaction behaviour between the different samples. Also shown is post-reaction HRTEM, which shows the nature of the graphite deposited (Fig. 2). The form of the carbon deposited has been reported to be a function of the hydrocarbon employed.12 For example, butane yields multiwalled carbon nanotubes (shown in Fig. 3). It has additionally been reported that the application of red mud in a fluidized bed can be used for large scale multiwalled carbon nanotube production from diluted ethylene/hydrogen.13 Subsequently, similar forms of carbon to those reported in the previous studies have also been observed to be produced upon reduction of red mud using ethanol.14 This was enacted to produce magnetically separable forms of carbon, which were applied to the demulsification of water–oil interfaces. A recent patent has disclosed the possibility of the reduction of red mud using hydrocarbons, generating syn gas which can then undergo the Fischer–Tropsch reaction using the magnetite resulting from the reduction process to yield a range of valuable products.15Thermal decomposition of acetic and formic acids has been accomplished on red mud.16 This reduces acidity and produces a range of higher hydrocarbons by various secondary reactions. This is of interest for the upgrading of bio-oil produced by fast pyrolysis of ligno-cellulosic biomass where the acidic components prevent its direct application as a fuel. It was shown that the hydrogen necessary for some conversions could be generated in situ from formic acidvia the formation of CO and H2O and a subsequent reverse water-gas shift reaction, as well as being supplied externally. The resultant phase was observed to be a non-alkaline magnetic material comprising magnetite and iron metal. Red mud has been reported to be an effective catalyst for the pyrolysis of hazelnut shell, a high lignin content agricultural waste of which >1 million tons were produced in 2008.17 It was reported that a wide range of pyrolysis oils were formed. However, a tin oxide catalyst was found to be more effective. The same group had previously reported the activity of red mud from the co-pyrolysis of lignite with polyethylene, with the latter functioning as a hydrogen donor medium.11,18 Although not strictly of catalytic relevance, it is of interest to note that the controlled reduction of red mud has been reported to generate heterogeneous Fenton systems based upon Fe/Fe3O4 composites.19 These composites are proposed to be active by the decomposition of H2O2 on the surface Fe2+ of Fe3O4. The presence of Fe metal was argued to be important in the production of Fe2+ species by the supply of electrons to Fe3+ species.| a Loss on ignition. b Metal components only, solid-liquid ratio: 5 g/200 ml aqua regia, residue 45%. c Composition of acid extracted silica. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Red mud 1 | SiO2 | Al2O3 | Fe2O3 | TiO2 | CaO | MgO | Na2O | K2O | P2O5 | MnO | LOI a |
| 14.60 | 23.51 | 36.79 | 0.74 | 1.18 | 0.07 | 6.08 | 0.02 | 0.15 | 0.12 | 16.47% | |
| Aluminium drossb21 | Al | Mg | Si | Pb | Zn | Ca | Na | K | Fe | Ti | Mn |
| 89.1 | 3.8 | 0.2 | 0.2 | 0.7 | 1.3 | 0.7 | 0.8 | 2.5 | ≪0.1 | 0.6 | |
| Fly ash 25 | SiO2 | Al2O3 | CaO | Fe2O3 | MgO | K2O | TiO2 | P2O5 | MnO | ||
| 37.04 | 15.74 | 16.12 | 7.23 | 2.59 | 0.91 | 0.76 | 0.29 | 0.20 | |||
| Raw blast furnace slag 51 | CaO | SiO2 | Al2O3 | MgO | Fe2O3 | TiO2 | MnO | ||||
| 40.09 | 34.58 | 14.78 | 5.29 | 1.53 | 0.78 | 0.27 | |||||
| Chicken egg shell 53 | CaCO3 | MgCO3 | Ca3(PO4)2 | Organic matter | |||||||
| 94 | 1 | 1 | 4 | ||||||||
| Shrimp shell 67 | C | H | N | S | O | Ca | P | ||||
| 25.93 | 3.92 | 8.29 | 0.69 | 30.68 | 22.68 | 0.86 | |||||
| Rice huskc77 | SiO2 | K2O | CaO | P2O5 | Al2O3 | Fe2O3 | |||||
| 99.76 | 0.01 | 0.01 | 0.07 | 0.10 | 0.03 | ||||||
| Rice husk ashc77 | SiO2 | K2O | CaO | P2O5 | Al2O3 | Fe2O3 | LOI a | ||||
| 95.62 | 0.17 | 0.12 | 0.14 | 0.10 | 0.01 | 3.75 | |||||
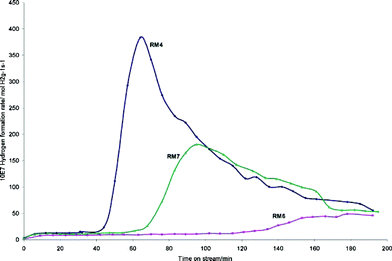 | ||
| Fig. 1 Mass normalised hydrogen formation rates during CH4 cracking at 800 °C over three red mud samples of Indian origin (reproduced from ref. 1 with kind permission from the Royal Society of Chemistry.). | ||
 | ||
| Fig. 2 Transmission electron micrograph showing the graphitic carbon formed by methane cracking over red mud at 800 °C (adapted from ref. 12). | ||
 | ||
| Fig. 3 Transmission electron micrograph showing carbon nanotubes formed by butane cracking over red mud at 800 °C (adapted from ref. 12). | ||
Whilst the activity of red mud has been demonstrated in a range of applications, a significant concern is the degree of variability in its composition, both with respect to its source and the time at which it is sourced. The degree of sensitivity of performance dependent upon composition is therefore a significant consideration, although this may be somewhat offset in some cases by its low cost. Pre-treatment procedures aimed at reducing this concern/selectively by extracting the active phases, or their precursors, may be possible.
(ii) Aluminium dross
Aluminium dross is a by-product of aluminium manufacture and recycling. It is generated by the reaction of molten aluminium with air where surface phases of aluminium oxide and nitride form. The dross is classified into “white” and “black” grades depending upon its metallic aluminium content, with the latter comprising a lower aluminium and higher salt content.20 Salt cake is also produced in secondary aluminium processes and contains a still lower aluminium content (typically 5–10% Al.) White dross, which typically comprises around 80 wt% metallic aluminium, is formed in the Hall–Heroult process of aluminium manufacture in which alumina obtained from the Bayer process is dissolved in molten cryolite and electrochemically processed to yield metallic aluminium.20 Black dross, which typically contains 5–20% Al, is a by-product of aluminium recycling and secondary processing of white dross. An example of the metal composition of an aqua regia leached dross is given in Table 2.21 Annually, around 5 million tonnes of dross are produced globally of which about four million tonnes are white dross.22 Whilst some dross is recycled for use as a deoxidizer in steel manufacture, much of the rest is disposed of by landfill where, for some waste products of the aluminium industry, there can be concerns over leaching of components and concomitant contamination of groundwater, as well as hydrolysis yielding noxious gases such as NH3. Therefore, there is great incentive to develop uses of dross and associated by-products. In terms of catalytic application, there have been a number of reports of dross being applied as an alternative source of aluminium for the preparation of catalytically active materials. Examples can be found which involve the direct use of dross for this purpose, as well those which detail the extraction of the aluminium containing components. The dross detailed in Table 2 has been directly applied to the synthesis of the catalytically interesting zeotype AlPO4-5 when reacted with phosphoric acid followed by adding triethylamine as a structure-directing agent.21Powder X-ray diffraction evidenced a number of additional phases including berlinite (non-porous AlPO4), quartz and alumina. The quartz and alumina phases are present in the original dross and the absence of aluminium metal in the powder pattern was interpreted as indicating that it was completely consumed in synthesizing the AlPO4-5. Complete consumption of aluminium metal from dross in the synthesis of AlPO4-5 has also been described in another study in which comparisons were made with materials prepared using Al(OH)3 and aluminium extracted from dross.23 When using dross directly, no impurity phases were evident in the resultant diffraction patterns. However, SEM examination indicated larger crystallite sizes when compared to the other two aluminium sources. This was ascribed to seeding by the impurities present in the dross during crystallisation. The same study also detailed the preparation of CrAPO-5 (the chromium containing analogue of AlPO4-5) using the same three aluminium precursors. Again, the dross derived sample comprised larger crystallites and the chromium content was observed to be lower than for the other two samples. Comparison of the activities for tetralin oxidation using tertiary butyl hydroperoxide as oxidant indicated the dross derived material to be less effective for tetralone production, which was attributed to its lower chromium content. Sulfuric acid leaching of dross followed by hydrolysis with aqueous ammonia has been demonstrated to yield aluminium hydroxide, which can then be calcined to yield η-Al2O3, a transition alumina of catalytic interest.24 In that study, washing the dross was shown to be beneficial – it enhanced alumina recovery and could also be used to recover salt for recycling.It is clear that dross can be used as an alternative source of aluminium for the preparation of materials of catalytic interest. However, when used directly, the presence of impurity phases or their effects on, for example, the crystallite size of the target material are considerations. Extraction of the aluminium component can be undertaken at the expense of increasing the number of stages in the process.
(iii) Fly ash
Fly ash is a residue generated by combustion that consists of fine particles with a composition which reflects that of the inorganic components of the particular fuel being burned and the combustion technology applied. It is therefore a heterogeneous material with a variable composition, dependent upon source and process operation, which predominantly comprises Al2O3 and SiO2 along with additional components such as Fe2O3, MgO, CaO, K2O and Na2O. The lighter particles produced consist of censospheres (hollow ceramic microspheres). An example of the composition of a coal fly ash from China is shown in Table 2.25 As would be expected, fly ash is a significant by-product of thermal power generation and its predominant disposal route is by means of landfill or dumping at sea. This results in obvious concerns which are particularly apparent when the scale of ash generation is considered. For example, in 2003 it was reported that 430 million tonnes of fly ash was generated globally.26 Utilisation of this residue is possible and various routes, including the manufacture of cement, concrete, ceramics, zeolites and adsorbents, have been documented.26 Among the high value applications of fly ash, its use as a catalyst and catalyst support is an active area of investigation which has been reviewed recently.26 Due to its silica and alumina content, several studies have examined the use of coal fly ash as a support.25,27–29 To enhance the surface area of the fly ash, in many cases thermal or chemical treatment has been undertaken prior to the loading of active phases. An example of this procedure involves the application of steam treated and nitric acid activated fly ash, which was used as a support for copper, nickel and iron catalysts, which were subsequently applied to the environmentally important selective catalytic reduction of NOx using NH3.25 As would be anticipated, the nature of the pre-treatment of fly ash will influence its acid–base properties. In a study illustrative of this, thermal treatment, acid treatment and a combination of both were compared for Ru loaded fly ash catalysts applied to ammonia decomposition. Thermal treatment was found to yield the most active catalyst, which was attributed to higher surface loading of Ru, stronger adsorption of NH3 and lower acidity.27 In relation to acidity, Ni loaded on fly ash was tested for CO2 reforming of methane and a comparison made between the effects of acid and base pretreatment upon resultant performance, with pre-treatment with CaO being observed to be most effective.29Calcium oxide treatment of thermally treated fly ash has also been shown to be effective for the generation of active catalysts for the base catalysed Knoevenagel reaction in which the resultant catalyst was found to comprise quartz, mullite and calcium silicate phases.30 Fly ash has been used as a catalyst after base treatment for transesterification reactions. Catalysts prepared by potassium nitrate wet impregnation on fly ash followed by calcination were tested for sunflower oil transesterification. Under optimum conditions up to 87.5% oil conversion was obtained.31 Another study using potassium nitrate impregnated fly ash reported a similar conversion (82.3%).32Since silica and alumina are major components of fly ash, it is perhaps not surprising that a number of studies have investigated the application of fly ash for the preparation of zeolites. The sodalite unit based zeolites A and Faujasite, as well as zeolite P1, have been reported to be formed by treatment of coal fly ash by alkaline solutions in a process which has been described as being analogous to the formation of natural zeolites from volcanic deposits.33 For example, a hydrothermal method has been employed which was based on a NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fly ash (1:3) composition to generate zeolites which were applied to heavy crude residue cracking and phenol alkylation with tertiary-butyl alcohol.34,35 The performance of the catalysts generated from fly ash, which had XRD patterns predominantly assigned to NaX zeolite, was reported to be comparable to commercial zeolite for both these reactions. Zeolite Y prepared from fly ash was reported to show good activity for cumene cracking in another study,36 whilst fly ash generated zeolite and amorphous silica alumina catalysts have been prepared and found to be active for pyrolysis of waste plastic.37,38
:fly ash (1:3) composition to generate zeolites which were applied to heavy crude residue cracking and phenol alkylation with tertiary-butyl alcohol.34,35 The performance of the catalysts generated from fly ash, which had XRD patterns predominantly assigned to NaX zeolite, was reported to be comparable to commercial zeolite for both these reactions. Zeolite Y prepared from fly ash was reported to show good activity for cumene cracking in another study,36 whilst fly ash generated zeolite and amorphous silica alumina catalysts have been prepared and found to be active for pyrolysis of waste plastic.37,38
The iron oxide content in fly ash has been utilised for certain applications. In a recent study, amorphous iron hydroxide was obtained from a high iron containing fly ash by means of HCl treatment followed by precipitation with NaOH. The resultant catalyst comprised amorphous iron hydroxide supported on acidified fly ash and showed almost total degradation of methyl orange in a photo-Fenton process.39 In an earlier study on methane steam reforming and the water gas shift reaction using fly ash catalyst, the activity shown was attributed to the high iron content in the fly ash.40 In a study reporting the production of industrial grade multiwalled carbon nanotubes, an iron component has purposely been added to the fly ash.28 In this study, a 5 wt% Fe impregnated fly ash catalyst reportedly generated an 82.5% yield with respect to metal loading when using ethylene as a carbon source.
Thus it is seen that the composition of coal fly ash makes as a catalyst support phase and also a feedstock for the preparation of zeolites. In instances where the iron oxide content is high, it can be extracted or the fly ash by itself can be used for applications where iron/iron oxide is active. Also, in addition to coal fly ash, wood fly ash and municipal incinerator fly ash have been used as catalysts, mainly for oxidation applications.41–44
(iv) Others
In addition to the materials discussed above, there are other wastes that have been investigated as catalysts in a number of studies. Waste sludges from sewage treatment and industrial sources (tannery, electroplating, textile dyeing, steel electropolishing, using different mechanical and thermal pre-treatment) have been applied to propane oxidation in which the iron, copper and chromium contents impart activity.45 The waste generated from the processing of the negative electrode for Ni metal hydride batteries was subjected to a series of chemical treatments to isolate NiO, CeO2 and LaCoO3.46 Among these components, NiO showed the highest activity for methane reforming and good stability, which was attributed to sintering resistance resulting from the small amounts of La and Co present. Steel dust after acid treatment showed good activity for Fenton like oxidation of polychlorinated dibenzo-p-dioxins (PCDDs).47 A chromium containing catalyst was prepared by spark erosion of wastewater from an electroplating plant that was rich in chromium. This catalyst was tested for VOC oxidation and showed up to 74% conversion for ethanol oxidation.48 In another study, copper contained in printed circuit boards was leached and deposited on a TiO2 suspension cathode using an electrokinetic process. Prior to this step, the printed circuit board was subjected to supercritical water oxidation for removal of brominated flame retardants. The catalyst showed good photocatalytic activity for methylene blue degradation.49Blast furnace and steel slags are major waste products originating from iron manufacture and processing.50,51 It has been reported that the global production of slag during 2008 was in the range of 240–290 million tons.51 The composition of a typical raw blast furnace slag is reported in Table 2.51 Whilst the SiO2 and Al2O3 content appears to be of interest for the preparation of aluminium rich zeolites, the presence of such large amounts of CaO is reported to be problematic. It has been reported that materials containing in excess of 15 wt% CaO are unsuitable for zeolite synthesis because of the formation of calcium silicate, which leads to inhibition of zeolite nucleation.50 In order to circumvent this problem, as an alternative to acid treatment to eliminate the undesirable components for zeolite synthesis,52 a two stage procedure has been enacted leading to the production of a hydroxyapatite – zeolite X composite material. In the early stages of the preparation, the magnesium and calcium components were reacted with H3PO4 and then NaOH was added. After prolonged aging (2 days) the composite material resulted, and P1 zeolite could be further formed at the expense of the Faujasite component by modification of aging/crystallization time.50 It was reported that the minor components present in the slag had little effect on the synthesis of the composite material. In an extension to this study, the synthesis of a hydrotalcite-like compound and separate zeolite phase was successfully achieved.51 On treatment of the slag with HCl, the dissolved phases and residual silica were used to prepare a hydrotalcite-like compound and zeolite by further chemical treatment of the leaching solution and residual silica respectively. The process steps, along with those employed for the preparation of the hydroxyapatite-zeolite composite, are summarized in Fig. 4.51
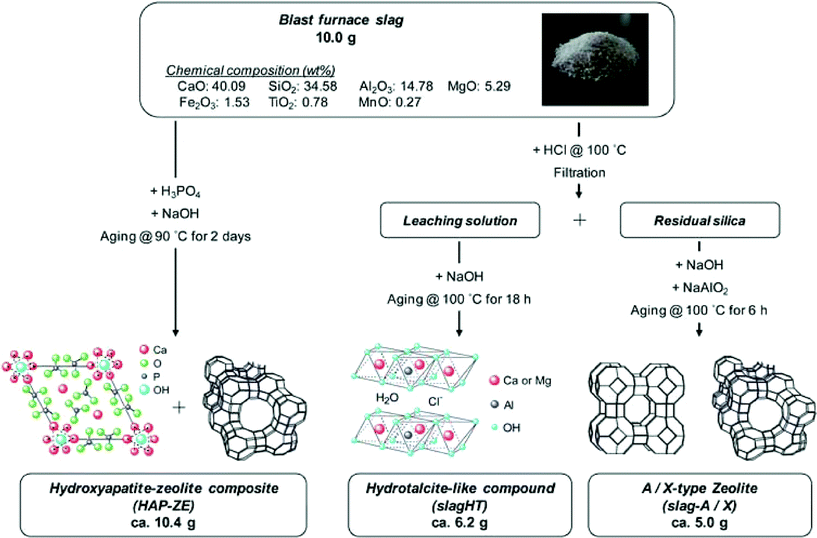 | ||
| Fig. 4 Possible blast furnace slag conversion processes (reproduced from ref. 51 with kind permission from the Royal Society of Chemistry). | ||
Wastes originating from biological sources
(i) Shells
Recently, there have been a number of reports detailing the application of waste shells to the preparation of catalysts. It has been reported that the shell constitutes about 10% by mass of a hen egg and that around 4 million tonnes per annum of egg shell waste is produced in China alone.53 Disposal of these shells is mainly by landfill dumping. There is interest in their use as they can be thermally decomposed at high temperatures to generate CaO from the decomposition of CaCO3 which is their major consitutent, as reported in Table 2.53CaO based catalysts are currently of interest as solid bases for transesterification to produce biodiesel.54 Within the literature, it has been reported that the catalyst generated by high temperature decomposition of chicken egg shells is active for biodiesel production from methanol and soybean oil. Using 3 wt% egg shell derived catalyst prepared by calcination at 1000 °C, a biodiesel yield of >95% after 3 h was obtained using a 9:1 molar ratio of ethanol to oil.53 The catalysts were found to be durable, maintaining their activity for 13 reaction runs, although total deactivation (reversible by calcination) occurred after 17 runs due to Ca(OH)2 formation. Textural considerations are of potential importance in such catalysts. This is illustrated in a study where a comparison of the application of catalysts derived from waste egg shells and waste mollusk shells (from golden apple snail and venus meretrix) have been compared for biodiesel production.55 Despite having a similar elemental composition, catalysts derived by calcination of egg shells were found to be more active for production of biodiesel from palm olein oil and methanol than those derived from golden snail shells which were in turn more active than those derived from venus meretrix. In all cases, highly crystalline CaO was reported to be the predominant phase and the order of activity was explained on the basis of surface area and particle size, with the egg shell derived catalyst having the highest area (1.1 m2 g−1). Using edible oil reactants as in the previous two studies, egg shells have also been applied to biodiesel production using karanja oil which is a non-edible feedstock with a high free fatty acid value.56 Following initial esterification with methanol catalysed by H2SO4 to reduce the acidity of the feedstock, egg shell derived catalysts were employed with a 95% yield of biodiesel being achieved. The resultant biodiesel was shown to meet the desired specifications, although low oxidation stability, which would necessitate the addition of antioxidants for long term storage, was a concern, as were the temperature constraints dictated by the cloud point and pour point values. A comparison of the behaviour of two different shells – chicken egg and quail egg – as precursors to catalysts for the transesterification of palm oil has been recently made.57 Quail egg shells were found to be more active and this was explained on the basis of the texture of the palisade layer of the shell leading to a greater number of strong base sites in the catalysts formed. Furthermore, removal of the dense cuticle layer with diluted acid prior to calcination resulted in catalysts which possessed high catalytic activities comparable to homogeneous potassium methoxide. Other waste sources of CaO which have been applied for biodiesel production include oyster shells58 and mud crab shells.59,60 The amount of waste generated in the latter case is exacerbated because the crabs moult shedding their shells in order to grow.
Crab shells are a source of chitin, poly-β(1→4)-N-acetyl-D-glucosamine. Its structure is shown in Fig. 5. Chitin is also found in the shells of other crustaceans, as well as in the exoskeletons of insects and as a component of fungal cell walls. It is, after cellulose, the second most abundant biopolymer with an estimated annual production rate of 1011 tonnes per year.61 It is therefore a highly abundant natural resource. For example, the estimated annual production of waste from the Malaysian fishery industry alone is of the order of 70 000 tonnes.62 There are a number of large scale applications of chitin, or its derivatives, which include those in the pharmaceutical industry, agriculture and the food industry. Generally, chitin is employed in its deacetylated form, chitosan, where the degree of deacetylation is often of the order of 90% or more.61 Chitosan is more stable in some environments and can be more easily cast for the various different applications. For some applications cross-linking is applied which increases stability under different solution conditions, e.g.ref. 63. The structure of chitosan is also shown in Fig. 5.
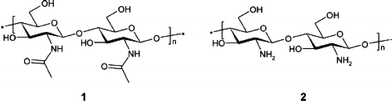 | ||
| Fig. 5 Structure of (1) chitin and (2) fully deacetylated chitin (reproduced from reference ref. 61 with kind permission from the American Chemical Society). Chitosan is formed by incomplete (approximately 90%) deacetylation of chitin and so is an intermediate of 1 and 2. | ||
A number of applications of chitin or chitosan based materials as supports for catalytic applications are detailed in the literature.64–66 Applications of shells as sources of chitin have also been reported. For example, incomplete carbonization of shrimp shells followed by loading of KF has been shown to yield active catalysts for biodiesel synthesis from rapeseed oil.67 The elemental composition of the shrimp shells used in this study is presented in Table 1 and it was reported that chitin constitutes 69% of the dry weight of shrimp shell. The carbonization process was reported to increase the surface area yielding a porous framework material. A similar pre-treatment of biont shells (i.e. incomplete carbonization and KF impregnation) was also reported to yield active catalysts for the same reaction.68 Since chitosan contains –NH2groups, it can be anticipated that it will have some activity for base catalysed processes. It has been shown to be an active catalyst for the solvent-free synthesis of jasminaldehyde by the condensation of 1-heptanal and benzaldehyde.69 It has also recently been reported to be active for asymmetric direct aldol reactions in water when applied in the form of an aerogel.70 It has been stated that chitosan aerogels with surface areas up to 350 m2 g−1 and high accessibility to the –NH2groups can be prepared. Surface modification of chitosan can also be enacted allowing different catalytic applications to become accessible.61 Prawn shells have also been used for the preparation of nitrogen doped mesoporous carbons.71 The shells were initially hydrothermally treated in an autoclave, and then calcined under nitrogen to yield an organic-inorganic composite from which the inorganic component (CaCO3) was removed by treatment with acetic acid.
It is clear that abundant natural resources such as egg shells and prawn shells, which are major waste products, are of interest for potential catalytic applications. This interest centres upon both the “inorganic” and “organic” components of these resources. In the former case, the wastes are an alternative source of CaO, a base catalyst, whereas in the latter they are a source of chitin/chitosan which is of potential application as both a support and/or a catalytic material in its own right. In selecting the most appropriate resource for the desired product, obviously attention should be paid to the composition of the shell in question. For example, crustacean shells in general have a much higher organic content than sea shells, meaning that they are a more appropriate starting point for the preparation of carbon based products.
(ii) Plants
The potential application of waste products originating from plant/tree sources to derive catalytic materials is widespread and varied. For example, it has been reported that almost one million tonnes per annum of microporous activated carbons are used in various applications, including as catalyst support materials, and that these are derived from coconut shells, wood and fruit stones. Corn stalks have recently been used for the preparation of microporous carbons with surface areas reported to be > 3000 m2 g−1 in a four step method, whereas the use of rice husk, pine cone needles and pine cone hull led to materials with lower surface area.72 A route to mesoporous carbon materials based upon pectin has been recently reported.73 This route is also of relevance to the application of waste materials, since pectin is a naturally occurring polysaccharide which can be derived from waste fruit skins.Rice husk is the indigestible hard protective coating of rice grains which is removed during processing. It is a major agricultural waste product which, amongst other things, can be used as a fuel generating ash as a waste product. Various estimates of annual global production of rice husks and rice husk ash can be found in the literature. For example, annual rice husk production of more than 40 million tonnes was reported in 199274 and it has recently been reported that about 70 million tonnes of rice husk ash is produced yearly worldwide.75 Rice husks are composed of cellulose, lignin and silica and it is the latter component that has generated most interest. Low levels of additional inorganic components can also be found in rice husk and, whilst the concentration of these may vary with location, these influences appear to be relatively small.76 Typical composition of the inorganic materials in rice husk and rice husk ash materials are presented in Table 2.77 Rice husks and their ash have attracted interest in terms of the extraction of the SiO2 component and this can act as a source for the synthesis of catalytically important materials such as zeolites and mesoporous silicates. The forms of silica which can be produced and their particle size control have attracted attention with chemical pre-treatment and/or controlled atmosphere calcination being employed as the methods of extraction in some studies. For example, the preparation of amorphous silica with a surface area of 321 m2 g−1 has been reported in one study which has involved a combination of chemical pre- and post-treatments and calcination procedures.78Silica supports from rice husk have been prepared using sol–gel method.79–81 The rice husk was dissolved in sodium hydroxide and the gel was formed by acid addition, which upon calcination yielded amorphous high surface area silica (626 m2 g−1).81 These supports were used with transition metal catalysts and tested for oxidation and Friedel–Crafts benzylation reactions. Silica obtained by chemical treatment, pyrolysis and calcination in air was high in purity and used as a support for transition metal catalysts.82,83 The supports prepared from rice husk gave better performance compared to commercial supports for CO2 hydrogenation and methane partial oxidation. This was attributed to better catalyst dispersion82 and higher catalyst surface area.83 The influence of the method of silica extraction upon the form/usefulness for zeolite synthesis has been investigated.84 Highly active amorphous silica suitable for the synthesis of zeolite A and zeolite Y was prepared by burning rice husk in air at 700 °C for 2 h, whereas calcination at 1000 °C led to the formation of cristobalite and tridymite in addition to amorphous material and was not a suitable source for zeolite synthesis. Cristobalite, tridymite and amorphous material were also formed in rice husk which was prepared by open field burning without temperature control for 24 h. This was again unsuitable for zeolite synthesis. Rice husk has been used as a silica source for the preparation of Pt–Fe/MCM-41 catalysts which have been applied to phenol oxidation.85 Both rice husk and rice husk ash derived silicas have been employed in the synthesis of MCM-41 materials in which hydrophobicity was subsequently varied using silanes. Rice husk ash obtained from an industry using rice husk as a fuel was used as a support for iron and tested for dye decolourisation using a Fenton-like process.86
Rice husk has also been used as a precursor for preparing carbon honeycomb monoliths which have potential as catalyst supports. The monoliths prepared by extrusion of carbonized rice husk along with clay binder had a high surface area after alkali activation (more than 300 m2 g−1) and good mechanical strength.87 Since rice husks also contain an organic component, routes to reacting the organic and silica components can be envisaged. For example, this can be used to offset/partially offset the amount of carbon reductant required to prepare Si from rice husk.76 Related to this idea, it has been reported that rice husk can be used to prepare high surface area (150 m2 g−1) β-SiC.88SiC is an interesting support material combining chemical inertness with high thermal stability. In this method, semi-ground rice husk was charred at 550 °C for 2 h in N2 and the resultant product was pyrolyzed by plasma processing.
In the above examples, the application of high volume waste plant derived materials to the generation of materials of interest for catalytic applications has been demonstrated. As for the earlier discussion of the use of shells, examples have been given of interest in both organic (fruit skins) and inorganic (rice husks) components of waste. In the case of rice husks potential reactions between both inorganic and organic components can be exploited in the synthesis of materials.
Conclusion
High volume waste materials from both large scale industrial and natural sources are attracting increasing interest in terms of catalysis. In some instances, the materials can be directly applied as catalysts themselves, whereas in others they are used as sources of components from which materials of catalytic interest can be prepared. For materials directly applied as catalysts, consideration needs to be given to the potential influence of compositional variation upon activity where this may be an issue. The predominant interest in waste materials does not lie so much in the provision of solutions to disposal problems, since an obvious paradox from their use as/in the generation of catalysts arises in this respect. It lies in the availability of low cost alternatives to existing catalysts or as precursors in their preparation. Accordingly, the successful application of waste materials will be a balance between the economics of any associated activity differences and/or processing costs. As can be seen for the selected examples presented in this overview, it is clear that this is an interesting area of research which is increasing in importance.Acknowledgements
The authors would like to express their appreciation to the British Council for the generous award of a UKIERI project grant in the area of the utilization of red mud (project SA07/19). The authors would also like to thank Miss Caiomhe Moore and Mr Azlan Kamari for useful discussions.References
- M. Balakrishnan, V. S. Batra, J. S. J. Hargreaves, A. Monaghan, I. D. Pulford, J. L. Rico and S. Sushil, Green Chem., 2009, 11, 42–47 RSC.
- Y. Pontikes, C. Rathossi, P. Nikolopulos, G. N. Angelopoulos, D. Jayaseelan and W. E. Lee, Ceram. Int., 2009, 35, 401–407 CrossRef CAS.
- S. B. Wang, H. M. Ang and M. O. Tade, Chemosphere, 2008, 72, 1621–1635 CrossRef CAS.
- R. K. Paramguru, P. C. Rath and V. N. Misra, Miner. Proces. Extr. Metall. Rev., 2004, 26, 1–29 Search PubMed.
- S. Sushil and V. S. Batra, Appl. Catal., B, 2008, 81, 64–77 CrossRef CAS.
- K. C. Pratt and V. Christoverson, Fuel, 1982, 61, 460–462 CrossRef CAS.
- S. Yokoyama, M. Yamamoto, Y. Maekawa and T. Kotanigawa, Fuel, 1989, 68, 531–533 CrossRef CAS.
- J. A. Lagaretta, B. M. Caballero, I. Marco, M. J. Chomon and P. M. Uria, Fuel, 1997, 76, 1309–1313 CrossRef.
- S. Ordonez, H. Sastre and F. V. Diez, J. Hazard. Mater., 2001, 81, 103–114 CrossRef CAS.
- J. R. Paredes, S. Ordonez, A. Vega and F. V. Diez, Appl. Catal., B, 2004, 47, 37–45 CrossRef CAS.
- A. Sinag, M. Sungur and M. Canel, Energy Fuels, 2006, 20, 1609–1613 CrossRef CAS.
- S. Sushil, A. M. Alabdulrahman, M. Balakrishnan, V. S. Batra, R. A. Blackley, J. Clapp, J. S. J. Hargreaves, A. Monaghan, I. D. Pulford, J. L. Rico and W. Zhou, J. Hazard. Mater., 2010, 180, 409–418 CrossRef CAS.
- O. M. Dunens, K. J. MacKenzie and A. T. Harris, Carbon, 2010, 48, 2375–2377 CrossRef CAS.
- A. A. S. Oliveira, I. F. Teixeira, L. P. Ribeiro, J. C. Tristao, A. Dias and R. M. Lago, J. Braz. Chem. Soc., 2010, 21, 2184–2188.
- E. Krause and V. Rohm, US Patent 2009/0255371 A1, Oct 15 2009.
- E. Karimi, A. Gomez, S. W. Kycia and M. Schlaf, Energy Fuels, 2010, 24, 2747–2757 CrossRef CAS.
- Z. Gokdai, A. Sinag and T. Yumak, Biomass Bioenergy, 2010, 34, 402–410 CrossRef.
- A. Sinag, M. Sungur, M. Gullu and M. Canel, Energy Fuels, 2006, 20, 2093–2098 CrossRef CAS.
- R. C. C. Costa, F. C. C. Moura, P. E. F. Oliviera, F. Magalhaes, J. D. Ardisson and R. M. Lago, Chemosphere, 2010, 78, 1116–1120 CrossRef CAS.
- M. C. Shinzato and R. Hypolito, Waste Manage., 2005, 25, 37–46 CrossRef CAS.
- N. Murayama, N. Okajima, S. Yamaoka, H. Yamamoto and J. Shibata, J. Eur. Ceram. Soc., 2006, 26, 459–462 CrossRef CAS.
- http://www.smartwaste.co.uk/filelibrary/Cement_aluminium_dross.pdf accessed 21.09.10.
- J. Kim, K. Biswas, K.-W. Jhon, S.-Y. Jeong and W.-S. Ahn, J. Hazard. Mater., 2009, 169, 919–925 CrossRef CAS.
- B. R. Das, B. Dash, B. C. Tripathy, I. N. Bhattacharya and S. C. Dass, Miner. Eng., 2007, 20, 252–258 CrossRef CAS.
- X. Xuan, C. Yue, S. Li and Q. Yao, Fuel, 2003, 82, 575–579 CrossRef CAS.
- S. Wang, Environ. Sci. Technol., 2008, 42, 7055–7063 CrossRef CAS.
- L. Li, S. Wang, Z. Zhu, X. Yao and Z. Yan, Fuel Process. Technol., 2008, 89, 1106–1112 CrossRef CAS.
- O. M. Dunens, K. J. MacKenzie and A. T. Harris, Environ. Sci. Technol., 2009, 43, 7889–7894 CrossRef CAS.
- S. Wang and G. Q. Lu, Stud. Surf. Sci. Catal., 2007, 167, 275–280 CAS.
- D. Jain, C. Khatri and A. Rani, Fuel Process. Technol., 2010, 91, 1015–1021 CrossRef CAS.
- M. S. Kotwal, P. S. Niphadkar, S. S. Deshpande, V. V. Bokade and P. N. Joshi, Fuel, 2009, 88, 1773–1778 CrossRef CAS.
- O. O. Babajide, L. Petrik, N. Musyoka and B. Amigun, AIChE Annual Meeting, 2010.
- G. G. Hollman, G. Steenbruggen and M. Janssen-Jurkovicova, Fuel, 1999, 78, 1225–1230 CrossRef CAS.
- K. Ojha and N. C. Pradhan, Indian J. Eng. Mater. Sci., 2001, 8, 100–103 Search PubMed.
- K. Ojha, N. C. Pradhan and A. N. Samanta, J. Chem. Technol. Biotechnol., 2006, 81, 659–666 CrossRef CAS.
- P. Kumar, Y. Oumi, T. Sano and K. Yamana, J. Ceram. Soc. Jpn., 2001, 109, 968–973 CAS.
- S. S. Kim, J. H. Kim and S. H. Chung, J. Ind. Eng. Chem., 2003, 9, 287–293 Search PubMed.
- J.-G. Na, B.-H. Jeong, S. H. Chung and S.-S. Kim, Journal of Material Cycles and Waste Management, 2006, 8, 126–132 Search PubMed.
- Y. Li and F.-S. Zhang, Chem. Eng. J., 2010, 158, 148–153 CrossRef CAS.
- W. J. Chen, F. R. Sheu and R. L. Savage, Fuel Process. Technol., 1987, 16, 279–288 CrossRef CAS.
- J. G. P. Born, P. Mulder and R. Louw, Environ. Sci. Technol., 1993, 27, 1849–1863 CrossRef CAS.
- J. R. Kastner, K. C. Das and N. D. Melear, J. Hazard. Mater., 2002, 95, 81–90 CrossRef CAS.
- J. R. Kastner, K. C. Das, Q. Buquoi and N. D. Melear, Environ. Sci. Technol., 2003, 37, 2568–2574 CrossRef CAS.
- J. R. Kastner, Q. Buquoi, R. Ganagavaram and K. C. Das, Environ. Sci. Technol., 2005, 39, 1835–1842 CrossRef CAS.
- F. Klose, P. Scholz, G. Kreisel, B. Ondruschka, R. Kneise and U. Knopf, Appl. Catal., B, 2000, 28, 209–221 CrossRef CAS.
- T. Kanamori, M. Matsuda and M. Miyake, J. Hazard. Mater., 2009, 169, 240–245 CrossRef CAS.
- J.-M. Lee, J.-H. Kim, Y.-Y. Chang and Y.-S. Chang, J. Hazard. Mater., 2009, 163, 222–230 CrossRef CAS.
- O. E. Lebedeva and A. G. Sarmurzina, Appl. Catal., B, 2000, 26, L1–L3 CrossRef CAS.
- F.-R. Xiu and F.-S. Zhang, J. Hazard. Mater., 2009, 172, 1458–1463 CrossRef CAS.
- Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2009, 19, 7263–7272 RSC.
- Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2010, 20, 5052–5062 RSC.
- Y. Kuwahara, T. Ohmichi, K. Mori, I. Katayama and H. Yamashita, J. Mater. Sci., 2008, 43, 2407–2410 CrossRef CAS.
- Z. Wei, C. Xu and B. Li, Bioresour. Technol., 2009, 100, 2883–2885 CrossRef CAS.
- Z. Helwani, M. R. Othman, N. Aziz, J. Kim and W. J. N. Fernando, Appl. Catal., A, 2009, 363, 1–10 CrossRef CAS.
- N. Viriya-empikul, P. Krasae, B. Puttasawat, B. Yoosuk, N. Chollacoop and K. Faungnawakij, Bioresour. Technol., 2010, 101, 3765–3767 CrossRef CAS.
- Y. C. Sharma, B. Singh and J. Korstad, Energy Fuels, 2010, 24, 3223–3231 CrossRef CAS.
- Y. B. Cho and G. Seo, Bioresour. Technol., 2010, 101, 8515–8519 CrossRef CAS.
- N. Nakatani, H. Takamori, K. Takeda and H. Sakugawa, Bioresour. Technol., 2009, 100, 1510–1513 CrossRef CAS.
- P.-L. Boey, G. P. Maniam and S. A. Hamid, J. Oleo Sci., 2009, 58, 499–502 Search PubMed.
- P.-L. Boey, G. P. Maniam and S. A. Hamid, Bioresour. Technol., 2009, 100, 6362–6368 CrossRef CAS.
- D. J. Macquarrie and J. J. E. Hardy, Ind. Eng. Chem. Res., 2005, 44, 8499–8520 CrossRef CAS.
- A. Kamari, personal communication.
- A. Kamari, I. D. Pulford and J. S. J. Hargreaves, Colloids Surf., B, 2011, 82, 71–80 CrossRef CAS.
- S. E. S. Leonhardt, A. Stolle, B. Ondruschka, G. Cravotto, C. De Leo, K. D. Jandt and T. F. Keller, Appl. Catal., A, 2010, 379, 30–37 CrossRef CAS.
- D. Wei, Y. Ye, X. Jia, C. Yuan and W. Qian, Carbohydr. Res., 2010, 345, 74–81 CrossRef CAS.
- A. Primo and F. Quignard, Chem. Commun., 2010, 46, 5593–5595 RSC.
- L. Yang, A. Zhang and X. Zheng, Energy Fuels, 2009, 23, 3859–3865 CrossRef CAS.
- J. Xie, X. Zheng, A. Dong, Z. Xiao and J. Zhang, Green Chem., 2009, 11, 355–364 RSC.
- N. Sudheesh, S. K. Sharma and R. S. Shulka, J. Mol. Catal. A: Chem., 2010, 321, 77–82 CrossRef CAS.
- A. Ricci, L. Bernardi, C. Gioia, S. Verucci, M. Robitzer and F. Quignard, Chem. Commun., 2010, 46, 6288–6290 RSC.
- R. J. White, M. Antonetti and M.-M. Titirici, J. Mater. Chem., 2009, 19, 8645–8650 RSC.
- F. Zhang, H. Ma, J. Chen, G.-D. Li, Y. Zhang and J.-S. Chen, Bioresour. Technol., 2008, 99, 4803–4808 CrossRef CAS.
- R. J. White, V. L. Budarin and J. H. Clark, Chem.–Eur. J., 2010, 16, 1326–1335 CrossRef CAS.
- R. Conradt, P. Pimkhaokham and U. Leela-Adisorn, J. Non-Cryst. Solids, 1992, 145, 75–79 CrossRef CAS and reference therein.
- http://www.ricehuskash.com/product.htm accessed on 21.09.10.
- L. P. Hunt, J. P. Dismukes, J. A. Amick, A. Schei and K. Larsen, J. Electrochem. Soc., 1984, 131, 1683–1686 CAS.
- S. Chiarakorn, T. Areerob and N. Grisdanurak, Sci. Technol. Adv. Mater., 2007, 8, 110–115 CrossRef CAS.
- N. Yalcin and V. Sevinc, Ceram. Int., 2001, 27, 219–224 CrossRef CAS.
- F. Adam, K. Kandasamy and S. Balakrishnan, J. Colloid Interface Sci., 2006, 304, 137–143 CrossRef CAS.
- F. Adam and A. Iqbal, Chem. Eng. J., 2010, 160, 742–750 CrossRef CAS.
- F. Adam and R. Thankappan, Chem. Eng. J., 2010, 160, 249–258 CrossRef CAS.
- F.-W. Chang, M.-S. Kuo, M.-T. Tsay and M.-C. Hsieh, Appl. Catal., A, 2003, 247, 309–320 CrossRef CAS.
- W. S. Chen, F. W. Chang, L. S. Roselin, T. C. Ou and S. C. Lai, J. Mol. Catal. A: Chem., 2010, 318, 36–43 CrossRef CAS.
- H. Hamdan, M. N. M. Muhid, S. Endud, E. Listiorini and Z. Ramli, J. Non-Cryst. Solids, 1997, 211, 126–131 CrossRef CAS.
- J. Chumee, N. Grisdanurak, A. Neramittagapong and J. Wittayakun, Sci. Technol. Adv. Mater., 2009, 10, 015006 CrossRef.
- N. K. Daud and B. H. Hameed, J. Hazard. Mater., 2010, 176, 938–944 CrossRef CAS.
- Z. R. Ismagilov, N. V. Shikina, I. P. Andrievskaya, N. A. Rudina, Z. A. Mansurov, M. M. Burkitbaev, M. A. Biisenbaev and A. A. Kurmanbekov, Catal. Today, 2009, 147, S58–S65 CrossRef CAS.
- S. K. Singh, K. M. Parida, B. C. Mohanty and S. B. Rao, React. Kinet. Catal. Lett., 1995, 54, 29–34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |